
Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanoparticle Synthesis
2.3. Cells and Treatments
2.4. Immunoblotting
2.5. Microscopy
2.6. Inducible mKeima Assay System
2.6.1. Generation of Stable Cell Lines
2.6.2. Detection of mKeima Processing
2.7. Long-Lived Protein Degradation Assay (LLPD)
2.8. Autophagic LDH Sequestration Assay
2.9. Detection of Reactive Oxygen Species (ROS)
2.10. Glutathione Measurement (GSH)
2.11. Statistical Analysis
3. Results
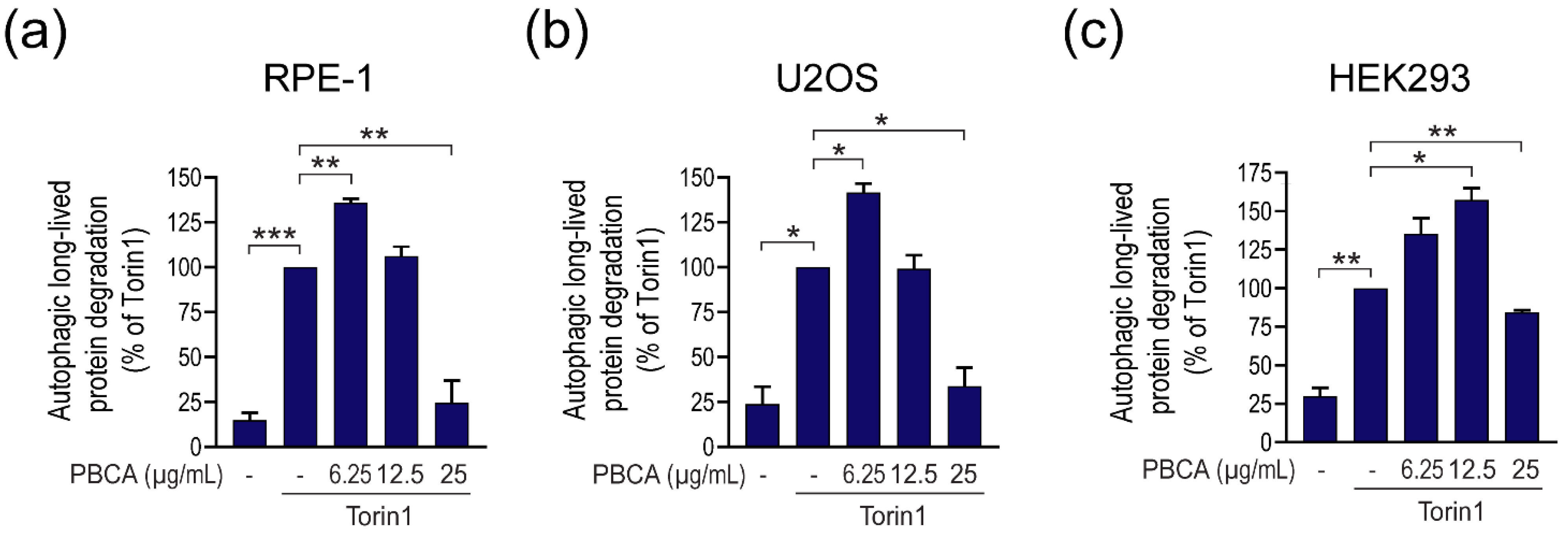
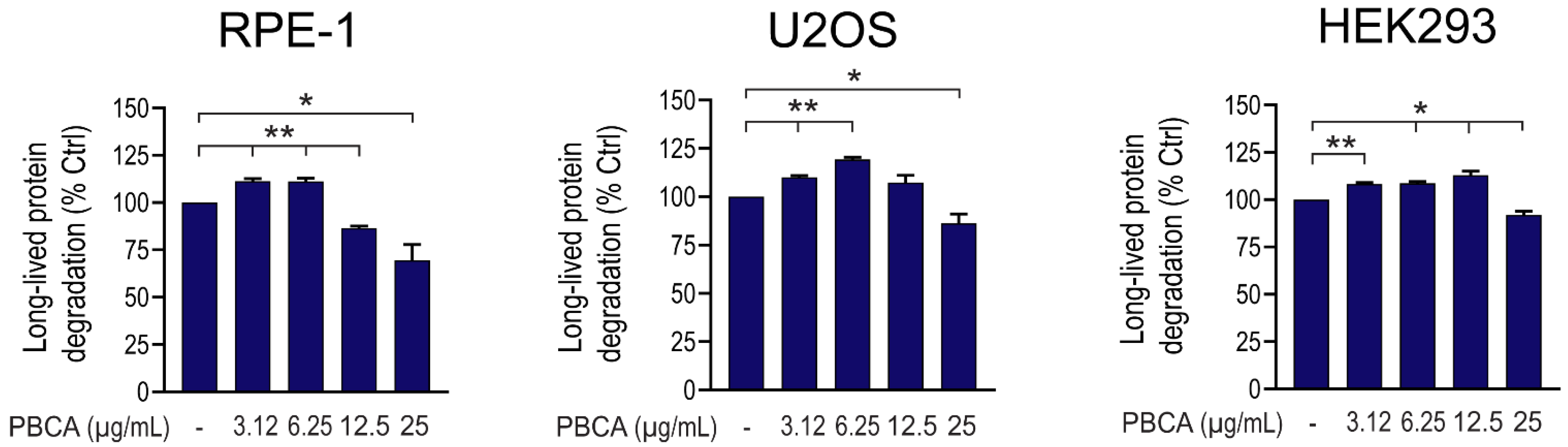
3.1. PBCA NPs Trigger Concentration-Dependent, Divergent Effects on Autophagy
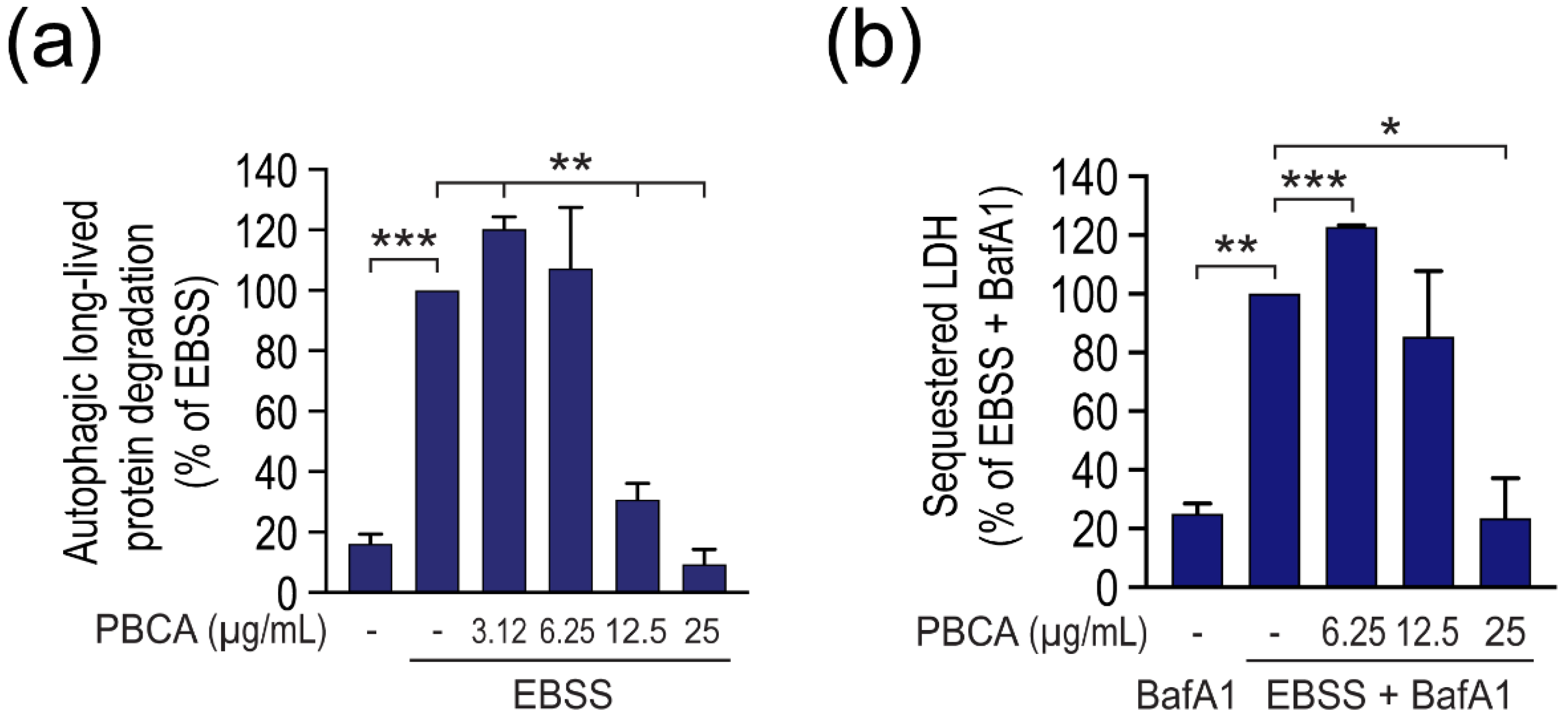
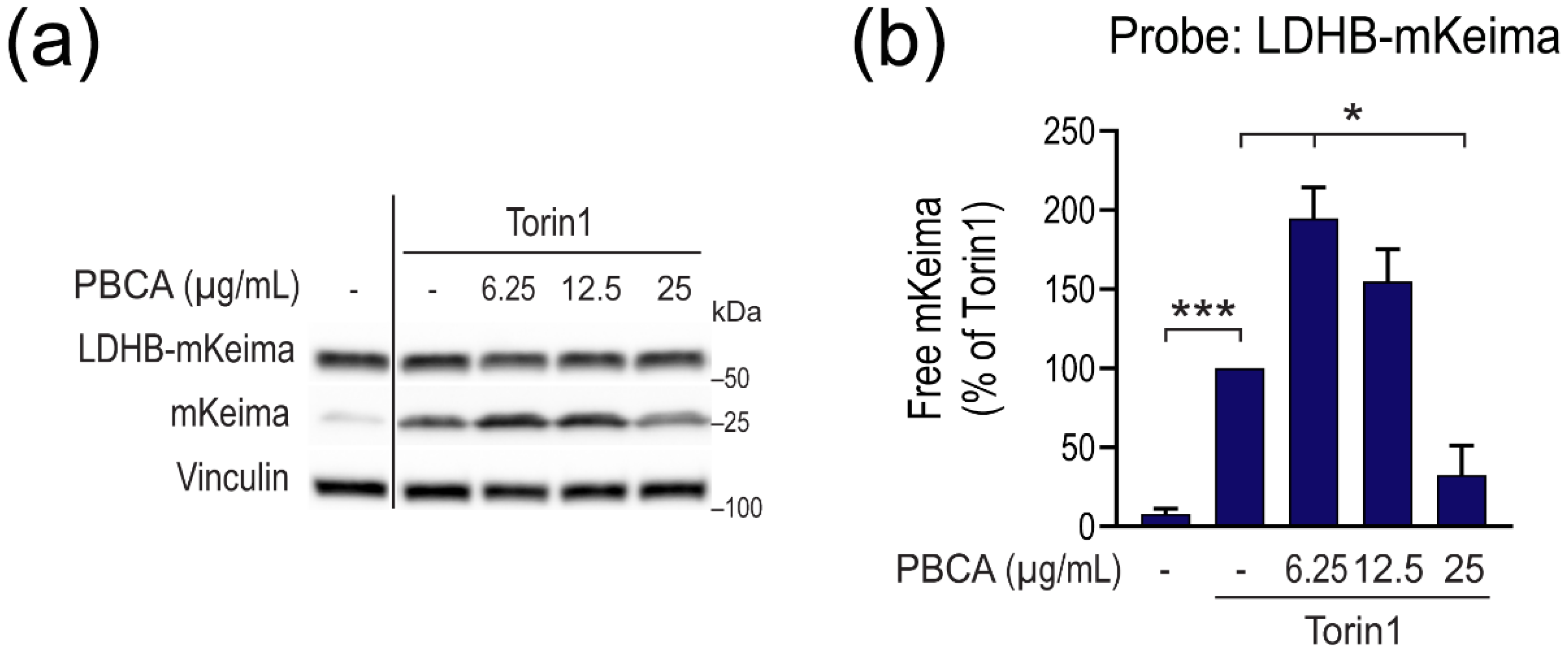
3.2. High Concentrations of PBCA Inhibit Autophagic LC3 Flux and LC3 Lipidation, Whereas Low Concentrations Activate Canonical, Yet LC3 Flux-Independent Autophagy
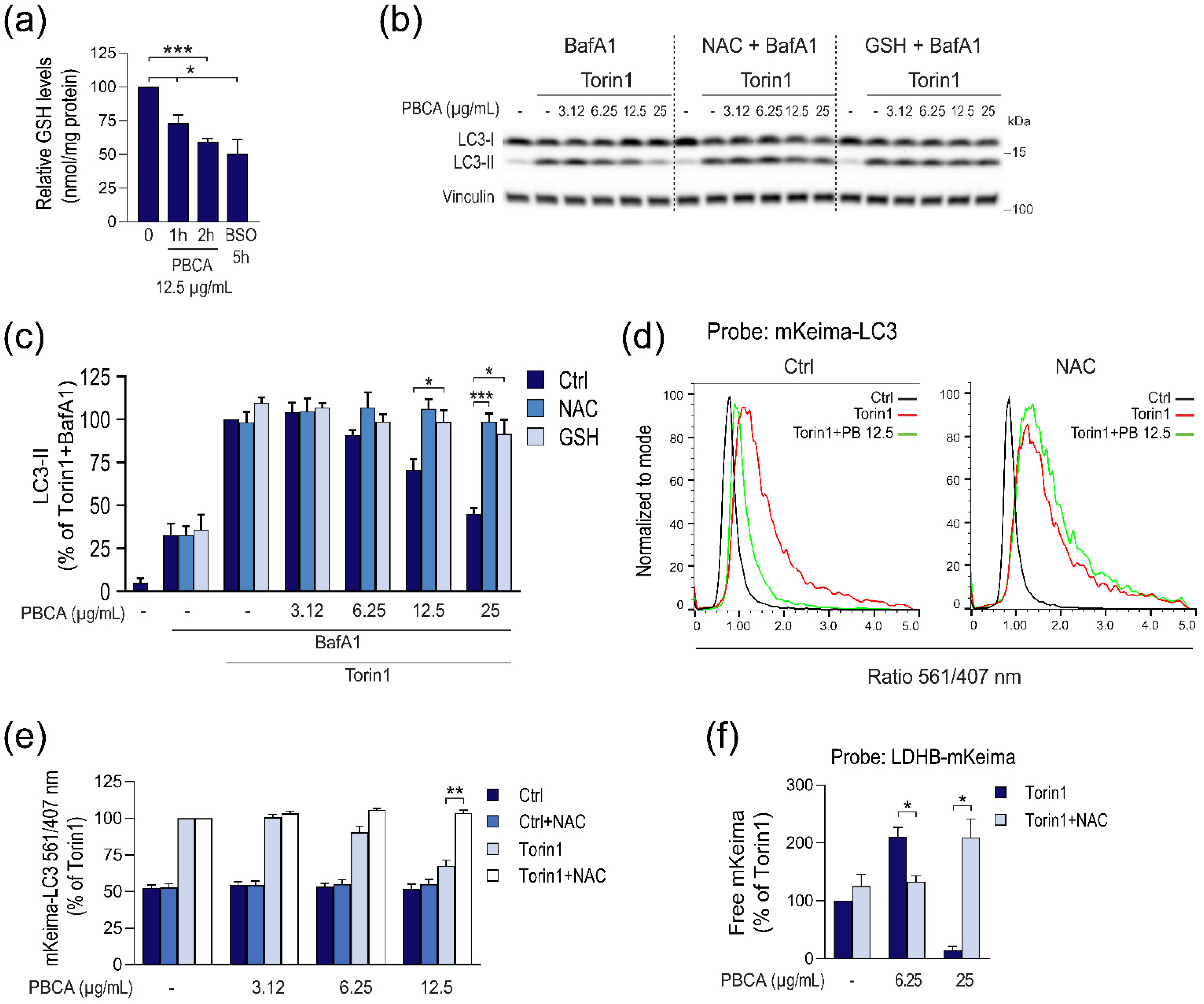
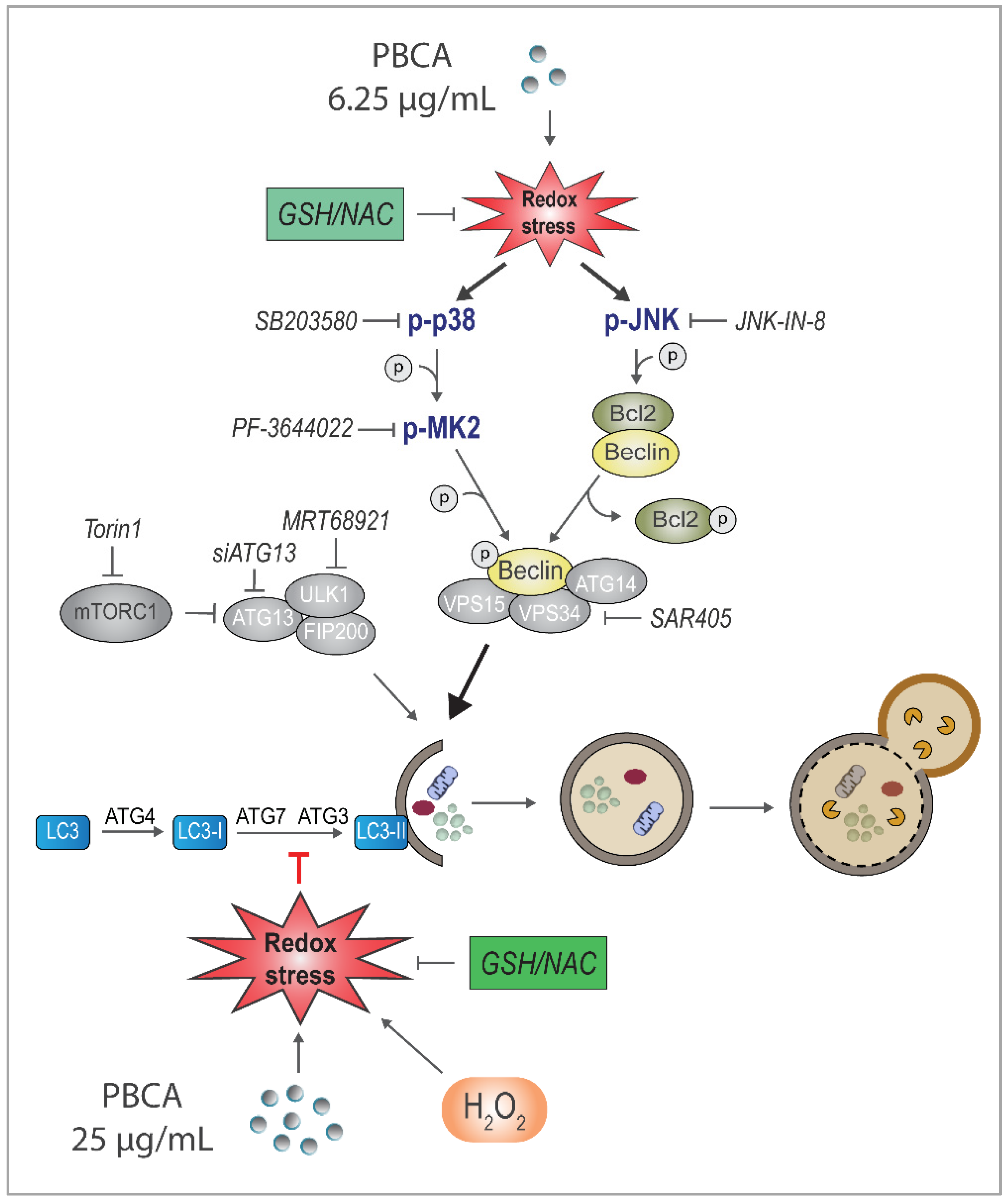
3.3. PBCA-Induced Redox Imbalance Is Required for Activation of Autophagy at Low PBCA Concentrations, and Mediates Inhibition of LC3 Lipidation and Autophagy, as Well as Subsequent Cytotoxicity at High PBCA Concentrations
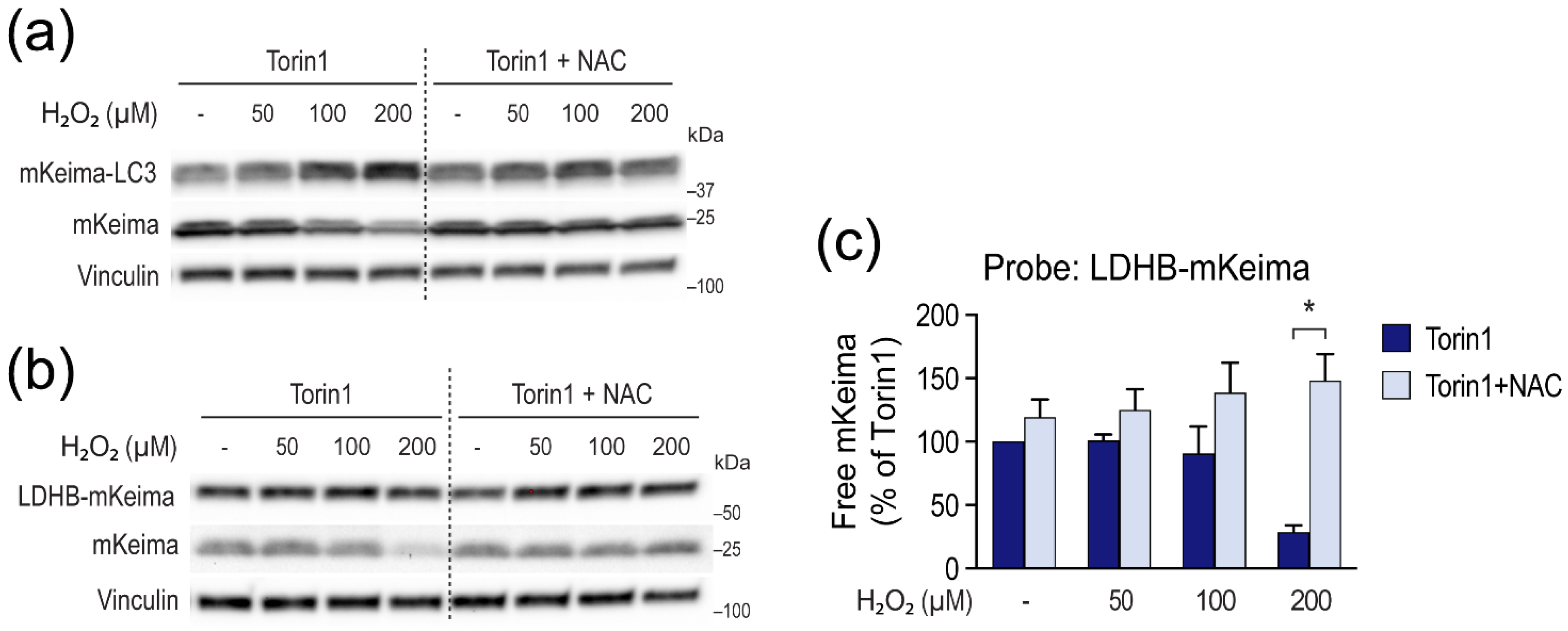
3.4. H2O2 Inhibits LC3 Lipidation and Autophagic Degradation
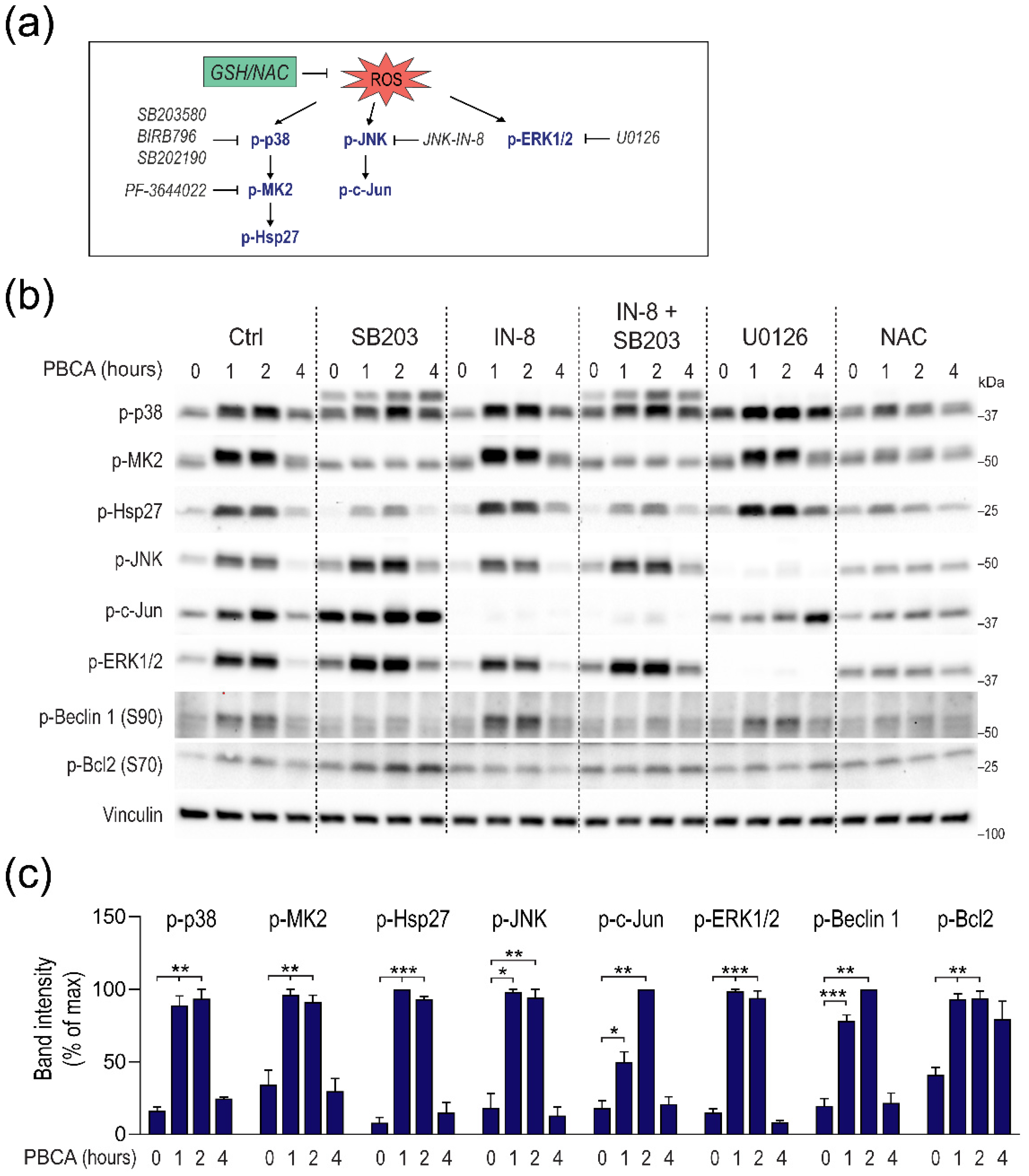
3.5. PBCA-Induced Redox Imbalance Induces Phosphorylation of Beclin-1 and Bcl-2 via p38 and JNK
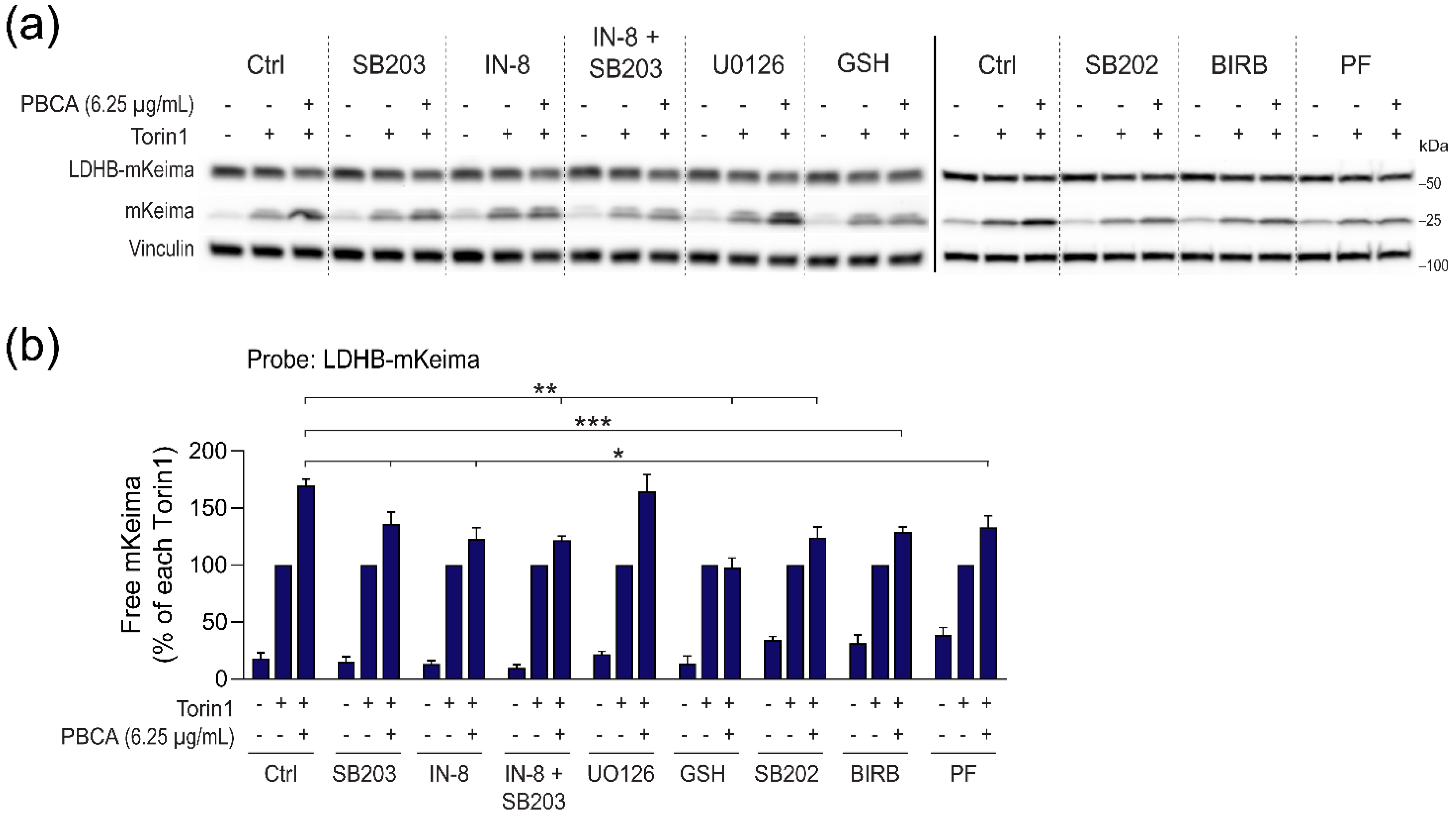
3.6. PBCA Activates Autophagy Downstream of p38 and JNK Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mørch, Ý.; Hansen, R.; Berg, S.; Åslund, A.K.; Glomm, W.R.; Eggen, S.; Schmid, R.; Johnsen, H.; Kubowicz, S.; Snipstad, S.; et al. Nanoparticle-stabilized microbubbles for multimodal