
Role of Endocytosis Proteins in Gefitinib-Mediated EGFR Internalisation in Glioma Cells

 ,
,  , ,
, , 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies
2.2. Cell Culture
2.3. Plasmid Transfection
2.4. Fluorescent Quantification of EGFR Endocytosis
2.5. Cell Surface Biotinylation and Endocytosis Assays
2.6. Immunoblotting
2.7. Rab5 Activation Assay
2.8. Confocal Microscopy and Image Analysis
2.9. Spheroid Migration Assays
2.10. Statistical Analysis
3. Results
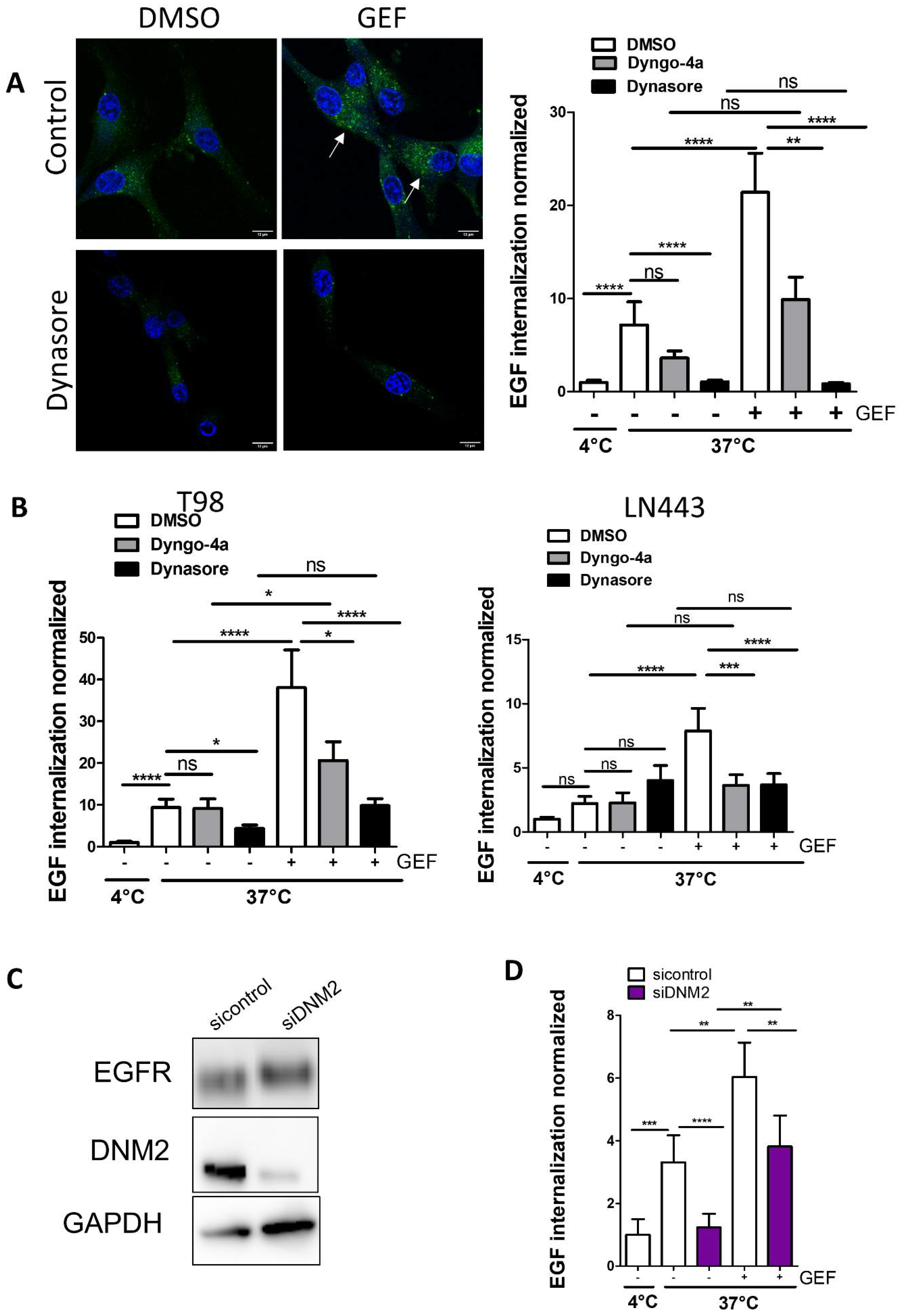
3.1. Knock-Down of DNM2 Decreases Gefitinib-Mediated EGF Endocytosis
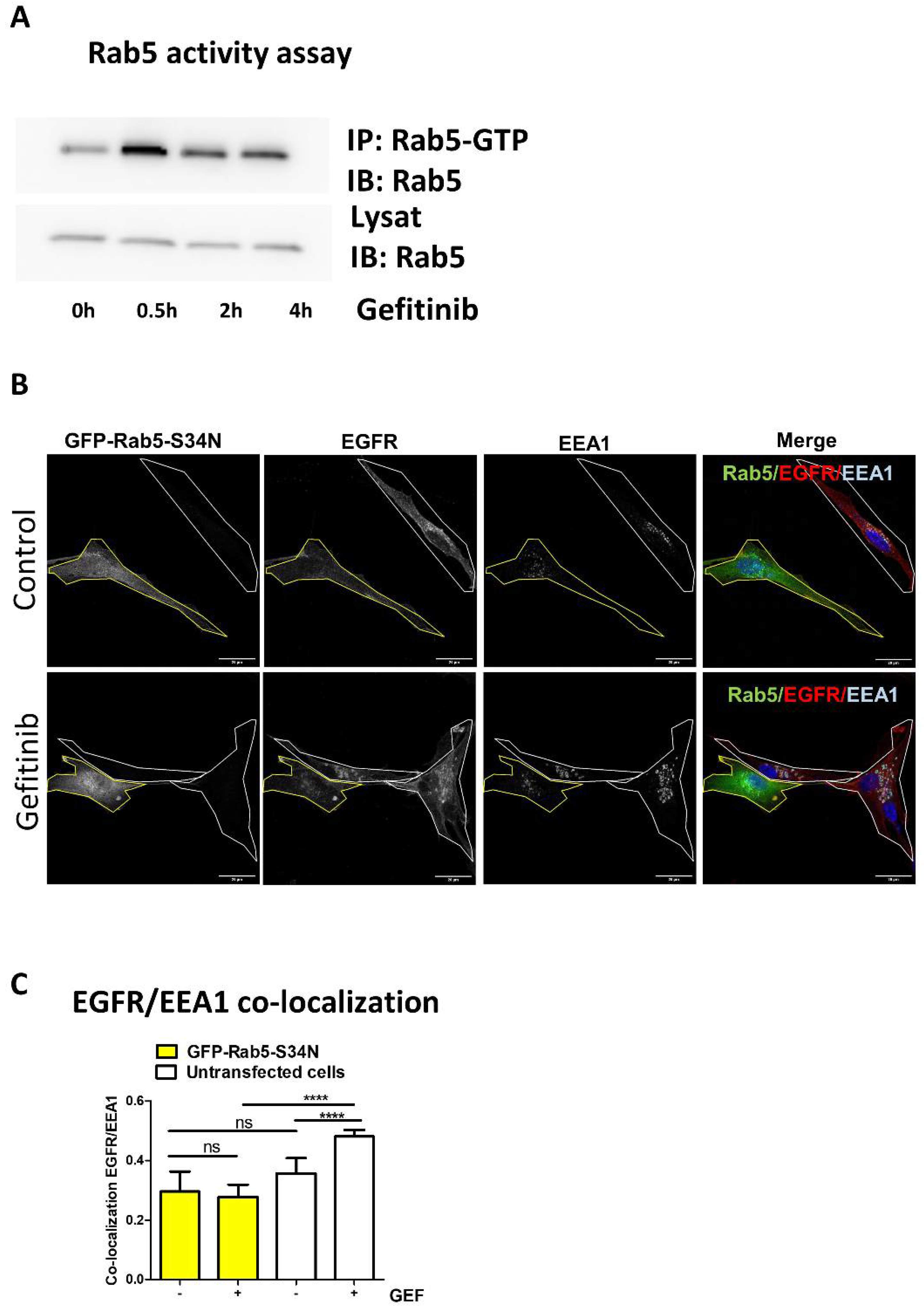
3.2. Gefitinib Activates Rab5 to Promote EGFR Endocytosis
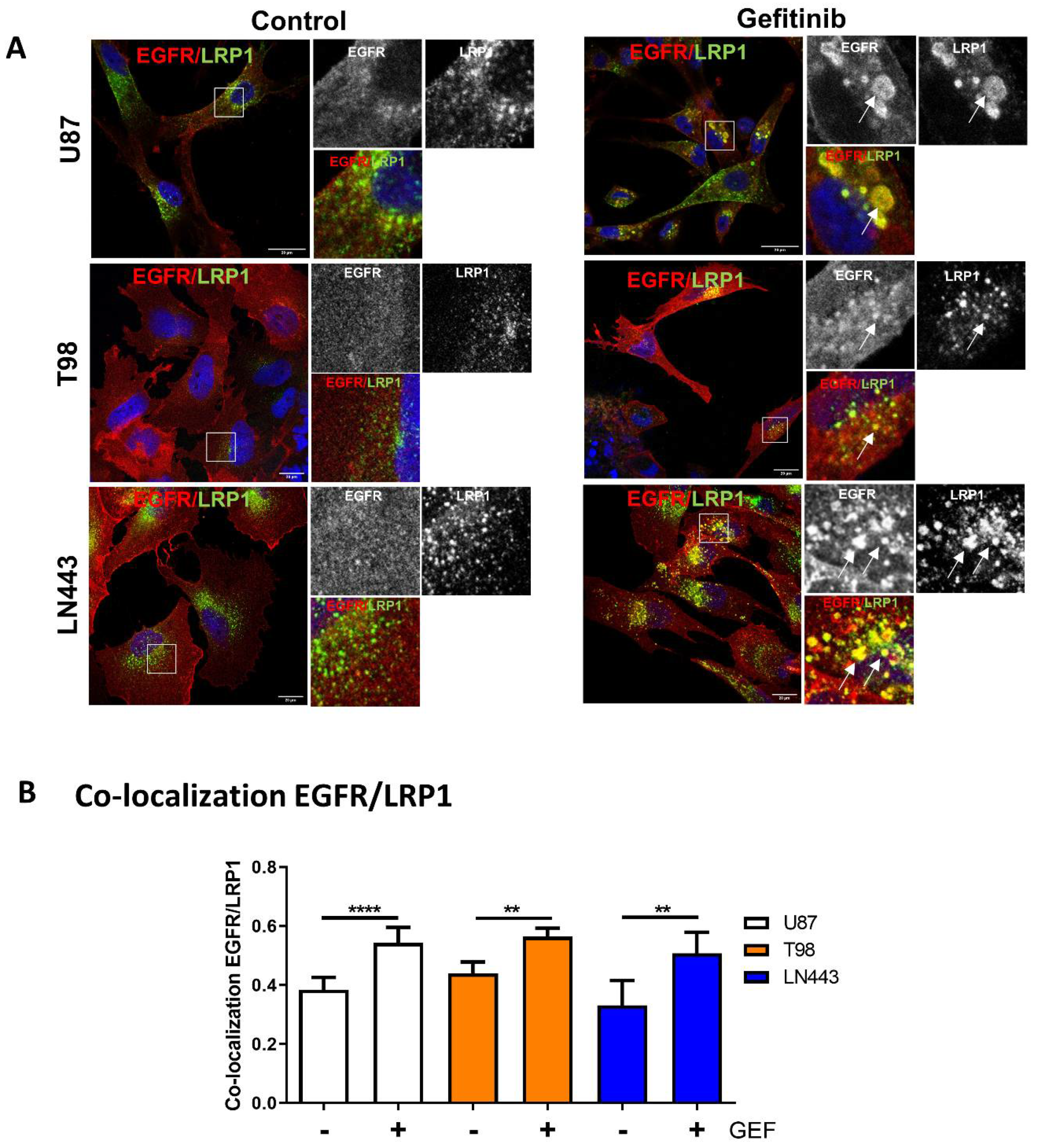
3.3. EGFR Is Recruited into LRP1 Rich Endosomes upon Gefitinib Treatment
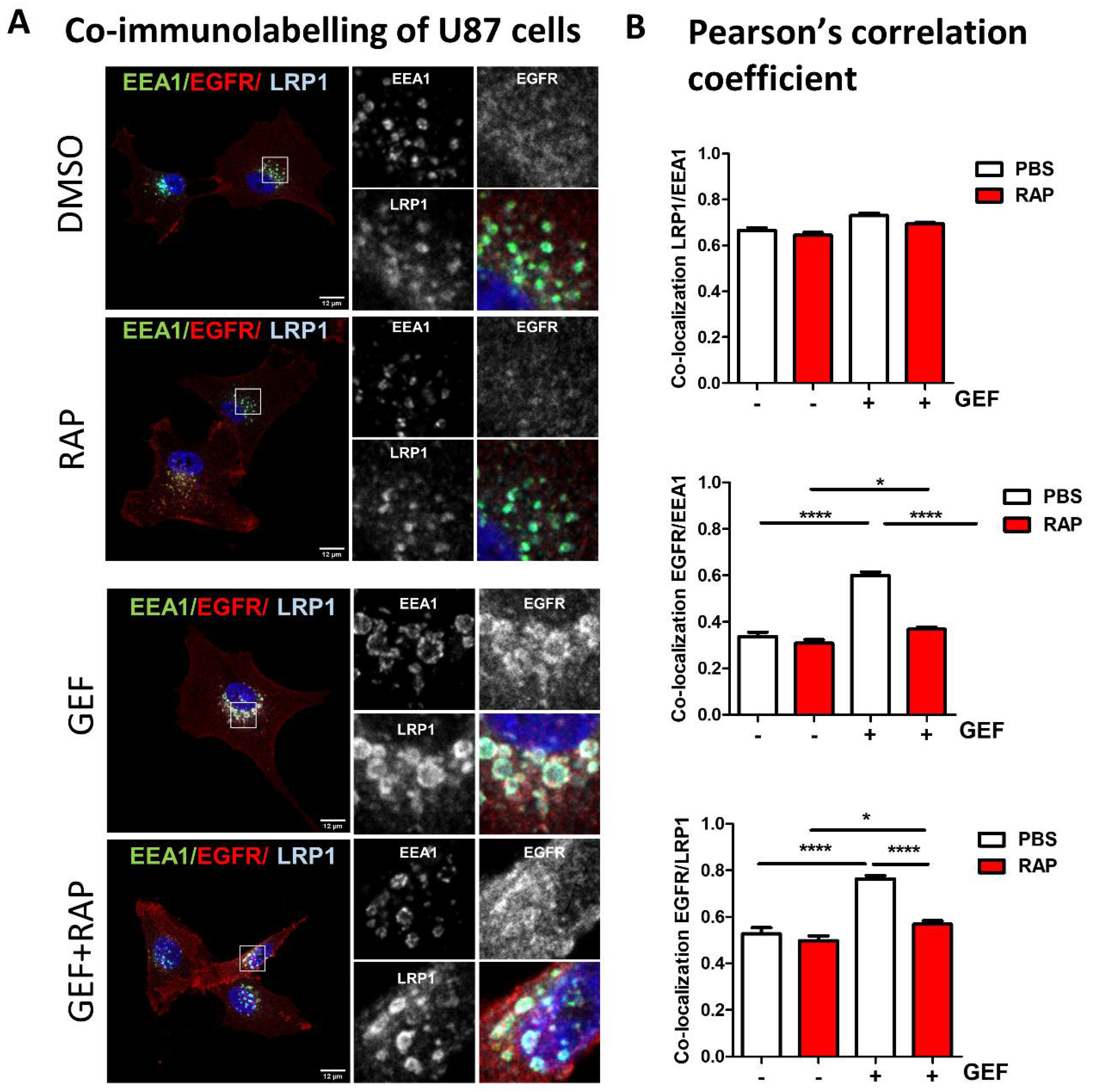
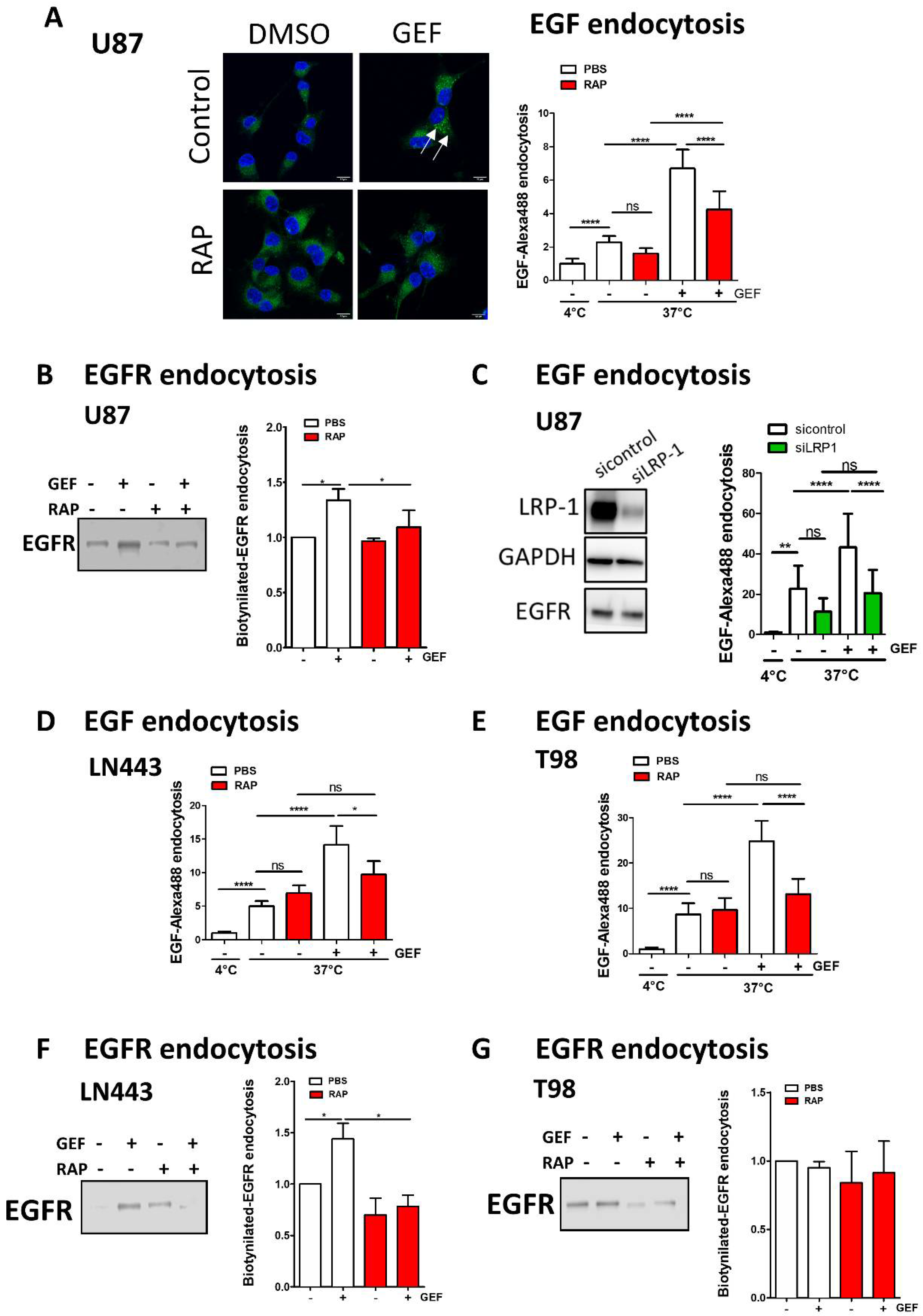
3.4. LRP-1 Is Involved in Gefitinib-Mediated EGFR Endocytosis
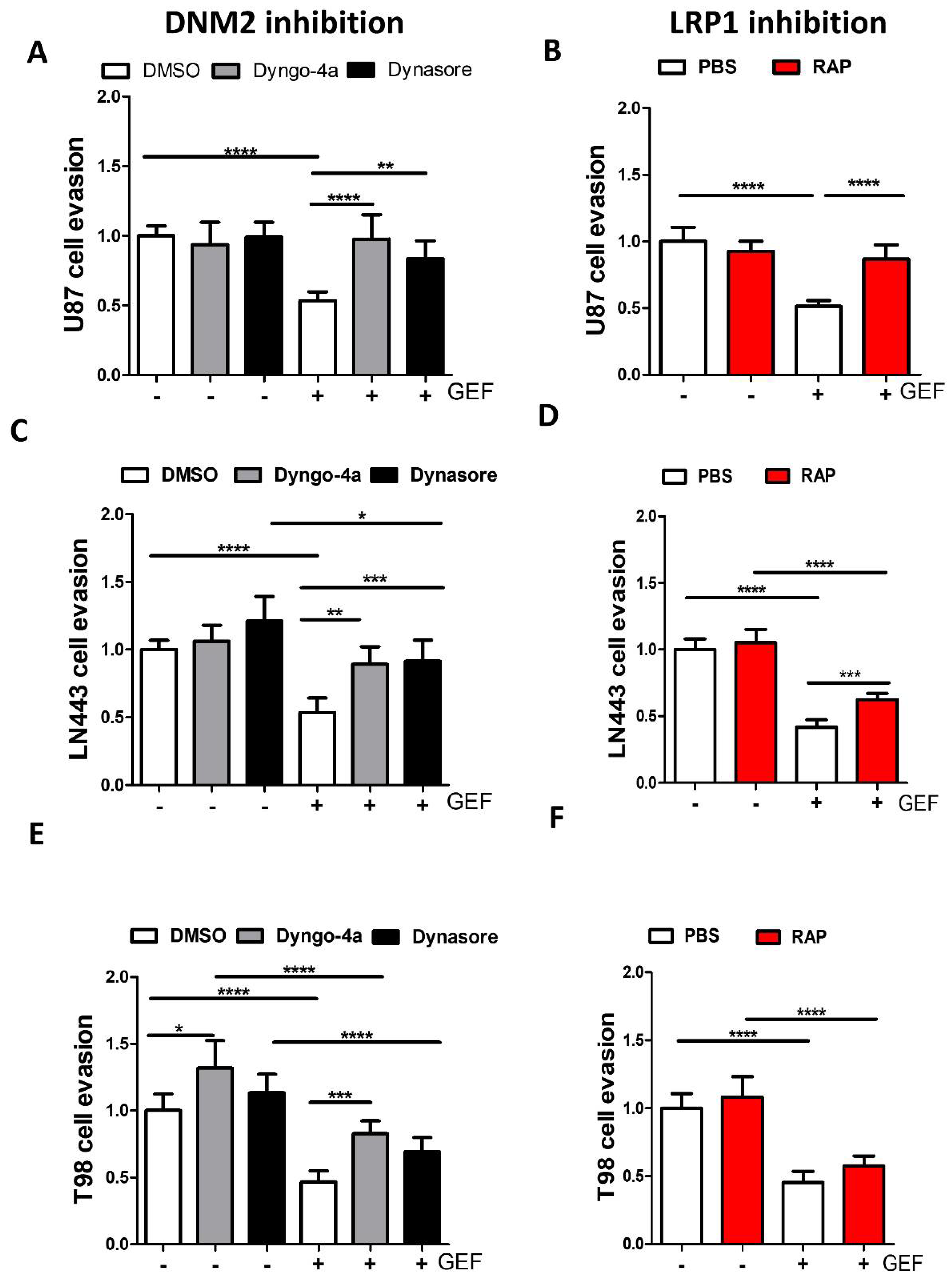
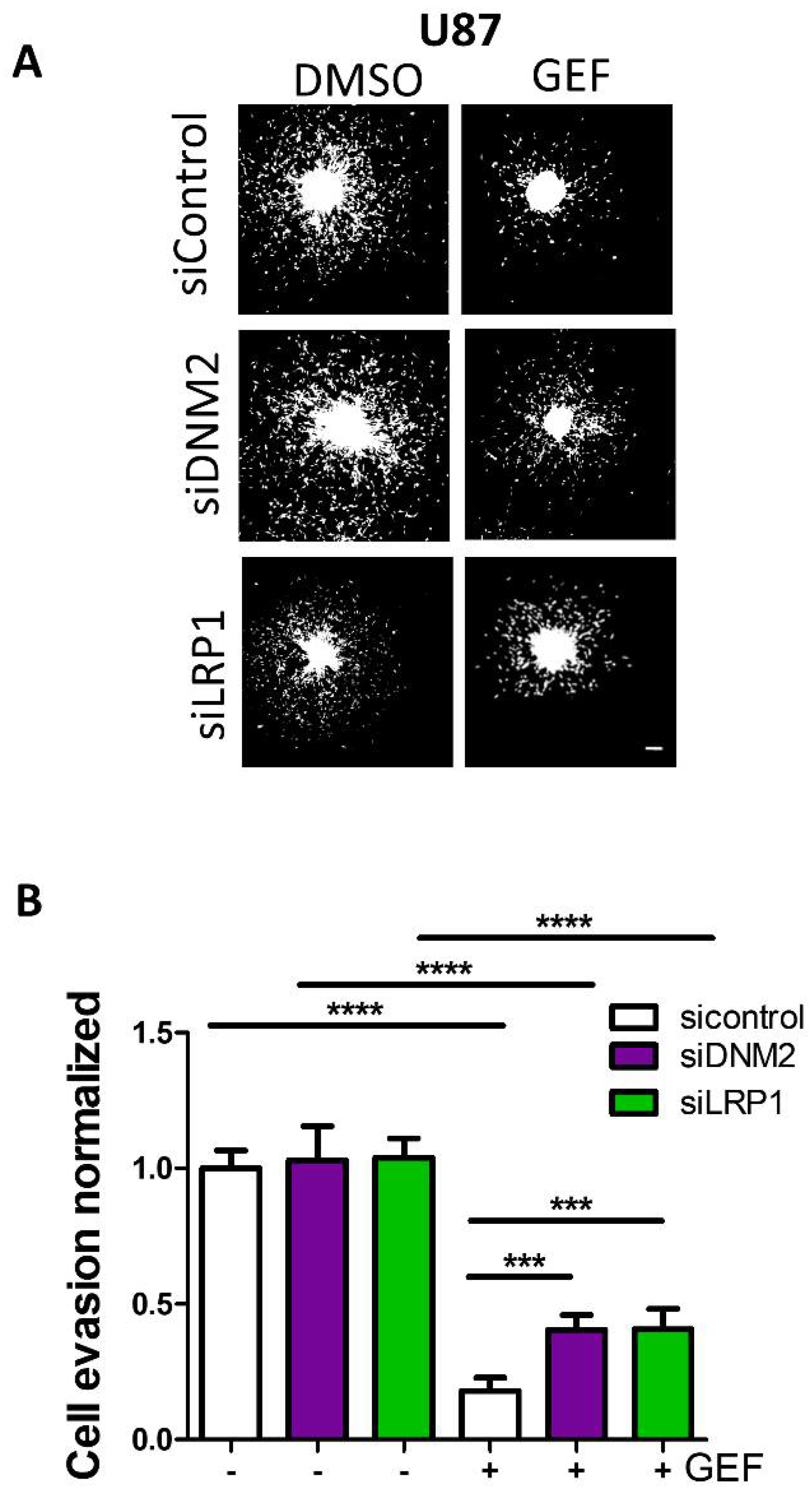
3.5. Endocytosis Is Critical for Gefitinib-Mediated Inhibition of GBM Cell Dissemination from 3D Spheroids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.; Weiss, W.A. Epidermal growth factor receptor (EGFR) and EGFRvIII in glioblastoma (GBM): Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncol. 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksen, L.; Grandal, M.V.; Knudsen, S.L.J.; van Deurs, B.; Grøvdal, L.M. Internalization Mechanisms of the Epidermal Growth Factor Receptor after Activation with Different Ligands. PLoS ONE 2013, 8, e58148. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Argenzio, E.; Tosoni, D.; Cavallaro, E.; Polo, S.; Di Fiore, P.P. Clathrin-Mediated Internalization Is Essential for Sustained EGFR Signaling but Dispensable for Degradation. Dev. Cell 2008, 15, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas, A.; Futter, C.E.; Eden, E.R. EGF receptor trafficking: Consequences for signaling and cancer. Trends Cell Biol. 2014, 24, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovic, M.; Sharma, M.; Rahajeng, J.; Caplan, S. The early endosome: A busy sorting station for proteins at the crossroads. Histol. Histopathol. 2010, 25, 99–112. [Google Scholar] [PubMed]
