
Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drug Treatments
2.3. Virus Stock Production and Infections
2.4. MT Regrowth Assays
2.5. Generation of AKAP450 KO SNB19 Lines
2.6. RNA Isolation, Reverse Transcription-Quantitative PCR (RT-qPCR), and Data Analysis
2.7. Immunofluorescent (IF) Staining and Microscopy
2.8. Antibodies
2.9. Centrosome Intensity Quantification and Statistics
3. Results
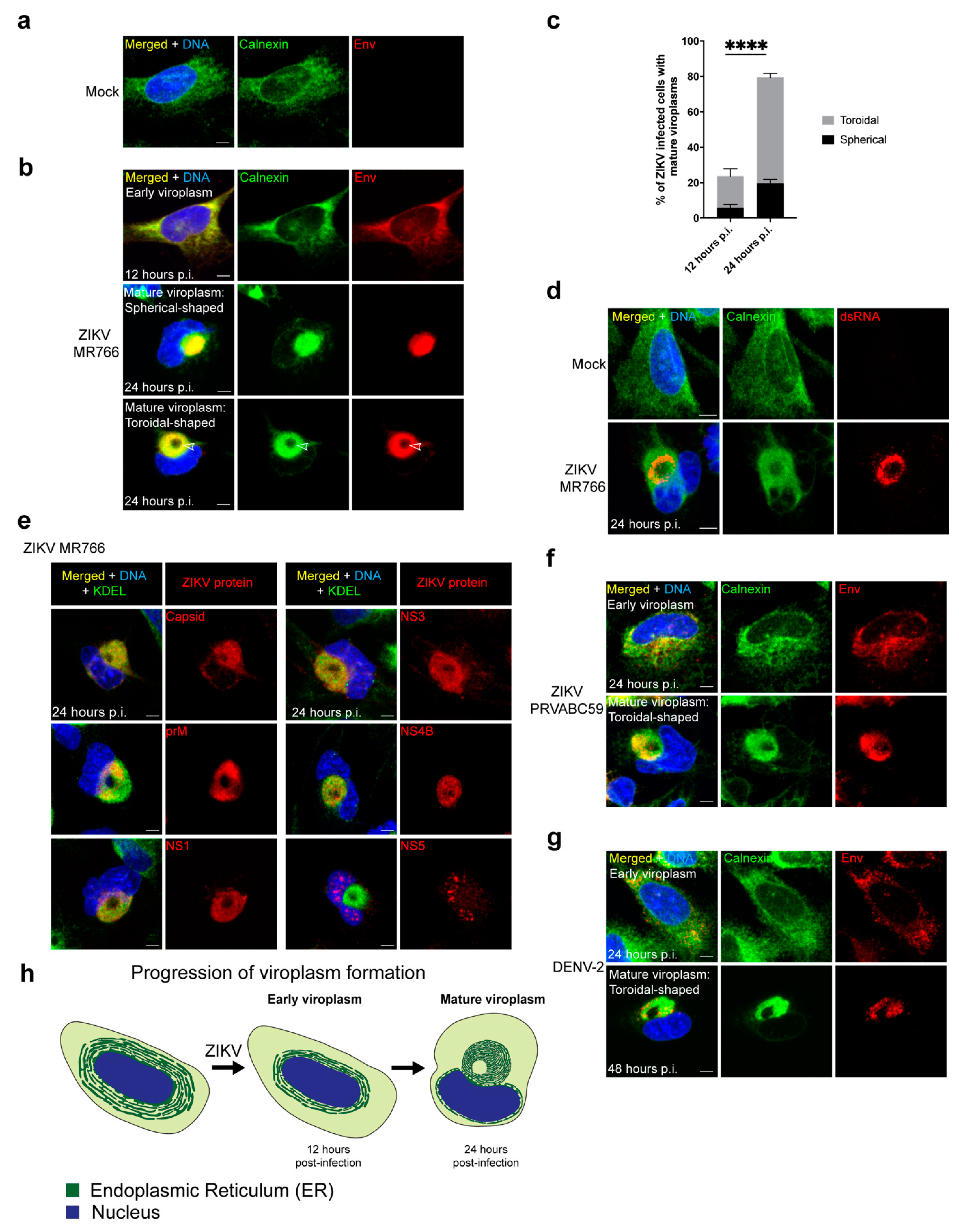
3.1. ZIKV Reorganizes the ER into a Compact Toroidal-Shaped Viroplasm
3.2. MTs Reorganize at the Viroplasm during ZIKV Infection
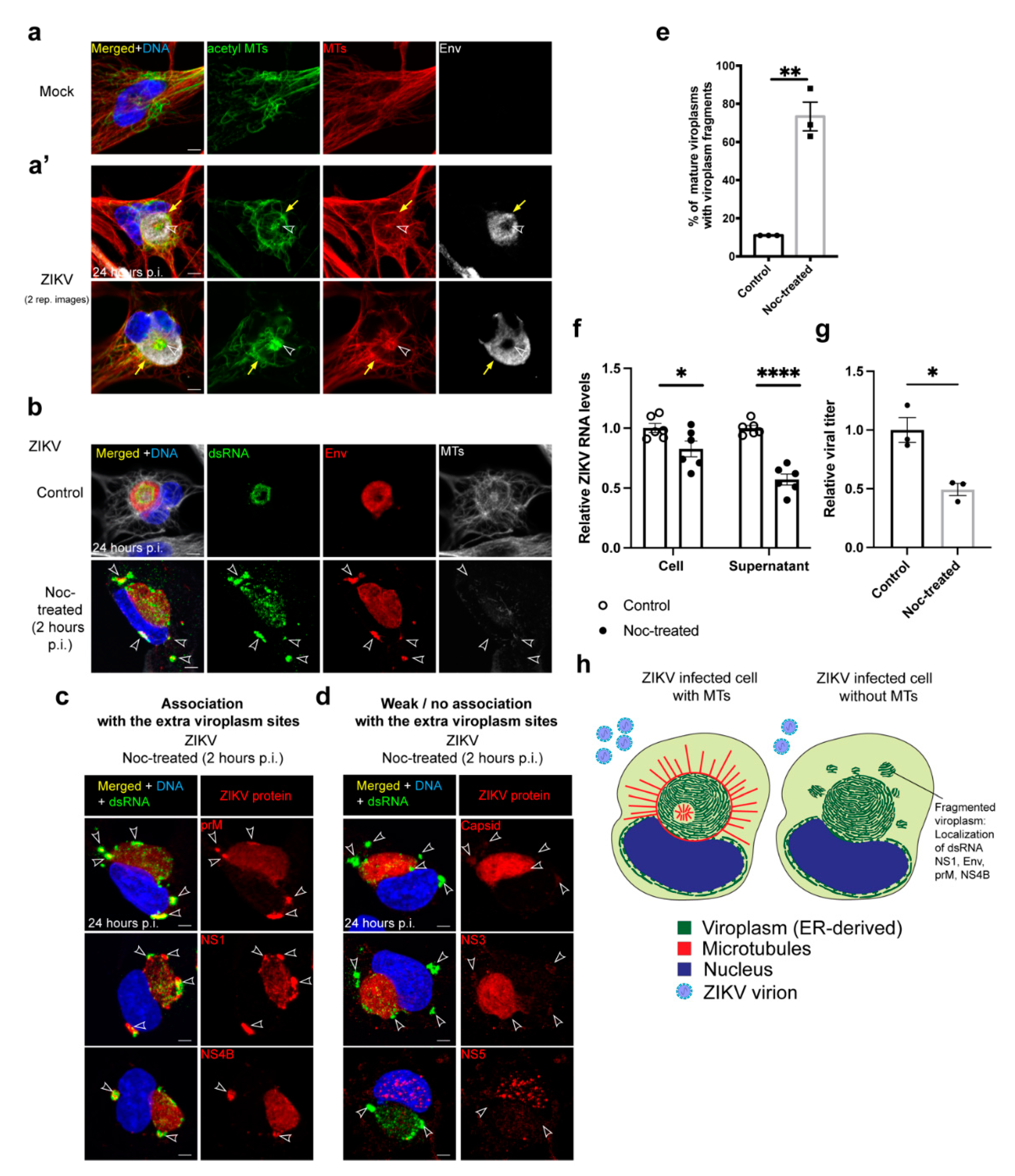
3.3. MTs Are Necessary for ZIKV Viroplasm Organization
3.4. MTs Are Required for Efficient ZIKV Virus Production
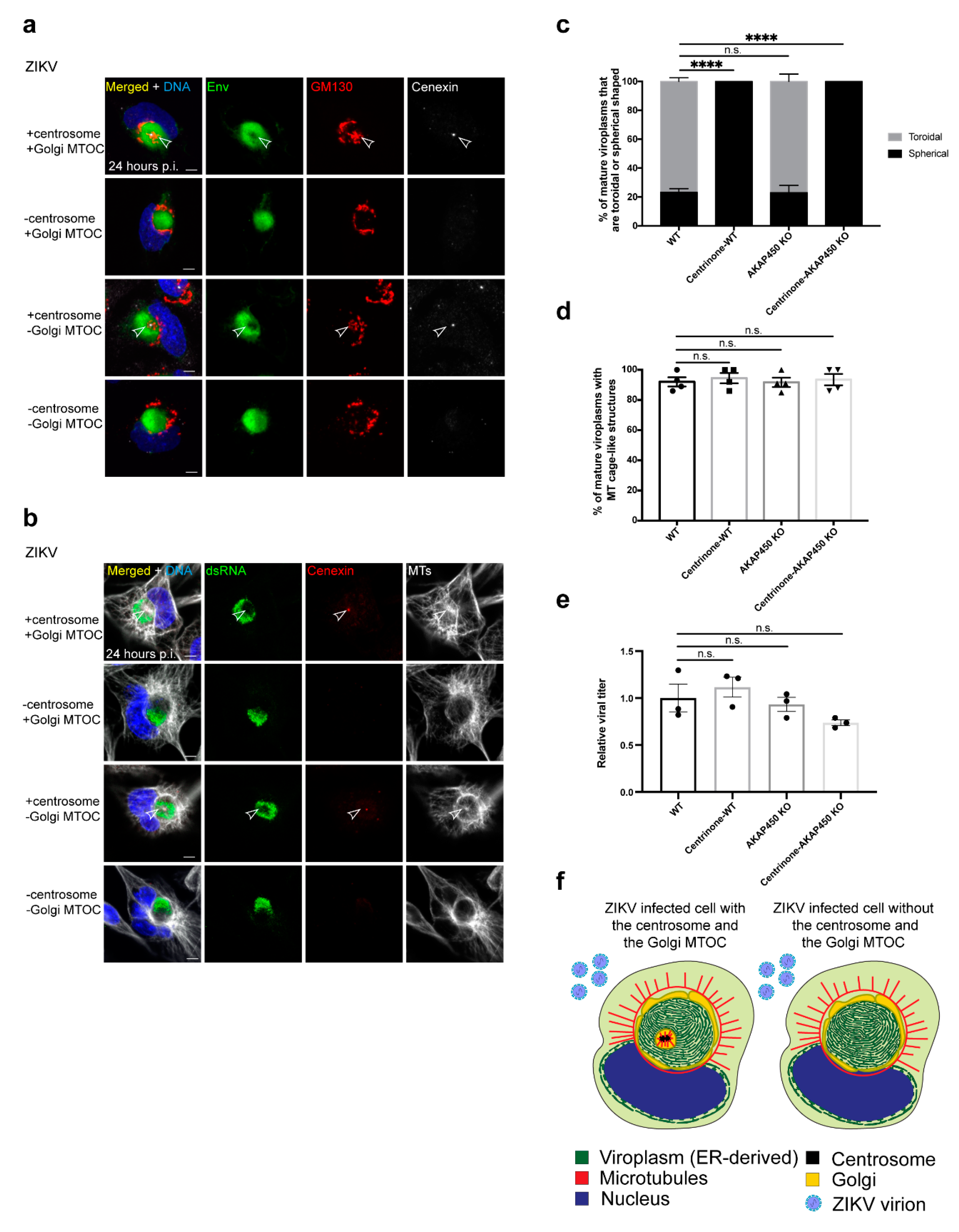
3.5. The ZIKV Viroplasm Is Organized in Conjunction with the Centrosome and the Golgi MTOC
3.6. The Centrosome and the Golgi MTOC Nucleate the MTs Associated with the Viroplasm
3.7. The Centrosome Is Required to Form a Toroidal Viroplasm
3.8. Virus Production IS Not Significantly Dependent on the Centrosome and Golgi MTOC
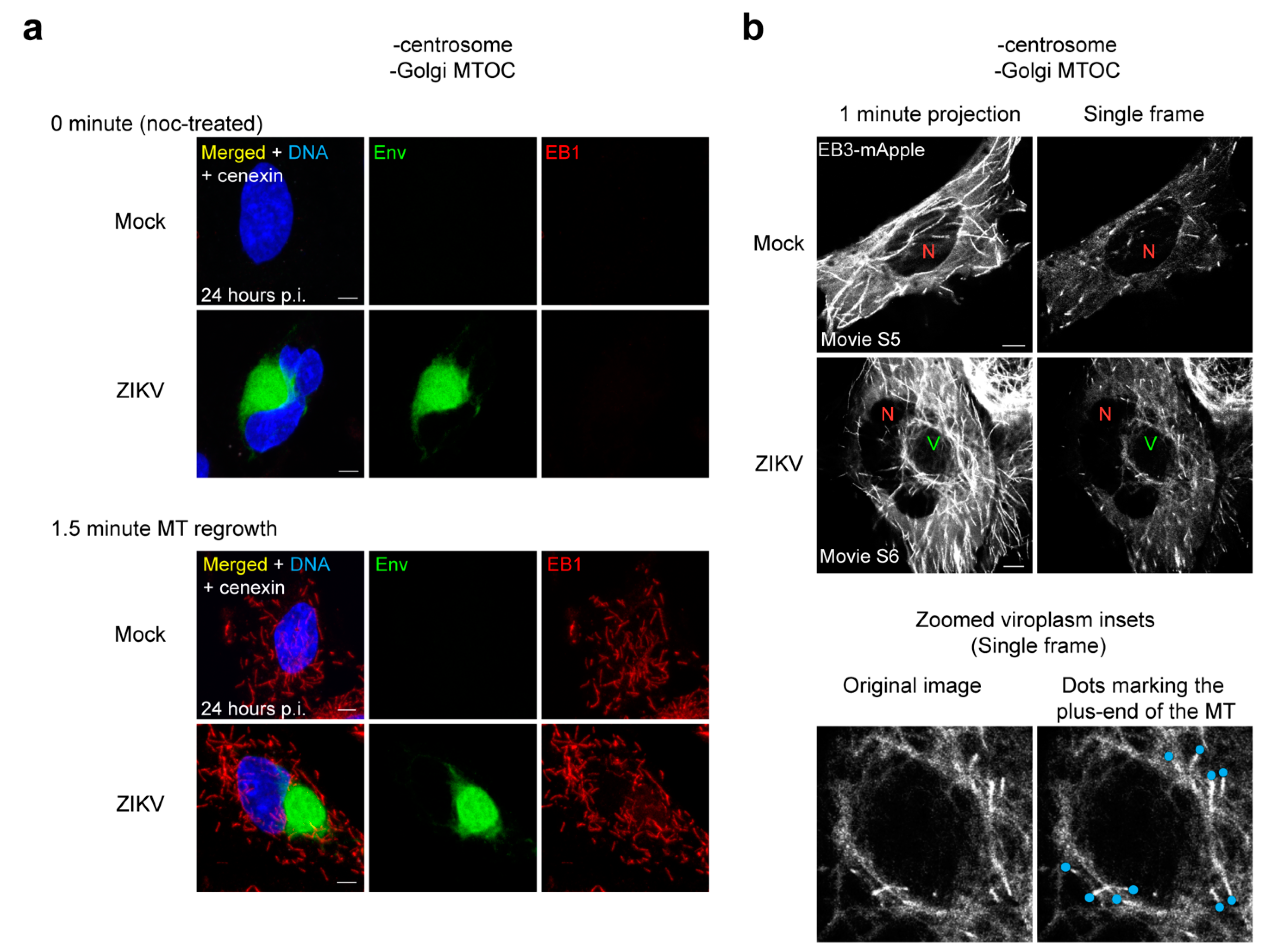
3.9. MTs Are Anchored at the ZIKV Viroplasm
4. Discussion
4.1. MT Requirements for ZIKV Viroplasm Organization and Virus Production
4.2. Impacts of ZIKV on the Centrosome
4.3. The Requirements of the Centrosome and Golgi MTOCs for ZIKV Infection
4.4. Methods to Remove MTOCs to Test Viral Infection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Musso, D.; Ko, A.I.; Baud, D. Zika virus infection—After the pandemic. N. Engl. J. Med. 2019, 381, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vipotnik, T.V.; Vodušek, V.F.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatnagar, J.; Rabeneck, D.B.; Martines, R.B.; Reagan-Steiner, S.; Ermias, Y.; Estetter, L.B.; Suzuki, T.; Ritter, J.; Keating, M.K.; Hale, G.; et al. Zika Virus RNA Replication and Persistence in Brain and Placental Tissue. Emerg. Infect. Dis. 2017, 23, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Rothan, H.A.; Kumar, M. Role of Endoplasmic Reticulum-Associated Proteins in Flavivirus Replication and Assembly Complexes. Pathogens 2019, 8, 148. [Google Scholar] [CrossRef] [Green Version]
- Rajah, M.M.; Monel, B.; Schwartz, O. The entanglement between flaviviruses and ER-shaping proteins. PLoS Pathog. 2020, 16, e1008389. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef]