imaging and drug delivery. Contrast Media Mol. Imaging 2015, 10, 356–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baghirov, H.; Snipstad, S.; Sulheim, E.; Berg, S.; Hansen, R.; Thorsen, F.; Mørch, Y.; Davies, C.L.; Åslund, A.K.O. Ultrasound-mediated delivery and distribution of polymeric nanoparticles in the normal brain parenchyma of a metastatic brain tumour model. PLoS ONE 2018, 13, e0191102. [Google Scholar] [CrossRef] [Green Version]
- Melguizo, C.; Cabeza, L.; Prados, J.; Ortiz, R.; Caba, O.; Rama, A.R.; Delgado, Á.V.; Arias, J.L. Enhanced antitumoral activity of doxorubicin against lung cancer cells using biodegradable poly(butylcyanoacrylate) nanoparticles. Drug Des. Dev. Ther. 2015, 9, 6433–6444. [Google Scholar]
- Fusser, M.; Øverbye, A.; Pandya, A.D.; Mørch, Ý.; Borgos, S.E.; Kildal, W.; Snipstad, S.; Sulheim, E.; Fleten, K.G.; Askautrud, H.A.; et al. Cabazitaxel-loaded Poly(2-ethylbutyl cyanoacrylate) nanoparticles improve treatment efficacy in a patient derived breast cancer xenograft. J. Control. Release 2019, 293, 183–192. [Google Scholar] [CrossRef]
- Graf, A.; McDowell, A.; Rades, T. Poly(alkylcyanoacrylate) nanoparticles for enhanced delivery of therapeutics—Is there real potential? Expert Opin. Drug Deliv. 2009, 6, 371–387. [Google Scholar] [CrossRef] [PubMed]
- Vauthier, C.; Dubernet, C.; Fattal, E.; Pinto-Alphandary, H.; Couvreur, P. Poly(alkylcyanoacrylates) as biodegradable materials for biomedical applications. Adv. Drug Deliv. Rev. 2003, 55, 519–548. [Google Scholar] [CrossRef]
