
Growth Hormone and IGF1 Actions in Kidney Development and Function


Abstract
:1. Introduction
2. Normal GH-IGF1 Axis and Physiology
3. GH-IGF1: Axis or Independent Functions?
4. Observations from Knockout and Transgenic Animals
5. GH and IGF1 in Normal Renal Development
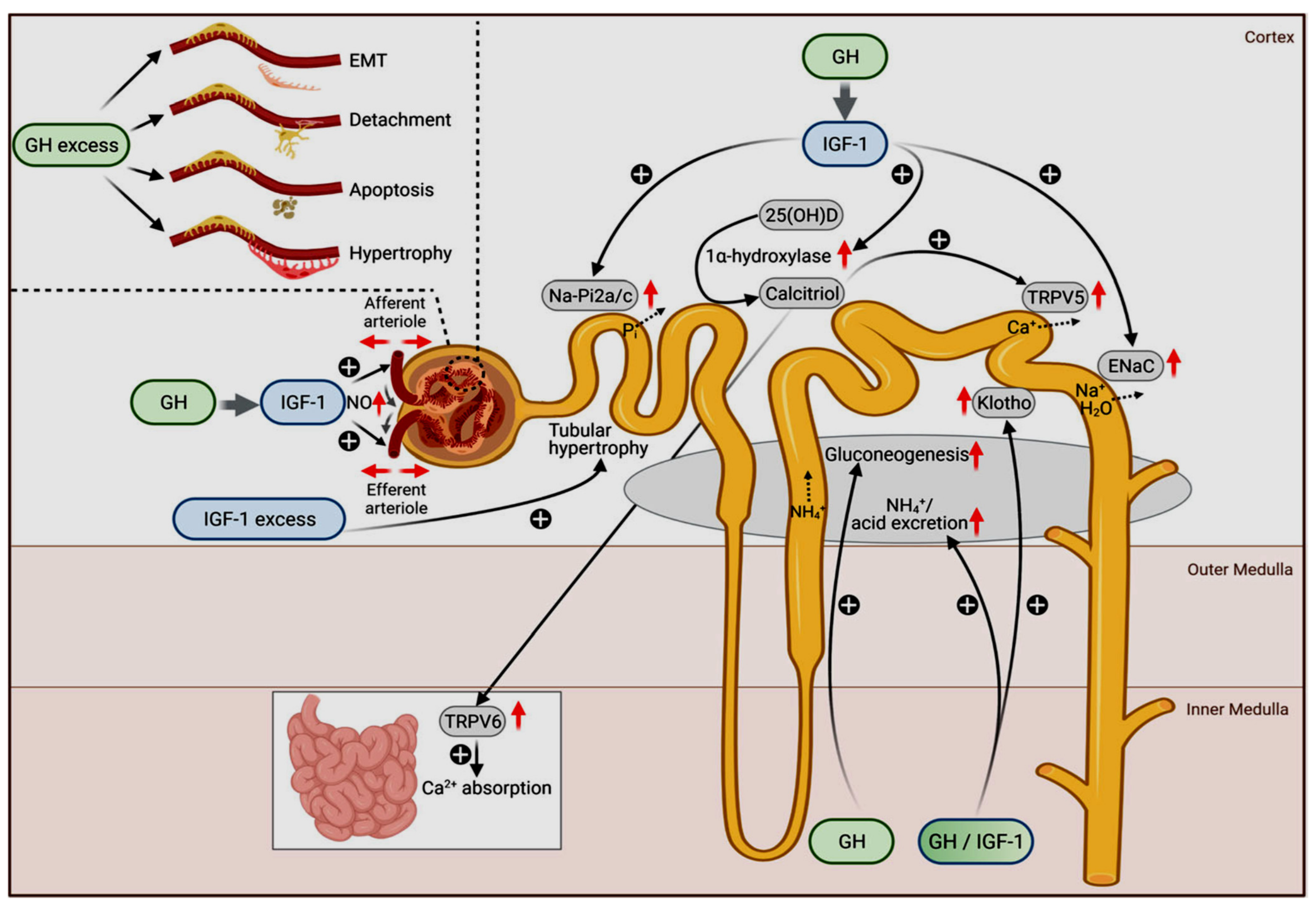
6. GH/IGF1 Effects on Normal Tubular and Glomerular Functions
7. GH/IGF1 Involvement in Kidney Diseases
Compensatory Renal Hypertrophy

{kind=link}
| KO/Human Mutation General Phenotype | KO/Kidney Phenotype | Ref. | |
|---|---|---|---|
| GH | Growth retardation | Disproportionally small kidneys | [17] |
| GHR/ GHBP | Growth retardation after birth, low IGF1, greater longevity | Disproportionally small kidneys Protection against diabetic nephropathy | [18] |
| JAK2 | Embryonic lethality due to a lack of hematopoiesis | NA | [19] |
| STAT5 | Abnormal postnatal growth, facial dysmorphism, immunodeficiency (h) perinatal death, dwarfism, anemia, immunodeficiency (m) | NA | [20,22] |
| IGF1 | Severe growth retardation, infertility, deficiencies in bone and muscle development, lethal respiratory failure | Proportionally small kidneys, decreased glomerular size and nephron number Liver specific IGF1 KO mice: compensatory remnant kidney hypertrophy after unilateral nephrectomy, no significant change in IGF1R phosphorylation (despite markedly decreased kidney IGF-1 levels) | [23,24,114] |
| IGF1R | Respiratory failure, low birth weight, developmental abnormalities, perinatal death | NA | [25] |
| SOCS2 | Gigantism, improved somatic growth in CKD model | No glomerulosclerosis development | [47] |
| IGFBP1 | indistinguishable from wild-type, no embryonic lethality | NA | [44] |
| IGFBP2 | minor gender specific changes in bone structure, minor changes in the weights of spleen and liver in adult males | NA | [43,45] |
| IGFBP3 | Normal | NA | [42] |
| IGFBP4 | mild 10%–15% reduction in prenatal growth | NA | [42] |
| IGFBP5 | Normal | NA | [42] |
| IGFBP6 | Normal | NA | [42] |
| General Phenotype | Kidney Phenotype | Ref. | |
|---|---|---|---|
| GH | Giant phenotype, organomegaly | Kidney hypertrophy, glomerular hyperthrophy, progressive albuminuria, glomerulosclerosis | [27,28,29] |
| IGF1 | Enhanced growth | Proportionately enlarged kidneys, glomerular hyperthrophy, no glomerulosclerosis | [30,31,32] |
| IGFBP1 | Low birth weight, postnatal growth retardation, disproportionally small brain, splenomegaly, hyperglycemia | Small kidneys, decreased nephron number; glomerulosclerosis without glomerular hypertrophy | [34,35,36,37] |
| IGFBP2 | Mild growth retardation, mildly reduced organs weight | NA | [38] |
| IGFBP3 | Increased spleen, liver, heart weight | Disproportionally small kidneys | [38,39,40] |
| IGFBP4 | Different tissues hypoplasia | [37] | |
| IGF2 | Disproportionately enlarged kidneys | [58] |
8. Diabetic Nephropathy
9. Chronic Kidney Disease (CKD)
10. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hartman, M.L.; Veldhuis, J.D.; Thorner, M.O. Normal control of growth hormone secretion. Horm. Res. 1993, 40, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Pang, A.L.-Y.; Chan, W.Y. Chapter 22—Molecular Basis of Diseases of the Endocrine System. In Molecular Pathology, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Rubinek, T.; Modan-Moses, D. Klotho and the growth hormone/insulin-like growth factor 1 axis: Novel insights into complex interactions. Vitam. Horm. 2016, 101, 85–118. [Google Scholar] [CrossRef] [PubMed]
- Parganas, E.; Wang, D.; Stravopodis, D.; Topham, D.J.; Marine, J.C.; Teglund, S.; Vanin, E.F.; Bodner, S.; Colamonici, O.R.; van Deursen, J.M.; et al. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 1998, 93, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.A.; Lindberg, K.; Hilton, D.J.; Nielsen, J.H.; Billestrup, N. Mechanism of inhibition of growth hormone receptor signaling by suppressor of cytokine signaling proteins. Mol. Endocrinol. 1999, 13, 1832–1843. [Google Scholar] [CrossRef]
- Frystyk, J.; Skjaerbaek, C.; Dinesen, B.; Orskov, H. Free insulin-like growth factors (IGF-I and IGF-II) in human serum. FEBS Lett. 1994, 348, 185–191. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef]
- Werner, H.; Bruchim, I. The insulin-like growth factor-I receptor as an oncogene. Arch. Physiol. Biochem. 2009, 115, 58–71. [Google Scholar] [CrossRef]
- Baxter, R.C. Insulin-like growth factor (IGF)-binding proteins: Interactions with IGFs and intrinsic bioactivities. Am. J. Physiol. Metab. 2000, 278, E967–E976. [Google Scholar] [CrossRef]
- Mohan, S.; Baylink, D.J. IGF-binding proteins are multifunctional and act via IGF-dependent and -independent mechanisms. J. Endocrinol. 2002, 175, 19–31. [Google Scholar] [CrossRef] [Green Version]
- Salmon, W.D., Jr.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar]
- Green, H.; Morikawa, M.; Nixon, T. A dual effector theory of growth-hormone action. Differentiation 1985, 29, 195–198. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.L.; Le Roith, D. The growth hormone/insulin-like growth factor-I system: Implications for organ growth and development. Pediatr. Nephrol. 2000, 14, 544–549. [Google Scholar] [CrossRef]