- Ropidi, M.I.M.; Khazali, A.S.; Rashid, N.N.; Yusof, R. Endoplasmic reticulum: A focal point of Zika virus infection. J. Biomed. Sci. 2020, 27, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-Dimensional Architecture of Tick-Borne Encephalitis Virus Replication Sites and Trafficking of the Replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.M.; Westaway, E.G. Assembly and Maturation of the Flavivirus Kunjin Virus Appear To Occur in the Rough Endoplasmic Reticulum and along the Secretory Pathway, Respectively. J. Virol. 2001, 75, 10787–10799. [Google Scholar] [CrossRef] [Green Version]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Gao, N.; Wang, J.-L.; Tian, Y.-P.; Chen, Z.-T.; An, J. Vimentin is required for dengue virus serotype 2 infection but microtubules are not necessary for this process. Arch. Virol. 2008, 153, 1777–1781. [Google Scholar] [CrossRef]
- Shrivastava, N.; Sripada, S.; Kaur, J.; Shah, P.S.; Cecilia, D. Insights into the Internalization and Retrograde Trafficking of Dengue 2 Virus in BHK-21 Cells. PLoS ONE 2011, 6, e25229. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.S.; Ng, M.L. Involvement of microtubules in Kunjin virus replication. Arch. Virol. 1987, 97, 115–121. [Google Scholar] [CrossRef]
- Ruzek, D.; Vancová, M.; Tesařová, M.; Ahantarig, A.; Kopecký, J.; Grubhoffer, L. Morphological changes in human neural cells following tick-borne encephalitis virus infection. J. Gen. Virol. 2009, 90, 1649–1658. [Google Scholar] [CrossRef]
- Chu, J.; Ng, M. Trafficking mechanism of west Nile (Sarafend) virus structural proteins. J. Med. Virol. 2002, 67, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Foo, K.Y.; Chee, H.-Y. Interaction between flavivirus and cytoskeleton during virus replication. BioMed Res. Int. 2015, 2015, 427814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gao, W.; Li, J.; Wu, W.; Jiu, Y. The Role of Host Cytoskeleton in Flavivirus Infection. Virol. Sin. 2019, 34, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Rinkenberger, N.; Schoggins, J.W. Comparative analysis of viral entry for Asian and African lineages of Zika virus. Virology 2019, 533, 59–67. [Google Scholar] [CrossRef]
- Li, M.; Zhang, D.; Li, C.; Zheng, Z.; Fu, M.; Ni, F.; Liu, Y.; Du, T.; Wang, H.; Griffin, G.E.; et al. Characterization of Zika Virus Endocytic Pathways in Human Glioblastoma Cells. Front. Microbiol. 2020, 11, 242. [Google Scholar] [CrossRef]
- Whelan, J.N.; Parenti, N.A.; Hatterschide, J.; Renner, D.M.; Li, Y.; Reyes, H.M.; Dong, B.; Perez, E.R.; Silverman, R.H.; Weiss, S.R. Zika virus employs the host antiviral RNase L protein to support replication factory assembly. Proc. Natl. Acad. Sci. USA 2021, 118, e2101713118. [Google Scholar] [CrossRef]
- Hackett, B.A.; Cherry, S. Flavivirus internalization is regulated by a size-dependent endocytic pathway. Proc. Natl. Acad. Sci. USA 2018, 115, 4246–4251. [Google Scholar] [CrossRef] [Green Version]
- Buchwalter, R.A.; Chen, J.V.; Zheng, Y.; Megraw, T.L. Centrosome in Cell Division, Development and Disease. eLS 2016, 1–12. [Google Scholar] [CrossRef]