- Petibone, D.M.; Majeed, W.; Casciano, D.A. Autophagy function and its relationship to pathology, clinical applications, drug metabolism and toxicity. J. Appl. Toxicol. 2017, 37, 23–37. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Ye, X.; Zhao, T. The physiological roles of autophagy in the mammalian life cycle. Biol. Rev. Camb. Philos. Soc. 2019, 94, 503–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nel, A.; Xia, T.; Mädler, L.; Li, N. Toxic Potential of Materials at the Nanolevel. Science 2006, 311, 622. [Google Scholar] [CrossRef] [Green Version]
- Wen, T.; Liu, J.; He, W.; Yang, A. Nanomaterials and Reactive Oxygen Species (ROS). In Nanotechnology in Regenerative Medicine and Drug Delivery Therapy; Xu, H., Gu, N., Eds.; Springer: Singapore, 2020; pp. 361–387. [Google Scholar]
- Wang, Y.; Cai, R.; Chen, C. The Nano–Bio Interactions of Nanomedicines: Understanding the Biochemical Driving Forces and Redox Reactions. Acc. Chem. Res. 2019, 52, 1507–1518. [Google Scholar] [CrossRef] [PubMed]
- Macip, S.; Igarashi, M.; Berggren, P.; Yu, J.; Lee, S.W.; Aaronson, S.A. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol. Cell. Biol. 2003, 23, 8576–8585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pajares, M.; Cuadrado, A.; Engedal, N.; Jirsova, Z.; Cahova, M. The Role of Free Radicals in Autophagy Regulation: Implications for Ageing. Oxid. Med. Cell. Longev. 2018, 2018, 2450748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Sedlackova, L.; Kelly, G.; Korolchuk, V.I. The pROS of Autophagy in Neuronal Health. J. Mol. Biol. 2020, 432, 2546–2559. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a Post-Translational Modification that Regulates Autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. Regulation of autophagy by ROS: Physiology and pathology. Trends Biochem. Sci. 2011, 36, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Ohsumi, Y.; Yoshimori, T. Autophagosome formation in mammalian cells. Cell Struct. Funct. 