- Al-Akhrass, H.; Naves, T.; Vincent, F.; Magnaudeix, A.; Durand, K.; Bertin, F.; Melloni, B.; Jauberteau, M.-O.; Lalloué, F. Sortilin limits EGFR signaling by promoting its internalization in lung cancer. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Kondapalli, K.C.; Llongueras, J.P.; Capilla-González, V.; Prasad, H.; Hack, A.; Smith, C.; Guerrero-Cázares, H.; Quiñones-Hinojosa, A.; Rao, R. A leak pathway for luminal protons in endosomes drives oncogenic signalling in glioblastoma. Nat. Commun. 2015, 6, 6289. [Google Scholar] [CrossRef] [Green Version]
- Walsh, A.M.; Kapoor, G.S.; Buonato, J.M.; Mathew, L.K.; Bi, Y.; Davuluri, R.V.; Martinez-Lage, M.; Simon, M.C.; O’Rourke, D.M.; Lazzara, M.J. Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol. Cancer Res. MCR 2015, 13, 1227–1237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Wang, Z.; Zhang, Y.; Wang, Y.; Zhang, H.; Xie, S.; Xie, P.; Yu, R.; Zhou, X. Golgi phosphoprotein 3 sensitizes the tumour suppression effect of gefitinib on gliomas. Cell Prolif. 2019, 52, e12636. [Google Scholar] [CrossRef]
- Ying, H.; Zheng, H.; Scott, K.; Wiedemeyer, R.; Yan, H.; Lim, C.; Huang, J.; Dhakal, S.; Ivanova, E.; Xiao, Y.; et al. Mig-6 controls EGFR trafficking and suppresses gliomagenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 6912–6917. [Google Scholar] [CrossRef] [Green Version]
- Grandal, M.V.; Zandi, R.; Pedersen, M.W.; Willumsen, B.M.; van Deurs, B.; Poulsen, H.S. EGFRvIII escapes down-regulation due to impaired internalization and sorting to lysosomes. Carcinogenesis 2007, 28, 1408–1417. [Google Scholar] [CrossRef] [Green Version]
- Cao, X.; Zhu, H.; Ali-Osman, F.; Lo, H.-W. EGFR and EGFRvIII undergo stress- and EGFR kinase inhibitor-induced mitochondrial translocalization: A potential mechanism of EGFR-driven antagonism of apoptosis. Mol. Cancer 2011, 10, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [Green Version]
- Tan, X.; Thapa, N.; Sun, Y.; Anderson, R.A. A Kinase-Independent Role for EGF Receptor in Autophagy Initiation. Cell 2015, 160, 145–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomas, A.; Vaughan, S.O.; Burgoyne, T.; Sorkin, A.; Hartley, J.A.; Hochhauser, D.; Futter, C.E. WASH and Tsg101/ALIX-dependent diversion of stress-internalized EGFR from the canonical endocytic pathway. Nat. Commun. 2015, 6, 7324. [Google Scholar] [CrossRef] [Green Version]