- Conduit, P.T.; Wainman, A.; Raff, J.W. Centrosome function and assembly in animal cells. Nat. Rev. Mol. Cell Biol. 2015, 16, 611–624. [Google Scholar] [CrossRef]
- Sanchez, A.D.; Feldman, J.L. Microtubule-organizing centers: From the centrosome to non-centrosomal sites. Curr. Opin. Cell Biol. 2016, 44, 93–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muroyama, A.; Lechler, T. Microtubule organization, dynamics and functions in differentiated cells. Development 2017, 144, 3012–3021. [Google Scholar] [CrossRef] [Green Version]
- Tillery, M.M.L.; Blake-Hedges, C.; Zheng, Y.; Buchwalter, R.A.; Megraw, T.L. Centrosomal and Non-Centrosomal Microtubule-Organizing Centers (MTOCs) in Drosophila melanogaster. Cells 2018, 7, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, J.; Lüders, J. Microtubule-Organizing Centers: Towards a Minimal Parts List. Trends Cell Biol. 2018, 28, 176–187. [Google Scholar] [CrossRef]
- Sallee, M.D.; Feldman, J.L. Microtubule organization across cell types and states. Curr. Biol. 2021, 31, R506–R511. [Google Scholar] [CrossRef]
- Wu, J.; Akhmanova, A. Microtubule-Organizing Centers. Annu. Rev. Cell Dev. Biol. 2017, 33, 51–75. [Google Scholar] [CrossRef]
- Martin, M.; Akhmanova, A. Coming into Focus: Mechanisms of Microtubule Minus-End Organization. Trends Cell Biol. 2018, 28, 574–588. [Google Scholar] [CrossRef]
- Weiner, A.T.; Thyagarajan, P.; Shen, Y.; Rolls, M.M. To nucleate or not, that is the question in neurons. Neurosci. Lett. 2021, 751, 135806. [Google Scholar] [CrossRef]
- Kelliher, M.T.; Saunders, H.A.; Wildonger, J. Microtubule control of functional architecture in neurons. Curr. Opin. Neurobiol. 2019, 57, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Sanders, A.A.; Kaverina, I. Nucleation and dynamics of Golgi-derived microtubules. Frontiers in neuroscience 2015, 9, 431. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Kaverina, I. Golgi as an MTOC: Making microtubules for its own good. Histochem. Cell Biol. 2013, 140, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, A.; Meservey, L.; Nguyen, H.; Fu, M.-M. Golgi Outposts Nucleate Microtubules in Cells with Specialized Shapes. Trends Cell Biol. 2020, 30, 792–804. [Google Scholar] [CrossRef]
- Jayaraman, D.; Bae, B.-I.; Walsh, C.A. The Genetics of Primary Microcephaly. Annu. Rev. Genom. Hum. Genet. 2018, 19, 177–200. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, R.S.; Schoborg, T.; Rusan, N.M. Same but different: Pleiotropy in centrosome-related microcephaly. Mol. Biol. Cell 2018, 29, 241–246. [Google Scholar] [CrossRef]
- Jean, F.; Stuart, A.; Tarailo-Graovac, M. Dissecting the Genetic and Etiological Causes of Primary Microcephaly. Front. Neurol. 2020, 11, 570830. [Google Scholar] [CrossRef] [PubMed]