- Le Roith, D.; Bondy, C.; Yakar, S.; Liu, J.L.; Butler, A. The somatomedin hypothesis: 2001. Endocr. Rev. 2001, 22, 53–74. [Google Scholar] [CrossRef]
- Roberts, C.T., Jr.; Lasky, S.R.; Lowe, W.L., Jr.; Seaman, W.T.; LeRoith, D. Molecular cloning of rat insulin-like growth factor I complementary deoxyribonucleic acids: Differential messenger ribonucleic acid processing and regulation by growth hormone in extrahepatic tissues. Mol. Endocrinol. 1987, 1, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Schimpff, R.M.; Donnadieu, M.; Duval, M. Serum somatomedin activity measured as sulphation factor in peripheral, hepatic and renal veins in normal mongrel dogs: Early effects of intravenous injection of growth hormone. Acta Endocrinol. 1980, 93, 155–161. [Google Scholar] [CrossRef]
- Guler, H.P.; Zapf, J.; Scheiwiller, E.; Froesch, E.R. Recombinant human insulin-like growth factor I stimulates growth and has distinct effects on organ size in hypophysectomized rats. Proc. Natl. Acad. Sci. USA 1988, 85, 4889–4893. [Google Scholar] [CrossRef] [Green Version]
- Guler, H.P.; Schmid, C.; Zapf, J.; Froesch, E.R. Effects of recombinant insulin-like growth factor I on insulin secretion and renal function in normal human subjects. Proc. Natl. Acad. Sci. USA 1989, 86, 2868–2872. [Google Scholar] [CrossRef] [Green Version]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef] [Green Version]
- List, E.O.; Sackmann-Sala, L.; Berryman, D.E.; Funk, K.; Kelder, B.; Gosney, E.S.; Okada, S.; Ding, J.; Cruz-Topete, D.; Kopchick, J.J. Endocrine parameters and phenotypes of the growth hormone receptor gene disrupted (GHR−/−) mouse. Endocr. Rev. 2011, 32, 356–386. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, H.; Cumano, A.; Müller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodnik, L.; Jasper, H.; Tepper, A.; et al. Growth hormone insensitivity associated with a STAT5b mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef]
- Landau, D.; London, L.; Bandach, I.; Segev, Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS ONE 2018, 13, e0196684. [Google Scholar] [CrossRef]
- Cui, Y.; Riedlinger, G.; Miyoshi, K.; Tang, W.; Li, C.; Deng, C.X.; Robinson, G.W.; Hennighausen, L. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol. Cell. Biol. 2004, 24, 8037–8047. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.L.; Yakar, S.; LeRoith, D. Conditional knockout of mouse insulin-like growth factor-1 gene using the Cre/loxP system. Proc. Soc. Exp. Boil. Med. 2000, 223, 344–351. [Google Scholar] [CrossRef]
- Rogers, S.A.; Powell-Braxton, L.; Hammerman, M.R. Insulin-like growth factor I regulates renal development in rodents. Dev. Genet. 1999, 24, 293–298. [Google Scholar] [CrossRef]
- Liu, J.P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Louvi, A.; Accili, D.; Efstratiadis, A. Growth-promoting interaction of IGF-II with the insulin receptor during mouse embryonic development. Dev. Biol. 1997, 189, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Holzenberger, M.; Dupont, J.; Ducos, B.; Leneuve, P.; Geloen, A.; Even, P.C.; Cervera, P.; Le Bouc, Y. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 2003, 421, 182–187. [Google Scholar] [CrossRef]
- Rotem-Grunbaum, B.; Landau, D. Genetic renal disease classification by hormonal axes. Pediatr. Nephrol. 2020, 35, 2211–2219. [Google Scholar] [CrossRef]
- Kamenický, P.; Mazziotti, G.; Lombès, M.; Giustina, A.; Chanson, P. Growth hormone, insulin-like growth factor-1, and the kidney: Pathophysiological and clinical implications. Endocr. Rev. 2014, 35, 234–281. [Google Scholar] [CrossRef] [Green Version]
- Striker, L.J.; Doi, T.; Striker, G.E. Transgenic mice in renal research. Adv. Nephrol. Necker Hosp. 1991, 20, 91–108. [Google Scholar] [PubMed]
- Pesce, C.M.; Striker, L.J.; Peten, E.; Elliot, S.J.; Striker, G.E. Glomerulosclerosis at both early and late stages is associated with increased cell turnover in mice transgenic for growth hormone. Lab. Investig. 1991, 65, 601–605. [Google Scholar] [PubMed]
- Mathews, L.S.; Hammer, R.E.; Behringer, R.R.; D’Ercole, A.J.; Bell, G.I.; Brinster, R.L.; Palmiter, R.D. Growth enhancement of transgenic mice expressing human insulin-like growth factor I. Endocrinology 1988, 123, 2827–2833. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Striker, L.J.; Quaife, C.; Conti, F.G.; Palmiter, R.; Behringer, R.; Brinster, R.; Striker, G.E. Progressive glomerulosclerosis develops in transgenic mice chronically expressing growth hormone and growth hormone releasing factor but not in those expressing insulin like growth factor-1. Am. J. Pathol. 1988, 131, 398–403. [Google Scholar]
- Doi, T.; Striker, L.J.; Gibson, C.C.; Agodoa, L.Y.; Brinster, R.L.; Striker, G.E. Glomerular lesions in mice transgenic for growth hormone and insulin like growth factor-I. I. Relationship between increased glomerular size and mesangial sclerosis. Am. J. Pathol. 1990, 137, 541–552. [Google Scholar]