2002, 27, 421–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, M.; Le, W.-D. The Role of Nanomaterials in Autophagy. In Autophagy: Biology and Diseases: Basic Science; Qin, Z.-H., Ed.; Springer: Singapore, 2019; pp. 273–286. [Google Scholar]
- Szwed, M.; Sønstevold, T.; Øverbye, A.; Engedal, N.; Grallert, B.; Mørch, Ý.; Sulheim, E.; Iversen, T.G.; Skotland, T.; Sandvig, K. Small variations in nanoparticle structure dictate differential cellular stress responses and mode of cell death. Nanotoxicology 2019, 13, 761–782. [Google Scholar] [CrossRef] [PubMed]
- Sønstevold, T.; Engedal, N.; Mørch, Ý.; Iversen, T.G.; Skotland, T.; Sandvig, K.; Torgersen, M.L. Structural Variants of poly(alkylcyanoacrylate) Nanoparticles Differentially Affect LC3 and Autophagic Cargo Degradation. J. Biomed. Nanotechnol. 2020, 16, 432–445. [Google Scholar] [CrossRef] [PubMed]
- Sulheim, E.; Iversen, T.-G.; To Nakstad, V.; Klinkenberg, G.; Sletta, H.; Schmid, R.; Hatletveit, A.R.; Wågbø, A.M.; Sundan, A.; Skotland, T.; et al. Cytotoxicity of Poly(alkyl cyanoacrylate) Nanoparticles. Int. J. Mol. Sci. 2017, 18, 2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopitz, J.; Kisen, G.O.; Gordon, P.B.; Bohley, P.; Seglen, P.O. Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol. 1990, 111, 941–953. [Google Scholar] [CrossRef]
- Luhr, M.; Szalai, P.; Engedal, N. The Lactate Dehydrogenase Sequestration Assay—A Simple and Reliable Method to Determine Bulk Autophagic Sequestration Activity in Mammalian Cells. J. Vis. Exp. 2018, 137, 57971. [Google Scholar]