- Megraw, T.L.; Sharkey, J.T.; Nowakowski, R. Cdk5rap2 exposes the centrosomal root of microcephaly syndromes. Trends Cell Biol. 2011, 21, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris-Rosendahl, D.J.; Kaindl, A.M. What next-generation sequencing (NGS) technology has enabled us to learn about primary autosomal recessive microcephaly (MCPH). Mol. Cell. Probes 2015, 29, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naveed, M.; Kazmi, S.K.; Amin, M.; Asif, Z.; Islam, U.; Shahid, K.; Tehreem, S. Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genet. Res. 2018, 100, e7. [Google Scholar] [CrossRef] [Green Version]
- Nano, M.; Basto, R. Consequences of Centrosome Dysfunction During Brain Development. Adv. Exp. Med. Biol. 2017, 1002, 19–45. [Google Scholar] [CrossRef] [PubMed]
- Faheem, M.; Naseer, M.I.; Rasool, M.; Chaudhary, A.G.; Kumosani, T.A.; Ilyas, A.M.; Pushparaj, P.N.; Ahmed, F.; Algahtani, H.A.; Al-Qahtani, M.H.; et al. Molecular genetics of human primary microcephaly: An overview. BMC Med. Genom. 2015, 8 (Suppl. S1), S4. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Yang, W.; Hao, H.; Wang, W.; Lin, S.; Shi, P.; Huang, Y.; Li, B.; Sun, Y.; Liu, Z.; et al. The Golgi microtubules regulate single cell durotaxis. EMBO Rep. 2021, 22, e51094. [Google Scholar] [CrossRef]
- Wu, J.; de Heus, C.; Liu, Q.; Bouchet, B.P.; Noordstra, I.; Jiang, K.; Hua, S.; Martin, M.; Yang, C.; Grigoriev, I.; et al. Molecular Pathway of Microtubule Organization at the Golgi Apparatus. Dev. Cell 2016, 39, 44–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero, S.; Cardenas, J.; Bornens, M.; Ríos, R.M. Microtubule nucleation at the cis-side of the Golgi apparatus requires AKAP450 and GM130. EMBO J. 2009, 28, 1016–1028. [Google Scholar] [CrossRef]
- Efimov, A.; Kharitonov, A.; Efimova, N.; Loncarek, J.; Miller, P.M.; Andreyeva, N.; Gleeson, P.; Galjart, N.; Maia, A.R.; McLeod, I.X.; et al. Asymmetric CLASP-Dependent Nucleation of Noncentrosomal Microtubules at the trans-Golgi Network. Dev. Cell 2007, 12, 917–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trogden, K.P.; Zhu, X.; Lee, J.S.; Wright, C.V.; Gu, G.; Kaverina, I. Regulation of Glucose-Dependent Golgi-Derived Microtubules by cAMP/EPAC2 Promotes Secretory Vesicle Biogenesis in Pancreatic β Cells. Curr. Biol. 2019, 29, 2339–2350.e5. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.M.; Folkmann, A.W.; Maia, A.R.R.; Efimova, N.; Efimov, A.; Kaverina, I. Golgi-derived CLASP-dependent microtubules control Golgi organization and polarized trafficking in motile cells. Nat. Cell Biol. 2009, 11, 1069–1080. [Google Scholar] [CrossRef] [Green Version]