- Blutke, A.; Schneider, M.R.; Wolf, E.; Wanke, R. Growth hormone (GH)-transgenic insulin-like growth factor 1 (IGF1)-deficient mice allow dissociation of excess GH and IGF1 effects on glomerular and tubular growth. Physiol. Rep. 2016, 4, e12709. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, K.; Barron, D.; Lewitt, M.S.; Murphy, L.J. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology 1995, 136, 4029–4034. [Google Scholar] [CrossRef]
- Doublier, S.; Seurin, D.; Fouqueray, B.; Verpont, M.C.; Callard, P.; Striker, L.J.; Striker, G.E.; Binoux, M.; Baud, L. Glomerulosclerosis in mice transgenic for human insulin-like growth factor-binding protein-1. Kidney Int. 2000, 57, 2299–2307. [Google Scholar] [CrossRef] [Green Version]
- Doublier, S.; Amri, K.; Seurin, D.; Moreau, E.; Merlet-Benichou, C.; Striker, G.E.; Gilbert, T. Overexpression of human insulin-like growth factor binding protein-1 in the mouse leads to nephron deficit. Pediatr. Res. 2001, 49, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.R.; Lahm, H.; Wu, M.; Hoeflich, A.; Wolf, E. Transgenic mouse models for studying the functions of insulin-like growth factor-binding proteins. FASEB J. 2000, 14, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Hoeflich, A.; Wu, M.; Mohan, S.; Föll, J.; Wanke, R.; Froehlich, T.; Arnold, G.J.; Lahm, H.; Kolb, H.J.; Wolf, E. Overexpression of insulin-like growth factor-binding protein-2 in transgenic mice reduces postnatal body weight gain. Endocrinology 1999, 140, 5488–5496. [Google Scholar] [CrossRef]
- Murphy, L.J.; Molnar, P.; Lu, X.; Huang, H. Expression of human insulin-like growth factor-binding protein-3 in transgenic mice. J. Mol. Endocrinol. 1995, 15, 293–303. [Google Scholar] [CrossRef]
- Modric, T.; Silha, J.V.; Shi, Z.; Gui, Y.; Suwanichkul, A.; Durham, S.K.; Powell, D.R.; Murphy, L.J. Phenotypic manifestations of insulin-like growth factor-binding protein-3 overexpression in transgenic mice. Endocrinology 2001, 142, 1958–1967. [Google Scholar] [CrossRef]
- Silha, J.V.; Gui, Y.; Mishra, S.; Leckstrom, A.; Cohen, P.; Murphy, L.J. Overexpression of gly56/gly80/gly81-mutant insulin-like growth factor-binding protein-3 in transgenic mice. Endocrinology 2005, 146, 1523–1531. [Google Scholar] [CrossRef] [Green Version]
- Ning, Y.; Schuller, A.G.; Bradshaw, S.; Rotwein, P.; Ludwig, T.; Frystyk, J.; Pintar, J.E. Diminished growth and enhanced glucose metabolism in triple knockout mice containing mutations of insulin-like growth factor binding protein-3, -4, and -5. Mol. Endocrinol. 2006, 20, 2173–2186. [Google Scholar] [CrossRef]
- Wood, T.L.; Rogler, L.E.; Czick, M.E.; Schuller, A.G.; Pintar, J.E. Selective alterations in organ sizes in mice with a targeted disruption of the insulin-like growth factor binding protein-2 gene. Mol. Endocrinol. 2000, 14, 1472–1482. [Google Scholar] [CrossRef]
- Leu, J.I.; Crissey, M.A.; Craig, L.E.; Taub, R. Impaired hepatocyte DNA synthetic response posthepatectomy in insulin-like growth factor binding protein 1-deficient mice with defects in C/EBP beta and mitogen-activated protein kinase/extracellular signal-regulated kinase regulation. Mol. Cell. Biol. 2003, 23, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- DeMambro, V.E.; Clemmons, D.R.; Horton, L.G.; Bouxsein, M.L.; Wood, T.L.; Beamer, W.G.; Canalis, E.; Rosen, C.J. Gender-specific changes in bone turnover and skeletal architecture in igfbp-2-null mice. Endocrinology 2008, 149, 2051–2061. [Google Scholar] [CrossRef]
- Gray, A.; Aronson, W.J.; Barnard, R.J.; Mehta, H.; Wan, J.; Said, J.; Cohen, P.; Galet, C. Global Igfbp1 deletion does not affect prostate cancer development in a c-Myc transgenic mouse model. J. Endocrinol. 2011, 211, 297–304. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.; Assadi, M.H.; Abu Hilal, R.; Chen, Y.; Rabkin, R.; Segev, Y. SOCS2 Silencing Improves Somatic Growth without Worsening Kidney Function in CKD. Am. J. Nephrol. 2020, 51, 520–526. [Google Scholar] [CrossRef]
- Stratikopoulos, E.; Szabolcs, M.; Dragatsis, I.; Klinakis, A.; Efstratiadis, A. The hormonal action of IGF1 in postnatal mouse growth. Proc. Natl. Acad. Sci. USA 2008, 105, 19378–19383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjögren, K.; Liu, J.L.; Blad, K.; Skrtic, S.; Vidal, O.; Wallenius, V.; LeRoith, D.; Törnell, J.; Isaksson, O.G.; Jansson, J.O.; et al. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7088–7092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindström, N.O.; McMahon, J.A.; Guo, J.; Tran, T.; Guo, Q.; Rutledge, E.; Parvez, R.K.; Saribekyan, G.; Schuler, R.E.; Liao, C.; et al. Conserved and divergent features of human and mouse kidney organogenesis. J. Am. Soc. Nephrol. 2018, 29, 785–805. [Google Scholar] [CrossRef] [PubMed]
- Chin, E.; Zhou, J.; Bondy, C.A. Renal growth hormone receptor gene expression: Relationship to renal insulin-like growth factor system. Endocrinology 1992, 131, 3061–3066. [Google Scholar] [CrossRef]
- Simard, M.; Manthos, H.; Giaid, A.; Lefèbvre, Y.; Goodyer, C.G. Ontogeny of growth hormone receptors in human tissues: An immunohistochemical study. J. Clin. Endocrinol. Metab. 1996, 81, 3097–3102. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.A.; Ryan, G.; Hammerman, M.R. Insulin-like growth factors I and II are produced in the metanephros and are required for growth and development in vitro. J. Cell Biol. 1991, 113, 1447–1453. [Google Scholar] [CrossRef]