- Dittmann, K.; Mayer, C.; Fehrenbacher, B.; Schaller, M.; Raju, U.; Milas, L.; Chen, D.J.; Kehlbach, R.; Rodemann, H.P. Radiation-induced Epidermal Growth Factor Receptor Nuclear Import Is Linked to Activation of DNA-dependent Protein Kinase. J. Biol. Chem. 2005, 280, 31182–31189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwang, Y.; Yarden, Y. p38 MAP kinase mediates stress-induced internalization of EGFR: Implications for cancer chemotherapy. EMBO J. 2006, 25, 4195–4206. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.; King, P.J.; Antonescu, C.N.; Sugiyama, M.G.; Bhamra, A.; Surinova, S.; Angelopoulos, N.; Kragh, M.; Pedersen, M.W.; Hartley, J.A.; et al. Targeting of EGFR by a combination of antibodies mediates unconventional EGFR trafficking and degradation. Sci. Rep. 2020, 10, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Keir, S.T.; Chandramohan, V.; Hemphill, C.D.; Grandal, M.M.; Melander, M.C.; Pedersen, M.W.; Horak, I.D.; Kragh, M.; Desjardins, A.; Friedman, H.S.; et al. Sym004-induced EGFR elimination is associated with profound anti-tumor activity in EGFRvIII patient-derived glioblastoma models. J. Neurooncol. 2018, 138, 489–498. [Google Scholar] [CrossRef] [Green Version]
- Liao, H.-J.; Carpenter, G. Cetuximab/C225-Induced Intracellular Trafficking of Epidermal Growth Factor Receptor. Cancer Res. 2009, 69, 6179–6183. [Google Scholar] [CrossRef] [Green Version]
- Blandin, A.-F.; Cruz Da Silva, E.; Mercier, M.-C.; Glushonkov, O.; Didier, P.; Dedieu, S.; Schneider, C.; Devy, J.; Etienne-Selloum, N.; Dontenwill, M.; et al. Gefitinib induces EGFR and α5β1 integrin co-endocytosis in glioblastoma cells. Cell. Mol. Life Sci. 2021, 78, 2949–2962. [Google Scholar] [CrossRef]
- Coker, E.A.; Mitsopoulos, C.; Tym, J.E.; Komianou, A.; Kannas, C.; Di Micco, P.; Villasclaras Fernandez, E.; Ozer, B.; Antolin, A.A.; Workman, P.; et al. canSAR: Update to the cancer translational research and drug discovery knowledgebase. Nucleic Acids Res. 2019, 47, D917–D922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrot, G.; Langlois, B.; Devy, J.; Jeanne, A.; Verzeaux, L.; Almagro, S.; Sartelet, H.; Hachet, C.; Schneider, C.; Sick, E.; et al. LRP-1--CD44, a new cell surface complex regulating tumor cell adhesion. Mol. Cell. Biol. 2012, 32, 3293–3307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandin, A.