- An, H.; Harper, J.W. Systematic analysis of ribophagy in human cells reveals bystander flux during selective autophagy. Nat. Cell Biol. 2018, 20, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Engedal, N.; Sønstevold, T.; Beese, C.J.; Selladurai, S.; Melcher, T.; Simensen, J.E.; Frankel, L.B.; Urbanucci, A.; Torgersen, M.L. Measuring autophagic cargo flux with Keima-based probes. In Methods Molecular Biology; Springer Nature: Berlin/Heidelberg, Germany, 2021; p. 2445. [Google Scholar]
- Heinz, N.; Schambach, A.; Galla, M.; Maetzig, T.; Baum, C.; Loew, R.; Schiedlmeier, B. Retroviral and transposon-based tet-regulated all-in-one vectors with reduced background expression and improved dynamic range. Hum. Gene Ther. 2011, 22, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Katayama, H.; Kogure, T.; Mizushima, N.; Yoshimori, T.; Miyawaki, A. A Sensitive and Quantitative Technique for Detecting Autophagic Events Based on Lysosomal Delivery. Chem. Biol. 2011, 18, 1042–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Violot, S.; Carpentier, P.; Blanchoin, L.; Bourgeois, D. Reverse pH-Dependence of Chromophore Protonation Explains the Large Stokes Shift of the Red Fluorescent Protein mKeima. J. Am. Chem. Soc. 2009, 131, 10356–10357. [Google Scholar] [CrossRef] [PubMed]
- Luhr, M.; Sætre, F.; Engedal, N. The Long-lived Protein Degradation Assay: An Efficient Method for Quantitative Determination of the Autophagic Flux of Endogenous Proteins in Adherent Cell Lines. Bio-Protocol 2018, 8, e2836. [Google Scholar] [CrossRef] [Green Version]
- Singh, V.; Gera, R.; Purohit, M.P.; Patnaik, S.; Ghosh, D. Fluorometric Estimation of Glutathione in Cultured Microglial Cell Lysate. Bio-Protocol 2017, 7, e2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Drew, R.; Miners, J.O. The effects of buthionine sulphoximine (BSO) on glutathione depletion and xenobiotic biotransformation. Biochem. Pharmacol. 