- Lindenbergh-Kortleve, D.J.; Rosato, R.R.; van Neck, J.W.; Nauta, J.; van Kleffens, M.; Groffen, C.; Zwarthoff, E.C.; Drop, S.L. Gene expression of the insulin-like growth factor system during mouse kidney development. Mol. Cell. Endocrinol. 1997, 132, 81–91. [Google Scholar] [CrossRef] [Green Version]
- Bondy, C.A.; Werner, H.; Roberts, C.T., Jr.; LeRoith, D. Cellular pattern of insulin-like growth factor-I (IGF-I) and type I IGF receptor gene expression in early organogenesis: Comparison with IGF-II gene expression. Mol. Endocrinol. 1990, 4, 1386–1398. [Google Scholar] [CrossRef]
- Chin, E.; Bondy, C. Insulin-like growth factor system gene expression in the human kidney. J. Clin. Endocrinol. Metab. 1992, 75, 962–968. [Google Scholar] [CrossRef]
- Daughaday, W.H.; Rotwein, P. Insulin-like growth factors I and II. Peptide, messenger ribonucleic acid and gene structures, serum, and tissue concentrations. Endocr. Rev. 1989, 10, 68–91. [Google Scholar] [CrossRef]
- Wolf, E.; Kramer, R.; Blum, W.F.; Föll, J.; Brem, G. Consequences of postnatally elevated insulin-like growth factor-II in transgenic mice: Endocrine changes and effects on body and organ growth. Endocrinology 1994, 135, 1877–1886. [Google Scholar] [CrossRef] [Green Version]
- Caidahl, K.; Edén, S.; Bengtsson, B.A. Cardiovascular and renal effects of growth hormone. Clin. Endocrinol. 1994, 40, 393–400. [Google Scholar] [CrossRef]
- Jørgensen, J.O.; Pedersen, S.A.; Thuesen, L.; Jørgensen, J.; Ingemann-Hansen, T.; Skakkebaek, N.E.; Christiansen, J.S. Beneficial effects of growth hormone treatment in GH-deficient adults. Lancet 1989, 333, 1221–1225. [Google Scholar] [CrossRef]
- Falkheden, T.; Sjoegren, B. Extracellular fluid volume and renal function in pituitary insufficiency and acromegaly. Acta Endocrinol. 1964, 46, 80–88. [Google Scholar] [CrossRef]
- Ece, A.; Çetinkaya, S.; Ekşioğlu, S.; Şenel, S.; Özkasap, S.; Giniş, T.; Sen, V.; Şahin, C. Kidney growth and renal functions under the growth hormone replacement therapy in children. Ren. Fail. 2014, 36, 508–513. [Google Scholar] [CrossRef]
- Ikkos, D.; Ljunggren, H.; Luft, R. Glomerular filtration rate and renal plasma flow in acromegaly. Acta Endocrinol. 1956, 21, 226–236. [Google Scholar] [CrossRef]
- Grunenwald, S.; Tack, I.; Chauveau, D.; Bennet, A.; Caron, P. Impact of growth hormone hypersecretion on the adult human kidney. Ann. Endocrinol. 2011, 72, 485–495. [Google Scholar] [CrossRef]
- Hoogenberg, K.; Sluiter, W.J.; Dullaart, R.P. Effect of growth hormone and insulin-like growth factor I on urinary albumin excretion: Studies in acromegaly and growth hormone deficiency. Acta Endocrinol. 1993, 129, 151–157. [Google Scholar] [CrossRef]
- Manelli, F.; Bossoni, S.; Burattin, A.; Doga, M.; Solerte, S.B.; Romanelli, G.; Giustina, A. Exercise-induced microalbuminuria in patients with active acromegaly: Acute effects of slow-release lanreotide, a long-acting somatostatin analog. Metabolism 2000, 49, 634–639. [Google Scholar] [CrossRef]
- Klinger, B.; Laron, Z. Renal function in Laron syndrome patients treated by insulin-like growth factor-I. Pediatr. Nephrol. 1994, 8, 684–688. [Google Scholar] [CrossRef]
- Hirschberg, R.; Kopple, J.D. Evidence that insulin-like growth factor I increases renal plasma flow and glomerular filtration rate in fasted rats. J. Clin. Investig. 1989, 83, 326–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschberg, R.; Kopple, J.D.; Blantz, R.C.; Tucker, B.J. Effects of recombinant human insulin-like growth factor I on glomerular dynamics in the rat. J. Clin. Investig. 1991, 87, 1200–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tönshoff, B.; Nowack, R.; Kurilenko, S.; Blum, W.F.; Seyberth, H.W.; Mehls, O.; Ritz, E. Growth hormone-induced glomerular hyperfiltration is dependent on vasodilating prostanoids. Am. J. Kidney Dis. 1993, 21, 145–151. [Google Scholar] [CrossRef]
- Hale, L.J.; Welsh, G.I.; Perks, C.M.; Hurcombe, J.A.; Moore, S.; Hers, I.; Saleem, M.A.; Mathieson, P.W.; Murphy, A.J.; Jeansson, M.; et al. Insulin-like growth factor-II is produced by, signals to and is an important survival factor for the mature podocyte in man and mouse. J. Pathol. 2013, 230, 95–106. [Google Scholar] [CrossRef]
- Feld, S.; Hirschberg, R. Growth hormone, the insulin-like growth factor system, and the kidney. Endocr. Rev. 1996, 17, 423–480. [Google Scholar] [CrossRef] [Green Version]
- De Boer, H.; Blok, G.J.; Van der Veen, E.A. Clinical aspects of growth hormone deficiency in adults. Endocr. Rev. 1995, 16, 63–86. [Google Scholar] [CrossRef]
- Jørgensen, J.O. Human growth hormone replacement therapy: Pharmacological and clinical aspects. Endocr. Rev. 1991, 12, 189–207. [Google Scholar] [CrossRef]
- Boguszewski, M.C.S. Growth hormone deficiency and replacement in children. Rev. Endocr. Metab. Disord. 2021, 22, 101–108. [Google Scholar] [CrossRef]
- Ikkos, D.; Luft, R.; Sjogren, B. Body water and sodium in patients with acromegaly. J. Clin. Investig. 1954, 33, 989–994. [Google Scholar] [CrossRef] [Green Version]