-F.; Noulet, F.; Renner, G.; Mercier, M.-C.; Choulier, L.; Vauchelles, R.; Ronde, P.; Carreiras, F.; Etienne-Selloum, N.; Vereb, G.; et al. Glioma cell dispersion is driven by α5 integrin-mediated cell–matrix and cell–cell interactions. Cancer Lett. 2016, 376, 328–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sousa, L.P.; Lax, I.; Shen, H.; Ferguson, S.M.; De Camilli, P.; Schlessinger, J. Suppression of EGFR endocytosis by dynamin depletion reveals that EGFR signaling occurs primarily at the plasma membrane. Proc. Natl. Acad. Sci. USA 2012, 109, 4419–4424. [Google Scholar] [CrossRef] [Green Version]
- Kirchhausen, T.; Macia, E.; Pelish, H.E. Use of dynasore, the small molecule inhibitor of dynamin, in the regulation of endocytosis. Methods Enzymol. 2008, 438, 77–93. [Google Scholar]
- Robertson, M.J.; Deane, F.M.; Robinson, P.J.; McCluskey, A. Synthesis of Dynole 34-2, Dynole 2-24 and Dyngo 4a for investigating dynamin GTPase. Nat. Protoc. 2014, 9, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Park, R.J.; Shen, H.; Liu, L.; Liu, X.; Ferguson, S.M.; Camilli, P.D. Dynamin triple knockout cells reveal off target effects of commonly used dynamin inhibitors. J. Cell Sci. 2013, 126, 5305–5312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, M.A.; Roberts, R.L.; Gumusboga, A.; Highfield, H.; Alvarez-Dominguez, C.; Wells, A.; Stahl, P.D. Epidermal Growth Factor and Membrane Trafficking. J. Cell Biol. 2000, 151, 539–550. [Google Scholar] [CrossRef]
- Chen, P.-I.; Kong, C.; Su, X.; Stahl, P.D. Rab5 Isoforms Differentially Regulate the Trafficking and Degradation of Epidermal Growth Factor Receptors. J. Biol. Chem. 2009, 284, 30328–30338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinneen, J.L.; Ceresa, B.P. Continual expression of Rab5(Q79L) causes a ligand-independent EGFR internalization and diminishes EGFR activity. Traffic Cph. Den. 2004, 5, 606–615. [Google Scholar] [CrossRef]
- Ceresa, B.; Lotscher, M.; Schmid, S. Receptor and Membrane Recycling Can Occur with Unaltered Efficiency Despite Dramatic Rab5(Q79L)-induced Changes in Endosome Geometry. J. Biol. Chem. 2001, 276, 9649–9654. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, Z. Regulation of epidermal growth factor receptor endocytosis by wortmannin through activation of Rab5 rather than inhibition of phosphatidylinositol 3-kinase. EMBO Rep. 2001, 2, 842–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etique, N.; Verzeaux, L.; Dedieu, S.; Emonard, H. LRP-1: A checkpoint for the extracellular matrix proteolysis. BioMed. Res. Int. 2013, 2013, 152163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bu, G.; Geuze, H.J.; Strous, G.J.; Schwartz, A.L. 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 1995, 14, 2269–2280. [Google Scholar] [CrossRef]
- Bu, G.; Schwartz, A.L. RAP, a novel type of ER chaperone. Trends Cell Biol. 1998, 8, 272–276. [Google Scholar] [CrossRef]
- Maritzen, T.; Schachtner, H.; Legler, D.F. On the move: Endocytic trafficking in cell migration. Cell. Mol. Life Sci. CMLS 2015, 72, 2119–2134. [Google Scholar] [CrossRef] [Green Version]
- Wilson, B.J.; Allen, J.L.; Caswell, P.T. Vesicle trafficking pathways that direct cell migration in 3D matrices and in vivo. Traffic Cph. Den. 2018, 19, 899–909. [Google Scholar] [CrossRef] [Green Version]
- Díaz, J.; Mendoza, P.; Ortiz, R.; Díaz, N.; Leyton, L.; Stupack, D.; Quest, A.F.G.; Torres, V.A. Rab5 is required in metastatic cancer cells for Caveolin-1-enhanced Rac1 activation, migration and invasion. J. Cell Sci. 2014, 127, 2401–2406. [Google Scholar] [CrossRef] [Green Version]
- Eppinga, R.D.; Krueger, E.W.; Weller, S.G.; Zhang, L.; Cao, H.; McNiven, M.A. Increased expression of the large GTPase dynamin 2 potentiates metastatic migration and invasion of pancreatic ductal carcinoma. Oncogene 2012, 31, 1228–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, H.; Liu, K.; Guo, P.; Zhang, P.; Cheng, T.; McNiven, M.; Johnson, G.; Hu, B.; Cheng, S. Dynamin 2 Mediates PDGFRα-SHP-2-Promoted Glioblastoma Growth and Invasion. Oncogene 2012, 31, 2691–2702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razidlo, G.L.; Wang, Y.; Chen, J.; Krueger, E.W.; Billadeau, D.D.; McNiven, M.A. Dynamin 2 Potentiates Invasive Migration of Pancreatic Tumor Cells through Stabilization of the Rac1 GEF Vav1. Dev. Cell 2013, 24, 573–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, H.; Takeda, T.; Michiue, H.; Abe, T.; Takei, K. Actin bundling by dynamin 2 and cortactin is implicated in cell migration by stabilizing filopodia in human non-small cell lung carcinoma cells. Int. J. Oncol. 