- Vergarajauregui, S.; Becker, R.; Steffen, U.; Sharkova, M.; Esser, T.; Petzold, J.; Billing, F.; Kapiloff, M.S.; Schett, G.; Thievessen, I.; et al. AKAP6 orchestrates the nuclear envelope microtubule-organizing center by linking golgi and nucleus via AKAP9. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Klemm, L.C.; Denu, R.A.; Hind, L.E.; Rocha-Gregg, B.L.; Burkard, M.E.; Huttenlocher, A. Centriole and Golgi microtubule nucleation are dispensable for the migration of human neutrophil-like cells. Mol. Biol. Cell 2021, 3, 1545–1556. [Google Scholar] [CrossRef]
- Gavilan, M.P.; Gandolfo, P.; Balestra, F.R.; Arias, F.; Bornens, M.; Rios, R.M. The dual role of the centrosome in organizing the microtubule network in interphase. EMBO Rep. 2018, 19, e45942. [Google Scholar] [CrossRef]
- Kotadia, S.; Kao, L.R.; Comerford, S.A.; Jones, R.T.; Hammer, R.E.; Megraw, T.L. PP2A-dependent disruption of centrosome replication and cytoskeleton organization in Drosophila by SV40 small tumor antigen. Oncogene 2008, 27, 6334–6346. [Google Scholar] [CrossRef] [Green Version]
- Yun, C.; Cho, H.; Kim, S.-J.; Lee, J.-H.; Park, S.Y.; Chan, G.K.; Cho, H. Mitotic aberration coupled with centrosome amplification is induced by hepatitis B virus X oncoprotein via the Ras-mitogen-activated protein/extracellular signal-regulated kinase-mitogen-activated protein pathway. Mol. Cancer Res. 2004, 2, 159–169. [Google Scholar]
- Shumilov, A.; Tsai, M.-H.; Schlosser, Y.T.; Kratz, A.-S.; Bernhardt, K.; Fink, S.; Mizani, T.; Lin, X.; Jauch, A.; Mautner, J.; et al. Epstein–Barr virus particles induce centrosome amplification and chromosomal instability. Nat. Commun. 2017, 8, 14257. [Google Scholar] [CrossRef] [Green Version]
- Peloponese, J.-M.; Haller, K.; Miyazato, A.; Jeang, K.-T. Abnormal centrosome amplification in cells through the targeting of Ran-binding protein-1 by the human T cell leukemia virus type-1 Tax oncoprotein. Proc. Natl. Acad. Sci. USA 2005, 102, 18974–18979. [Google Scholar] [CrossRef] [Green Version]
- Duensing, S.; Lee, L.Y.; Duensing, A.; Basile, J.; Piboonniyom, S.-O.; Gonzalez, S.; Crum, C.P.; Münger, K. The human papillomavirus type 16 E6 and E7 oncoproteins cooperate to induce mitotic defects and genomic instability by uncoupling centrosome duplication from the cell division cycle. Proc. Natl. Acad. Sci. USA 2000, 97, 10002–10007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Luca, A.; Mangiacasale, R.; Severino, A.; Malquori, L.; Baldi, A.; Palena, A.; Mileo, A.M.; LaVia, P.; Paggi, M.G. E1A deregulates the centrosome cycle in a Ran GTPase-dependent manner. Cancer Res. 2003, 63, 1430–1437. [Google Scholar] [PubMed]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91, e00538-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furey, C.; Astar, H.; Walsh, D. Human cytomegalovirus exploits TACC3 to control microtubule dynamics and late stages of infection. J. Virol. 2021, 95, e0082121. [Google Scholar] [CrossRef]