- Kamenický, P.; Maione, L.; Chanson, P. Cardiovascular complications of acromegaly. Ann. Endocrinol. 2020, 82, 206–209. [Google Scholar] [CrossRef]
- Hirschberg, R.; Brunori, G.; Kopple, J.D.; Guler, H.P. Effects of insulin-like growth factor I on renal function in normal men. Kidney Int. 1993, 43, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.L.; Ginalska-Malinowska, M.; Romer, T.E.; Pucilowska, J.B.; Underwood, L.E. Effects of the infusion of insulin-like growth factor I in a child with growth hormone insensitivity syndrome (Laron dwarfism). N. Engl. J. Med. 1991, 324, 1483–1488. [Google Scholar] [CrossRef]
- Svensson, J.; Tivesten, A.; Sjögren, K.; Isaksson, O.; Bergström, G.; Mohan, S.; Mölne, J.; Isgaard, J.; Ohlsson, C. Liver-derived IGF-I regulates kidney size, sodium reabsorption, and renal IGF-II expression. J. Endocrinol. 2007, 193, 359–366. [Google Scholar] [CrossRef] [Green Version]
- Ho, K.Y.; Weissberger, A.J. The antinatriuretic action of biosynthetic human growth hormone in man involves activation of the renin-angiotensin system. Metabolism 1990, 39, 133–137. [Google Scholar] [CrossRef]
- Møller, J.; Møller, N.; Frandsen, E.; Wolthers, T.; Jørgensen, J.O.; Christiansen, J.S. Blockade of the renin-angiotensin-aldosterone system prevents growth hormone-induced fluid retention in humans. Am. J. Physiol. Content 1997, 272, E803–E808. [Google Scholar] [CrossRef]
- Møller, J.; Jørgensen, J.O.; Møller, N.; Hansen, K.W.; Pedersen, E.B.; Christiansen, J.S. Expansion of extracellular volume and suppression of atrial natriuretic peptide after growth hormone administration in normal man. J. Clin. Endocrinol. Metab. 1991, 72, 768–772. [Google Scholar] [CrossRef]
- Kamenicky, P.; Viengchareun, S.; Blanchard, A.; Meduri, G.; Zizzari, P.; Imbert-Teboul, M.; Doucet, A.; Chanson, P.; Lombès, M. Epithelial sodium channel is a key mediator of growth hormone-induced sodium retention in acromegaly. Endocrinology 2008, 149, 3294–3305. [Google Scholar] [CrossRef] [Green Version]
- Kamenicky, P.; Blanchard, A.; Frank, M.; Salenave, S.; Letierce, A.; Azizi, M.; Lombès, M.; Chanson, P. Body fluid expansion in acromegaly is related to enhanced epithelial sodium channel (ENaC) activity. J. Clin. Endocrinol. Metab. 2011, 96, 2127–2135. [Google Scholar] [CrossRef]
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Harkleroad, K.L.; Tong, Q.; Carattino, M.D.; Johnson, J.P.; Stockand, J.D.; Kleyman, T.R. Epithelial sodium channels are activated by furin-dependent proteolysis. J. Biol. Chem. 2004, 279, 18111–18114. [Google Scholar] [CrossRef] [Green Version]
- Shimomura, Y.; Lee, M.; Oku, J.; Bray, G.A.; Glick, Z. Sodium potassium dependent ATPase in hypophysectomized rats: Response to growth hormone, triiodothyronine, and cortisone. Metabolism 1982, 31, 213–216. [Google Scholar] [CrossRef]
- Nesbitt, T.; Drezner, M.K. Insulin-like growth factor-I regulation of renal 25-hydroxyvitamin D-1-hydroxylase activity. Endocrinology 1993, 132, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Bianda, T.; Glatz, Y.; Bouillon, R.; Froesch, E.R.; Schmid, C. Effects of short-term insulin-like growth factor-I (IGF-I) or growth hormone (GH) treatment on bone metabolism and on production of 1,25-dihydroxycholecalciferol in GH-deficient adults. J. Clin. Endocrinol. Metab. 1998, 83, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, B.A.; Edén, S.; Lönn, L.; Kvist, H.; Stokland, A.; Lindstedt, G.; Bosaeus, I.; Tölli, J.; Sjöström, L.; Isaksson, O.G. Treatment of adults with growth hormone (GH) deficiency with recombinant human GH. J. Clin. Endocrinol. Metab. 1993, 76, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Brixen, K.; Vahl, N.; Jørgensen, J.O.; Christiansen, J.S.; Mosekilde, L.; Hagen, C. Effects of 12 months of growth hormone (GH) treatment on calciotropic hormones, calcium homeostasis, and bone metabolism in adults with acquired GH deficiency: A double blind, randomized, placebo-controlled study. J. Clin. Endocrinol. Metab. 1996, 81, 3352–3359. [Google Scholar] [CrossRef] [Green Version]
- Saggese, G.; Baroncelli, G.I.; Bertelloni, S.; Cinquanta, L.; Di Nero, G. Effects of long-term treatment with growth hormone on bone and mineral metabolism in children with growth hormone deficiency. J. Pediatr. 1993, 122, 37–45. [Google Scholar] [CrossRef]
- Vaccarello, M.A.; Diamond, F.B., Jr.; Guevara-Aguirre, J.; Rosenbloom, A.L.; Fielder, P.J.; Gargosky, S.; Cohen, P.; Wilson, K.; Rosenfeld, R.G. Hormonal and metabolic effects and pharmacokinetics of recombinant insulin-like growth factor-I in growth hormone receptor deficiency/Laron syndrome. J. Clin. Endocrinol. Metab. 1993, 77, 273–280. [Google Scholar] [CrossRef]
- Manroa, P.; Kannan, S.; Hatipoglu, B.; Licata, A. Hypercalcemia and acromegaly-clarifying the connections. A case report and review of the literature. Endocr. Pract. 2014, 20, e86–e90. [Google Scholar] [CrossRef]
- Adema, A.Y.; de Roij van Zuijdewijn, C.L.M.; Hoenderop, J.G.; de Borst, M.H.; Ter Wee, P.M.; Heijboer, A.C.; Vervloet, M.G.; NIGRAM Consortium. Influence of exogenous growth hormone administration on circulating concentrations of α-klotho in healthy and chronic kidney disease subjects: A prospective, single-center open case-control pilot study. BMC Nephrol. 2018, 19, 327. [Google Scholar] [CrossRef]