2016, 49, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Destaing, O.; Ferguson, S.M.; Grichine, A.; Oddou, C.; Camilli, P.D.; Albiges-Rizo, C.; Baron, R. Essential Function of Dynamin in the Invasive Properties and Actin Architecture of v-Src Induced Podosomes/Invadosomes. PLoS ONE 2013, 8, e77956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Zhang, J.; Zhang, L.; Wang, Y.; Ma, H.; Wu, W.; Cui, J.; Wang, Y.; Ren, Z. Dynamin2 downregulation delays EGFR endocytic trafficking and promotes EGFR signaling and invasion in hepatocellular carcinoma. Am. J. Cancer Res. 2015, 5, 702–713. [Google Scholar]
- Khan, I.; Gril, B.; Steeg, P.S. Metastasis Suppressors NME1 and NME2 Promote Dynamin 2 Oligomerization and Regulate Tumor Cell Endocytosis, Motility, and Metastasis. Cancer Res. 2019, 79, 4689–4702. [Google Scholar] [CrossRef]
- Jian, Z.; Zhang, L.; Jin, L.; Lan, W.; Zhang, W.; Gao, G. Rab5 regulates the proliferation, migration and invasion of glioma cells via cyclin E. Oncol. Lett. 2020, 20, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Xie, S.; Wu, S.; Qi, Y.; Wang, Z.; Zhang, H.; Lu, D.; Wang, X.; Dong, Y.; Liu, G.; et al. Golgi phosphoprotein 3 promotes glioma progression via inhibiting Rab5-mediated endocytosis and degradation of epidermal growth factor receptor. Neuro-Oncology 2017, 19, 1628–1639. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Su, Y.; Li, T.; Yuan, W.; Mo, X.; Li, H.; He, Q.; Ma, D.; Han, W. CMTM7 knockdown increases tumorigenicity of human non-small cell lung cancer cells and EGFR-AKT signaling by reducing Rab5 activation. Oncotarget 2015, 6, 41092–41107. [Google Scholar] [CrossRef] [Green Version]
- Yuan, W.; Liu, B.; Wang, X.; Li, T.; Xue, H.; Mo, X.; Yang, S.; Ding, S.; Han, W. CMTM3 decreases EGFR expression and EGF-mediated tumorigenicity by promoting Rab5 activity in gastric cancer. Cancer Lett. 2017, 386, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Pan, B.; Xu, H.; Zhao, Z.; Shen, J.; Lu, J.; Yu, R.; Liu, H. Co-Delivery of GOLPH3 siRNA and gefitinib by cationic lipid-PLGA nanoparticles improves EGFR-targeted therapy for glioma. J. Mol. Med. Berl. Ger. 2019, 97, 1575–1588. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, V.; Vilbois, F.; Corti, M.; Marcote, M.J.; Tamura, K.; Karin, M.; Arkinstall, S.; Gruenberg, J. The stress-induced MAP kinase p38 regulates endocytic trafficking via the GDI:Rab5 complex. Mol. Cell 2001, 7, 421–432. [Google Scholar] [CrossRef]