- Procter, D.J.; Banerjee, A.; Nukui, M.; Kruse, K.; Gaponenko, V.; Murphy, E.A.; Komarova, Y.; Walsh, D. The HCMV Assembly Compartment Is a Dynamic Golgi-Derived MTOC that Controls Nuclear Rotation and Virus Spread. Dev. Cell 2018, 45, 83–100.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabriel, E.; Ramani, A.; Karow, U.; Gottardo, M.; Natarajan, K.; Gooi, L.M.; Goranci-Buzhala, G.; Krut, O.; Peters, F.; Nikolić, M.; et al. Recent Zika Virus Isolates Induce Premature Differentiation of Neural Progenitors in Human Brain Organoids. Cell Stem Cell 2017, 20, 397–406.e5. [Google Scholar] [CrossRef] [Green Version]
- Wolf, B.; Diop, F.; Ferraris, P.; Wichit, S.; Busso, C.; Missé, D.; Gönczy, P. Zika virus causes supernumerary foci with centriolar proteins and impaired spindle positioning. Open Biol. 2017, 7, 160231. [Google Scholar] [CrossRef] [Green Version]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [Green Version]
- Souza, B.S.F.; Sampaio, G.; Pereira, C.S.; Campos, G.S.; Sardi, S.I.; Freitas, L.A.R.; Figueira, C.P.; Paredes, B.D.; Nonaka, C.K.V.; Azevedo, C.M.; et al. Zika virus infection induces mitosis abnormalities and apoptotic cell death of human neural progenitor cells. Sci. Rep. 2016, 6, 39775. [Google Scholar] [CrossRef]
- Wen, F.; Armstrong, N.; Hou, W.; Cruz-Cosme, R.; Obwolo, L.A.; Ishizuka, K.; Ullah, H.; Luo, M.-H.; Sawa, A.; Tang, Q. Zika virus increases mind bomb 1 levels, causing degradation of pericentriolar material 1 (PCM1) and dispersion of PCM1-containing granules from the centrosome. J. Biol. Chem. 2019, 294, 18742–18755. [Google Scholar] [CrossRef] [PubMed]
- Kodani, A.; Knopp, K.; Di Lullo, E.; Retallack, H.; Kriegstein, A.; DeRisi, J.; Reiter, J. Zika virus alters centrosome organization to suppress the innate immune response. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wong, Y.L.; Anzola, J.V.; Davis, R.L.; Yoon, M.; Motamedi, A.; Kroll, A.; Seo, C.P.; Hsia, J.E.; Kim, S.K.; Mitchell, J.W.; et al. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science 2015, 348, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammack, C.; Ogden, S.C.; Madden, J.C., Jr.; Medina, A.; Xu, C.; Phillips, E.; Son, Y.; Cone, A.; Giovinazzi, S.; Didier, R.A.; et al. Zika Virus Infection Induces DNA Damage Response in Human Neural Progenitors That Enhances Viral Replication. J. Virol. 2019, 93, e00638-19. [Google Scholar] [CrossRef] [Green Version]