- Locher, R.; Egger, A.; Zwimpfer, C.; Sze, L.; Schmid, C.; Christ, E. Effect of Growth hormone replacement therapy on soluble Klotho in patients with Growth hormone deficiency. Clin. Endocrinol. 2015, 83, 593–595. [Google Scholar] [CrossRef]
- Andrukhova, O.; Smorodchenko, A.; Egerbacher, M.; Streicher, C.; Zeitz, U.; Goetz, R.; Shalhoub, V.; Mohammadi, M.; Pohl, E.E.; Lanske, B.; et al. FGF23 promotes renal calcium reabsorption through the TRPV5 channel. EMBO J. 2014, 33, 229–246. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, A.M.; Thomas, J.; Clewes, A.; Hopkins, M.T.; Guzder, R.; Ibrahim, H.; Durham, B.H.; Vora, J.P.; Fraser, W.D. Effects of growth hormone replacement on parathyroid hormone sensitivity and bone mineral metabolism. J. Clin. Endocrinol. Metab. 2003, 88, 2860–2868. [Google Scholar] [CrossRef] [Green Version]
- Quigley, R.; Baum, M. Effects of growth hormone and insulin-like growth factor I on rabbit proximal convoluted tubule transport. J. Clin. Investig. 1991, 88, 368–374. [Google Scholar] [CrossRef] [Green Version]
- Hirschberg, R.; Ding, H.; Wanner, C. Effects of insulin-like growth factor I on phosphate transport in cultured proximal tubule cells. J. Lab. Clin. Med. 1995, 126, 428–434. [Google Scholar]
- Jehle, A.W.; Forgo, J.; Biber, J.; Lederer, E.; Krapf, R.; Murer, H. IGF-I and vanadate stimulate Na/Pi-cotransport in OK cells by increasing type II Na/Pi-cotransporter protein stability. Pflugers Arch. 1998, 437, 149–154. [Google Scholar] [CrossRef]
- Xie, T.; Tian, P.; Wu, S.; Zhang, X.; Liu, T.; Gu, Y.; Sun, C.; Hu, F. Serum phosphate: Does it more closely reflect the true state of acromegaly? J. Clin. Neurosci. 2020, 71, 26–31. [Google Scholar] [CrossRef]
- Haffner, D.; Grund, A.; Leifheit-Nestler, M. Renal effects of growth hormone in health and in kidney disease. Pediatr. Nephrol. 2021, 36, 2511–2530. [Google Scholar] [CrossRef]
- Moskowitz, D.W.; Liu, W. Gene expression after uninephrectomy in the rat: Simultaneous expression of positive and negative growth control elements. J. Urol. 1995, 154, 1560–1565. [Google Scholar] [CrossRef]
- Flyvbjerg, A.; Bennett, W.F.; Rasch, R.; van Neck, J.W.; Groffen, C.A.; Kopchick, J.J.; Scarlett, J.A. Compensatory renal growth in uninephrectomized adult mice is growth hormone dependent. Kidney Int. 1999, 56, 2048–2054. [Google Scholar] [CrossRef] [Green Version]
- Mulroney, S.E.; Haramati, A.; Werner, H.; Bondy, C.; Roberts, C.T., Jr.; LeRoith, D. Altered expression of insulin-like growth factor-I (IGF-I) and IGF receptor genes after unilateral nephrectomy in immature rats. Endocrinology 1992, 130, 249–256. [Google Scholar] [CrossRef] [Green Version]
- McArdle, Z.; Schreuder, M.F.; Moritz, K.M.; Denton, K.M.; Singh, R.R. Physiology and pathophysiology of compensatory adaptations of a solitary functioning kidney. Front. Physiol. 2020, 11, 725. [Google Scholar] [CrossRef]
- Luque, V.; Escribano, J.; Grote, V.; Ferre, N.; Koletzko, B.; Gruszfeld, D.; Socha, P.; Langhendries, J.P.; Goyens, P.; Closa-Monasterolo, R. European childhood obesity project. Does insulin-like growth factor-1 mediate protein-induced kidney growth in infants? A secondary analysis from a randomized controlled trial. Pediatr. Res. 2013, 74, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haramati, A.; Lumpkin, M.D.; Mulroney, S.E. Early increase in pulsatile growth hormone release after unilateral nephrectomy in adult rats. Am. J. Physiol. Physiol. 1994, 266, F628–F632. [Google Scholar] [CrossRef] [PubMed]
- Mulroney, S.E.; Woda, C.; Johnson, M.; Pesce, C. Gender differences in renal growth and function after uninephrectomy in adult rats. Kidney Int. 1999, 56, 944–953. [Google Scholar] [CrossRef] [Green Version]
- Mok, K.-Y.; Sweeny, J.M.; Zheng, W.; Sandberg, K.; Mulroney, S.E. Gender differences in renal Ang II AT1 receptor regulation after uninephrectomy: GH dependence in the male rat. FASEB J. 1998, 12, A1154. [Google Scholar]
- Nordstrom, S.M.; Tran, J.L.; Sos, B.C.; Wagner, K.U.; Weiss, E.J. Liver-derived IGF-I contributes to GH-dependent increases in lean mass and bone mineral density in mice with comparable levels of circulating GH. Mol. Endocrinol. 2011, 25, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Landau, D.; Biada, J.; Chen, Y.; Sood, S.; Yakar, S.; Leroith, D.; Segev, Y.; Rabkin, R. A marked deficiency in circulating and renal IGF-I peptide does not inhibit compensatory renal enlargement in uninephrectomized mice. Growth Horm. IGF Res. 2011, 21, 279–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, B.A.; Johnson, R.J.; Alpers, C.E.; Eng, E.; Gordon, K.; Floege, J.; Couser, W.G.; Seidel, K. Cellular events in the evolution of experimental diabetic nephropathy. Kidney Int. 1995, 47, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flyvbjerg, A. Putative pathophysiological role of growth factors and cytokines in experimental diabetic kidney disease. Diabetologia 2000, 43, 1205–1223. [Google Scholar] [CrossRef] [Green Version]