- Macé, G.; Miaczynska, M.; Zerial, M.; Nebreda, A.R. Phosphorylation of EEA1 by p38 MAP kinase regulates μ opioid receptor endocytosis. EMBO J. 2005, 24, 3235–3246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, K.; Dai, Q.; Wei, J.; Shao, G.; Sun, A.; Yang, W.; Lin, Q. Stress-induced endocytosis and degradation of epidermal growth factor receptor are two independent processes. Cancer Cell Int. 2016, 16, 25. [Google Scholar] [CrossRef] [Green Version]
- Tomas, A.; Jones, S.; Vaughan, S.O.; Hochhauser, D.; Futter, C.E. Stress-Specific p38 MAPK activation is sufficient to drive EGFR endocytosis but not its nuclear translocation. J. Cell Sci. 2017, 130, 2481–2490. [Google Scholar]
- Vergarajauregui, S.; Miguel, A.S.; Puertollano, R. Activation of p38 Mitogen-Activated Protein Kinase Promotes Epidermal Growth Factor Receptor Internalization. Traffic Cph. Den. 2006, 7, 686–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Ozawa, T.; Oga, E.; Muraguchi, A.; Sakurai, H. Cisplatin-induced non-canonical endocytosis of EGFR via p38 phosphorylation of the C-terminal region containing Ser-1015 in non-small cell lung cancer cells. Oncol. Lett. 2018, 15, 9251–9256. [Google Scholar] [CrossRef] [Green Version]
- Soeda, A.; Lathia, J.; Williams, B.J.; Wu, Q.; Gallagher, J.; Androutsellis-Theotokis, A.; Giles, A.J.; Yang, C.; Zhuang, Z.; Gilbert, M.R.; et al. The p38 signaling pathway mediates quiescence of glioma stem cells by regulating epidermal growth factor receptor trafficking. Oncotarget 2017, 5, 33316–33328. [Google Scholar] [CrossRef]
- Baldwin, R.M.; Garratt-Lalonde, M.; Parolin, D.A.E.; Krzyzanowski, P.M.; Andrade, M.A.; Lorimer, I.A.J. Protection of glioblastoma cells from cisplatin cytotoxicity via protein kinase C ι -mediated attenuation of p38 MAP kinase signaling. Oncogene 2006, 25, 2909–2919. [Google Scholar] [CrossRef] [Green Version]
- Morello, V.; Cabodi, S.; Sigismund, S.; Camacho-Leal, M.P.; Repetto, D.; Volante, M.; Papotti, M.; Turco, E.; Defilippi, P. β1 integrin controls EGFR signaling and tumorigenic properties of lung cancer cells. Oncogene 2011, 30, 4087–4096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theret, L.; Jeanne, A.; Langlois, B.; Hachet, C.; David, M.; Khrestchatisky, M.; Devy, J.; Hervé, E.; Almagro, S.; Dedieu, S. Identification of LRP-1 as an endocytosis and recycling receptor for β1-integrin in thyroid cancer cells. Oncotarget 2017, 8, 78614–78632. [Google Scholar] [CrossRef] [Green Version]