1984, 33, 2989–2994. [Google Scholar] [CrossRef]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Corcelle, E.; Djerbi, N.; Mari, M.; Nebout, M.; Fiorini, C.; Fenichel, P.; Hofman, P.; Poujeol, P.; Mograbi, B. Control of the Autophagy Maturation Step by the MAPK ERK and p38: Lessons from Environmental Carcinogens. Autophagy 2007, 3, 57–59. [Google Scholar] [CrossRef] [Green Version]
- Lorin, S.; Pierron, G.; Ryan, K.M.; Codogno, P.; Djavaheri-Mergny, M. Evidence for the interplay between JNK and p53-DRAM signaling pathways in the regulation of autophagy. Autophagy 2010, 6, 153–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webber, J.L.; Tooze, S.A. Coordinated regulation of autophagy by p38α MAPK through mAtg9 and p38IP. EMBO J. 2010, 29, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moruno-Manchón, J.F.; Pérez-Jiménez, E.; Knecht, E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem. J. 2012, 449, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef]
- Wei, Y.; An, Z.; Zou, Z.; Sumpter, R.; Su, M.; Zang, X.; Sinha, S.; Gaestel, M.; Levine, B. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. eLife 2015, 4, e05289. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Bauvy, C.; Carpentier, S.; Levade, T.; Levine, B.; Codogno, P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J. Biol. Chem. 2009, 284, 2719–2728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Pattingre, S.; Sinha, S.; Bassik, M.; Levine, B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 2008, 30, 678–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fogel, A.I.; Dlouhy, B.J.; Wang, C.; Ryu, S.-W.; Neutzner, A.; Hasson, S.A.; Sideris, D.P.; Abeliovich, H.; Youle, R.J. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol. Cell. Biol. 2013, 33, 3675–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, N.; Usui, T.; Ohama, T.; Sato, K. Regulation of Beclin 1 Protein Phosphorylation and Autophagy by Protein Phosphatase 2A (PP2A) and Death-associated Protein Kinase 3 (DAPK3). J. Biol. Chem. 2016, 291, 10858–10866. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; She, H.; Zhang, T.; Xu, H.; Cheng, L.; Yepes, M.; Zhao, Y.; Mao, Z. p38 MAPK inhibits autophagy and promotes microglial inflammatory responses by phosphorylating ULK1. J. Cell Biol. 2018, 217, 315–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szalai, P.; Hagen, L.K.; Sætre, F.; Luhr, M.; Sponheim, M.; Øverbye, A.; Mills, I.G.; Seglen, P.O.; Engedal, N. Autophagic bulk sequestration of cytosolic cargo is independent of LC3, but requires GABARAPs. Exp. Cell Res. 2015, 333, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Luhr, M.; Torgersen, M.L.; Szalai, P.; Hashim, A.; Brech, A.; Staerk, J.; Engedal, N. The kinase PERK and the transcription factor ATF4 play distinct and essential roles in autophagy resulting from tunicamycin-induced ER stress. J. Biol. Chem. 2019, 294, 8197–8217. [Google Scholar] [CrossRef] [PubMed]
- Schläfli, A.M.; Tokarchuk, I.; Parejo, S.; Jutzi, S.; Berezowska, S.; Engedal, N.; Tschan, M.P. ALK inhibition activates LC3B-independent, protective autophagy in EML4-ALK positive lung cancer cells. Sci. Rep. 2021, 11, 9011. [Google Scholar] [CrossRef] [PubMed]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wu, X.-Q.; Deng, R.; Li, D.-D.; Tang, J.; Chen, W.-D.; Chen, J.-H.; Ji, J.; Jiao, L.; Jiang, S.; et al. CaMKII-mediated Beclin 1 phosphorylation regulates autophagy that promotes degradation of Id and neuroblastoma cell differentiation. Nat. Commun. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, Y.C.; Fang, C.; Russell, R.C.; Kim, J.H.; Fan, W.; Liu, R.; Zhong, Q.; Guan, K.L. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell 2013, 152, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canaparo, R.; Foglietta, F.; Limongi, T.; Serpe, L. Biomedical Applications of Reactive Oxygen Species Generation by Metal Nanoparticles. Materials 2020, 14, 53. [Google Scholar] [CrossRef] [PubMed]
- Erlichman, J.S.; Leiter, J.C. Complexity of the Nano-Bio Interface and the Tortuous Path of Metal Oxides in Biological Systems. Antioxidants 2021, 10, 547. [Google Scholar] [CrossRef] [PubMed]
- Ichimura, Y.; Kirisako, T.; Takao, T.; Satomi, Y.; Shimonishi, Y.; Ishihara, N.; Mizushima, N.; Tanida, I.; Kominami, E.; Ohsumi, M.; et al. A ubiquitin-like system mediates protein lipidation. Nature 2000, 408, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sønstevold, T.; Engedal, N.; Torgersen, M.L. Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy. Cells 2021, 10, 3432. https://doi.org/10.3390/cells10123432
Sønstevold T, Engedal N, Torgersen ML. Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy. Cells. 2021; 10(12):3432. https://doi.org/10.3390/cells10123432
Chicago/Turabian StyleSønstevold, Tonje, Nikolai Engedal, and Maria Lyngaas Torgersen. 2021. "Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy" Cells 10, no. 12: 3432. https://doi.org/10.3390/cells10123432
APA StyleSønstevold, T., Engedal, N., & Torgersen, M. L. (2021). Perturbation of Cellular Redox Homeostasis Dictates Divergent Effects of Polybutyl Cyanoacrylate (PBCA) Nanoparticles on Autophagy. Cells, 10(12), 3432. https://doi.org/10.3390/cells10123432