- Landau, D.; Segev, Y.; Eshet, R.; Flyvbjerg, A.; Phillip, M. Changes in the growth hormone-IGF-I axis in non-obese diabetic mice. Int. J. Exp. Diabetes Res. 2000, 1, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Mercado, M.; Molitch, M.E.; Baumann, G. Low plasma growth hormone binding protein in IDDM. Diabetes 1992, 41, 605–609. [Google Scholar] [CrossRef]
- Landau, D.; Israel, E.; Rivkis, I.; Kachko, L.; Schrijvers, B.F.; Flyvbjerg, A.; Phillip, M.; Segev, Y. The effect of growth hormone on the development of diabetic kidney disease in rats. Nephrol. Dial. Transplant. 2003, 18, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Muchaneta-Kubara, E.C.; Sayed-Ahmed, N.; Besbas, N.; Zhang, G.; Cope, G.H.; el Nahas, A.M. Experimental diabetic renal growth: Role of growth hormone and insulin-like growth factor-I. Nephrol. Dial. Transplant. 1994, 9, 1395–1401. [Google Scholar] [CrossRef]
- Segev, Y.; Landau, D.; Rasch, R.; Flyvbjerg, A.; Phillip, M. Growth hormone receptor antagonism prevents early renal changes in nonobese diabetic mice. J. Am. Soc. Nephrol. 1999, 10, 2374–2381. [Google Scholar] [CrossRef] [PubMed]
- Serri, O.; Beauregard, H.; Brazeau, P.; Abribat, T.; Lambert, J.; Harris, A.; Vachon, L. Somatostatin analogue, octreotide, reduces increased glomerular filtration rate and kidney size in insulin-dependent diabetes. JAMA 1991, 265, 888–892. [Google Scholar] [CrossRef]
- Segev, Y.; Eshet, R.; Rivkis, I.; Hayat, C.; Kachko, L.; Phillip, M.; Landau, D. Comparison between somatostatin analogues and ACE inhibitor in the NOD mouse model of diabetic kidney disease. Nephrol. Dial. Transplant. 2004, 19, 3021–3028. [Google Scholar] [CrossRef] [Green Version]
- Segev, Y.; Landau, D.; Marbach, M.; Shehadeh, N.; Flyvbjerg, A.; Phillip, M. Renal hypertrophy in hyperglycemic non-obese diabetic mice is associated with persistent renal accumulation of insulin-like growth factor I. J. Am. Soc. Nephrol. 1997, 8, 436–444. [Google Scholar] [CrossRef]
- Park, I.S.; Kiyomoto, H.; Alvarez, F.; Xu, Y.C.; Abboud, H.E.; Abboud, S.L. Preferential expression of insulin-like growth factor binding proteins-1, -3, and -5 during early diabetic renal hypertrophy in rats. Am. J. Kidney Dis. 1998, 32, 1000–1010. [Google Scholar] [CrossRef]
- Flyvbjerg, A.; Kessler, U.; Kiess, W. Increased kidney and liver insulin-like growth factor II/mannose-6-phosphate receptor concentration in experimental diabetes in rats. Growth Regul. 1994, 4, 188–193. [Google Scholar]
- Verzola, D.; Gandolfo, M.T.; Ferrario, F.; Rastaldi, M.P.; Villaggio, B.; Gianiorio, F.; Giannoni, M.; Rimoldi, L.; Lauria, F.; Miji, M.; et al. Apoptosis in the kidneys of patients with type II diabetic nephropathy. Kidney Int. 2007, 72, 1262–1272. [Google Scholar] [CrossRef] [Green Version]
- Reddy, G.R.; Pushpanathan, M.J.; Ransom, R.F.; Holzman, L.B.; Brosius, F.C., 3rd; Diakonova, M.; Mathieson, P.; Saleem, M.A.; List, E.O.; Kopchick, J.J.; et al. Identification of the glomerular podocyte as a target for growth hormone action. Endocrinology 2007, 148, 2045–2055. [Google Scholar] [CrossRef] [Green Version]
- Vasylyeva, T.L.; Ferry, R.J., Jr. Novel roles of the IGF-IGFBP axis in etiopathophysiology of diabetic nephropathy. Diabetes Res. Clin. Pract. 2007, 76, 177–186. [Google Scholar] [CrossRef] [Green Version]
- Fornoni, A.; Rosenzweig, S.A.; Lenz, O.; Rivera, A.; Striker, G.E.; Elliot, S.J. Low insulin-like growth factor binding protein-2 expression is responsible for increased insulin receptor substrate-1 phosphorylation in mesangial cells from mice susceptible to glomerulosclerosis. Endocrinology 2006, 147, 3547–3554. [Google Scholar] [CrossRef] [Green Version]
- Vasylyeva, T.L.; Chen, X.; Ferry, R.J., Jr. Insulin-like growth factor binding protein-3 mediates cytokine-induced mesangial cell apoptosis. Growth Horm. IGF Res. 2005, 15, 207–214. [Google Scholar] [CrossRef] [Green Version]
- Chitra, P.S.; Swathi, T.; Sahay, R.; Reddy, G.B.; Menon, R.K.; Kumar, P.A. Growth Hormone Induces Transforming Growth Factor-Beta-Induced Protein in Podocytes: Implications for Podocyte Depletion and Proteinuria. J. Cell. Biochem. 2015, 116, 1947–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishad, R.; Mukhi, D.; Tahaseen, S.V.; Mungamuri, S.K.; Pasupulati, A.K. Growth hormone induces Notch1 signaling in podocytes and contributes to proteinuria in diabetic nephropathy. J. Biol. Chem. 2019, 294, 16109–16122. [Google Scholar] [CrossRef] [PubMed]