- Boyé, K.; Pujol, N.; Alves, I.D.; Chen, Y.-P.; Daubon, T.; Lee, Y.-Z.; Dedieu, S.; Constantin, M.; Bello, L.; Rossi, M.; et al. The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 2017, 8, 1571. [Google Scholar] [CrossRef] [PubMed]
- Chew, H.Y.; De Lima, P.O.; Gonzalez Cruz, J.L.; Banushi, B.; Echejoh, G.; Hu, L.; Joseph, S.R.; Lum, B.; Rae, J.; O’Donnell, J.S.; et al. Endocytosis Inhibition in Humans to Improve Responses to ADCC-Mediating Antibodies. Cell 2020, 180, 895–914.e27. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.R.; Gaffney, D.; Barry, R.; Hu, L.; Banushi, B.; Wells, J.W.; Lambie, D.; Strutton, G.; Porceddu, S.V.; Burmeister, B.; et al. An Ex Vivo Human Tumor Assay Shows Distinct Patterns of EGFR Trafficking in Squamous Cell Carcinoma Correlating to Therapeutic Outcomes. J. Investig. Dermatol. 2019, 139, 213–223. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cruz Da Silva, E.; Choulier, L.; Thevenard-Devy, J.; Schneider, C.; Carl, P.; Rondé, P.; Dedieu, S.; Lehmann, M. Role of Endocytosis Proteins in Gefitinib-Mediated EGFR Internalisation in Glioma Cells. Cells 2021, 10, 3258. https://doi.org/10.3390/cells10113258
Cruz Da Silva E, Choulier L, Thevenard-Devy J, Schneider C, Carl P, Rondé P, Dedieu S, Lehmann M. Role of Endocytosis Proteins in Gefitinib-Mediated EGFR Internalisation in Glioma Cells. Cells. 2021; 10(11):3258. https://doi.org/10.3390/cells10113258
Chicago/Turabian StyleCruz Da Silva, Elisabete, Laurence Choulier, Jessica Thevenard-Devy, Christophe Schneider, Philippe Carl, Philippe Rondé, Stéphane Dedieu, and Maxime Lehmann. 2021. "Role of Endocytosis Proteins in Gefitinib-Mediated EGFR Internalisation in Glioma Cells" Cells 10, no. 11: 3258. https://doi.org/10.3390/cells10113258
APA StyleCruz Da Silva, E., Choulier, L., Thevenard-Devy, J., Schneider, C., Carl, P., Rondé, P., Dedieu, S., & Lehmann, M. (2021). Role of Endocytosis Proteins in Gefitinib-Mediated EGFR Internalisation in Glioma Cells. Cells, 10(11), 3258. https://doi.org/10.3390/cells10113258