- Coelho, S.V.A.; Neris, R.L.S.; Papa, M.P.; Schnellrath, L.C.; Meuren, L.M.; Tschoeke, D.A.; Leomil, L.; Verçoza, B.; Miranda, M.; Thompson, F.L.; et al. Development of standard methods for Zika virus propagation, titration, and purification. J. Virol. Methods 2017, 246, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.-K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.D.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.-Y.; Liu, S.-Q.; Deng, C.-L.; Zhang, Q.-Y.; Zhang, B. Detection of Zika virus by SYBR green one-step real-time RT-PCR. J. Virol. Methods 2016, 236, 93–97. [Google Scholar] [CrossRef] [PubMed]
- York, S.B.; Sun, L.; Cone, A.S.; Duke, L.C.; Cheerathodi, M.R.; Meckes, D.G., Jr. Zika Virus Hijacks Extracellular Vesicle Tetraspanin Pathways for Cell-to-Cell Transmission. mSphere 2021, 6, e0019221. [Google Scholar] [CrossRef]
- Chen, J.V.; Buchwalter, R.A.; Kao, L.-R.; Megraw, T.L. A Splice Variant of Centrosomin Converts Mitochondria to Microtubule-Organizing Centers. Curr. Biol. 2017, 27, 1928–1940.e6. [Google Scholar] [CrossRef] [Green Version]
- Dráberová, E.; D’Agostino, L.; Caracciolo, V.; Sládková, V.; Sulimenko, T.; Sulimenko, V.; Sobol, M.; Maounis, N.F.; Tzelepis, E.; Mahera, E.; et al. Overexpression and Nucleolar Localization of γ-Tubulin Small Complex Proteins GCP2 and GCP3 in Glioblastoma. J. Neuropathol. Exp. Neurol. 2015, 74, 723–742. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, D.B.; Pearson, C.G.; Yen, T.; Howell, B.J.; Salmon, E. Microtubule-dependent Changes in Assembly of Microtubule Motor Proteins and Mitotic Spindle Checkpoint Proteins at PtK1 Kinetochores. Mol. Biol. Cell 2001, 12, 1995–2009. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Denman, A.J.; MacKenzie, J.M. The IMPORTance of the Nucleus during Flavivirus Replication. Viruses 2017, 9, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, C.; Magiera, M.M. The tubulin code and its role in controlling microtubule properties and functions. Nat. Rev. Mol. Cell Biol. 2020, 21, 307–326. [Google Scholar] [CrossRef] [PubMed]
- Marthiens, V.; Rujano, M.A.; Pennetier, C.; Tessier, S.; Paul-Gilloteaux, P.; Basto, R. Centrosome amplification causes microcephaly. Nat. Cell Biol. 2013, 15, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Link, N.; Jang, G.M.; Sharp, P.P.; Zhu, T.; Swaney, D.L.; Johnson, J.; Von Dollen, J.; Ramage, H.R.; Satkamp, L.; et al. Comparative Flavivirus-Host Protein Interaction Mapping Reveals Mechanisms of Dengue and Zika Virus Pathogenesis. Cell 2018, 175, 1931–1945.e18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyaud, E.; Ranadheera, C.; Cheng, D.T.; Gonçalves, J.; Dyakov, B.; Laurent, E.M.; St-Germain, J.R.; Pelletier, L.; Gingras, A.