- Levin-Iaina, N.; Iaina, A.; Raz, I. The emerging role of NO and IGF-1 in early renal hypertrophy in STZ-induced diabetic rats. Diabetes Metab. Res. Rev. 2011, 27, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Bach, L.A.; Dean, R.; Youssef, S.; Cooper, M.E. Aminoguanidine ameliorates changes in the IGF system in experimental diabetic nephropathy. Nephrol. Dial. Transplant. 2000, 15, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Isshiki, K.; He, Z.; Maeno, Y.; Ma, R.C.; Yasuda, Y.; Kuroki, T.; White, G.S.; Patti, M.E.; Weir, G.C.; King, G.L. Insulin regulates SOCS2 expression and the mitogenic effect of IGF-1 in mesangial cells. Kidney Int. 2008, 74, 1434–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahesh, S.; Kaskel, F. Growth hormone axis in chronic kidney disease. Pediatr. Nephrol. 2008, 23, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Edmondson, S.R.; Baker, N.L.; Oh, J.; Kovacs, G.; Werther, G.A.; Mehls, O. Growth hormone receptor abundance in tibial growth plates of uremic rats: GH/IGF-I treatment. Kidney Int. 2000, 58, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Tönshoff, B.; Cronin, M.J.; Reichert, M.; Haffner, D.; Wingen, A.M.; Blum, W.F.; Mehls, O. Reduced concentration of serum growth hormone (GH)-binding protein in children with chronic renal failure: Correlation with GH insensitivity. The European Study Group for Nutritional Treatment of Chronic Renal Failure in Childhood. The German Study Group for Growth Hormone Treatment in Chronic Renal Failure. J. Clin. Endocrinol. Metab. 1997, 82, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Rabkin, R.; Sun, D.F.; Chen, Y.; Tan, J.; Schaefer, F. Growth hormone resistance in uremia, a role for impaired JAK/STAT signaling. Pediatr. Nephrol. 2005, 20, 313–318. [Google Scholar] [CrossRef]
- Schaefer, F.; Chen, Y.; Tsao, T.; Nouri, P.; Rabkin, R. Impaired JAK-STAT signal transduction contributes to growth hormone resistance in chronic uremia. J. Clin. Investig. 2001, 108, 467–475. [Google Scholar] [CrossRef]
- Troib, A.; Landau, D.; Kachko, L.; Rabkin, R.; Segev, Y. Epiphyseal growth plate growth hormone receptor signaling is decreased in chronic kidney disease-related growth retardation. Kidney Int. 2013, 84, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Sun, D.F.; Zheng, Z.; Tummala, P.; Oh, J.; Schaefer, F.; Rabkin, R. Chronic uremia attenuates growth hormone-induced signal transduction in skeletal muscle. J. Am. Soc. Nephrol. 2004, 15, 2630–2636. [Google Scholar] [CrossRef]
- Wiezel, D.; Assadi, M.H.; Landau, D.; Troib, A.; Kachko, L.; Rabkin, R.; Segev, Y. Impaired renal growth hormone JAK/STAT5 signaling in chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, 791–799. [Google Scholar] [CrossRef] [Green Version]
- Bach, L.A.; Hale, L.J. Insulin-like growth factors and kidney disease. Am. J. Kidney Dis. 2015, 65, 327–336. [Google Scholar] [CrossRef]
- Powell, D.R.; Durham, S.K.; Liu, F.; Baker, B.K.; Lee, P.D.; Watkins, S.L.; Campbell, P.G.; Brewer, E.D.; Hintz, R.L.; Hogg, R.J. The insulin-like growth factor axis and growth in children with chronic renal failure: A report of the Southwest Pediatric Nephrology Study Group. J. Clin. Endocrinol. Metab. 1998, 83, 1654–1661. [Google Scholar] [CrossRef] [Green Version]
- Ding, H.; Gao, X.L.; Hirschberg, R.; Vadgama, J.V.; Kopple, J.D. Impaired actions of insulin-like growth factor 1 on protein Synthesis and degradation in skeletal muscle of rats with chronic renal failure. Evidence for a postreceptor defect. J. Clin. Investig. 1996, 97, 1064–1075. [Google Scholar] [CrossRef] [Green Version]
- Tönshoff, B.; Kiepe, D.; Ciarmatori, S. Growth hormone/insulin-like growth factor system in children with chronic renal failure. Pediatr. Nephrol. 2005, 20, 279–289. [Google Scholar] [CrossRef]
- Challa, A.; Chan, W.; Krieg, R.J., Jr.; Thabet, M.A.; Liu, F.; Hintz, R.L.; Chan, J.C. Effect of metabolic acidosis on the expression of insulin-like growth factor and growth hormone receptor. Kidney Int. 1993, 44, 1224–1227. [Google Scholar] [CrossRef] [Green Version]
- Hochberg, Z. Mechanisms of steroid impairment of growth. Horm. Res. 2002, 58, 33–38. [Google Scholar] [CrossRef]
- Kawaguchi, H.; Hattori, M.; Ito, K. Somatic and renal effects of growth hormone in rats with chronic renal failure. Pediatr. Nephrol. 1997, 11, 280–284. [Google Scholar] [CrossRef]
- Miller, S.B.; Hansen, V.A.; Hammerman, M.R. Effects of growth hormone and IGF-I on renal function in rats with normal and reduced renal mass. Am. J. Physiol. Physiol. 1990, 259, F747–F751. [Google Scholar] [CrossRef]
- Tönshoff, B.; Heinrich, U.; Mehls, O. How safe is the treatment of uraemic children with recombinant human growth hormone? Pediatr. Nephrol. 1991, 5, 454–460. [Google Scholar] [CrossRef]
- Maxwell, H.; Nair, D.R.; Dalton, R.N.; Rigden, S.P.; Rees, L. Differential effects of recombinant human growth hormone on glomerular filtration rate and renal plasma flow in chronic renal failure. Pediatr. Nephrol. 1995, 9, 458–463. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gurevich, E.; Segev, Y.; Landau, D. Growth Hormone and IGF1 Actions in Kidney Development and Function. Cells 2021, 10, 3371. https://doi.org/10.3390/cells10123371
Gurevich E, Segev Y, Landau D. Growth Hormone and IGF1 Actions in Kidney Development and Function. Cells. 2021; 10(12):3371. https://doi.org/10.3390/cells10123371
Chicago/Turabian StyleGurevich, Evgenia, Yael Segev, and Daniel Landau. 2021. "Growth Hormone and IGF1 Actions in Kidney Development and Function" Cells 10, no. 12: 3371. https://doi.org/10.3390/cells10123371
APA StyleGurevich, E., Segev, Y., & Landau, D. (2021). Growth Hormone and IGF1 Actions in Kidney Development and Function. Cells, 10(12), 3371. https://doi.org/10.3390/cells10123371