-C.; Brumell, J.H.; et al. Global Interactomics Uncovers Extensive Organellar Targeting by Zika Virus. Mol. Cell. Proteom. 2018, 17, 2242–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubeva, V.A.; Nepomuceno, T.C.; De Gregoriis, G.; Mesquita, R.D.; Li, X.; Dash, S.; Garcez, P.P.; Suarez-Kurtz, G.; Izumi, V.; Koomen, J.; et al. Network of Interactions between ZIKA Virus Non-Structural Proteins and Human Host Proteins. Cells 2020, 9, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khadka, S.; Vangeloff, A.D.; Zhang, C.; Siddavatam, P.; Heaton, N.S.; Wang, L.; Sengupta, R.; Sahasrabudhe, S.; Randall, G.; Gribskov, M.; et al. A Physical Interaction Network of Dengue Virus and Human Proteins. Mol. Cell. Proteom. 2011, 10, 012187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Breton, M.; Meyniel-Schicklin, L.; Deloire, A.; Coutard, B.; Canard, B.; de Lamballerie, X.; Andre, P.; Rabourdin-Combe, C.; Lotteau, V.; Davoust, N. Flavivirus NS3 and NS5 proteins interaction network: A high-throughput yeast two-hybrid screen. BMC Microbiol. 2011, 11, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiou, C.-T.; Hu, C.-C.A.; Chen, P.-H.; Liao, C.-L.; Lin, Y.-L.; Wang, J.-J. Association of Japanese encephalitis virus NS3 protein with microtubules and tumour susceptibility gene 101 (TSG101) protein. J. Gen. Virol. 2003, 84, 2795–2805. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.L.; Hong, S.S. Flavivirus infection: Essential ultrastructural changes and association of Kunjin virus NS3 protein with microtubules. Arch. Virol. 1989, 106, 103–120. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchwalter, R.A.; Ogden, S.C.; York, S.B.; Sun, L.; Zheng, C.; Hammack, C.; Cheng, Y.; Chen, J.V.; Cone, A.S.; Meckes, D.G., Jr.; et al. Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers. Cells 2021, 10, 3335. https://doi.org/10.3390/cells10123335
Buchwalter RA, Ogden SC, York SB, Sun L, Zheng C, Hammack C, Cheng Y, Chen JV, Cone AS, Meckes DG Jr., et al. Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers. Cells. 2021; 10(12):3335. https://doi.org/10.3390/cells10123335
Chicago/Turabian StyleBuchwalter, Rebecca A., Sarah C. Ogden, Sara B. York, Li Sun, Chunfeng Zheng, Christy Hammack, Yichen Cheng, Jieyan V. Chen, Allaura S. Cone, David G. Meckes, Jr., and et al. 2021. "Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers" Cells 10, no. 12: 3335. https://doi.org/10.3390/cells10123335
APA StyleBuchwalter, R. A., Ogden, S. C., York, S. B., Sun, L., Zheng, C., Hammack, C., Cheng, Y., Chen, J. V., Cone, A. S., Meckes, D. G., Jr., Tang, H., & Megraw, T. L. (2021). Coordination of Zika Virus Infection and Viroplasm Organization by Microtubules and Microtubule-Organizing Centers. Cells, 10(12), 3335. https://doi.org/10.3390/cells10123335