
Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease




Abstract
:1. Introduction
2. Pathogenesis and Pathology
2.1. Neuropsychiatric Manifestations of PD
- -
- at least two supportive criteria (e.g., clear positive response to L-dopa, induced dyskinesia, rest tremor),
- -
- absence of absolute exclusion criteria (e.g., associated cerebellar signs, vertical ocular palsy, frontotemporal dementia or sensitive signs, no L-dopa response, isolated lower limb parkinsonism, treatment with dopamine receptor blockers), and
- -
- absence of red flags (e.g., rapid progression with, over the 5 first years, wheelchair use, bulbar dysfunction, severe dysautonomic dysfunction, recurrent falls during the first 3 years, inspiratory respiratory dysfunction, associated pyramidal signs or muscle contractures (neck, hands; feet), bilateral and symmetric evolution, absence of dopamine sensitivity and /or evolution, absence of non-motor signs).
2.2. Current Treatments for PD and Clinical Management
3. Autophagy
3.1. The Autophagy Machinery
3.2. Neuronal Autophagy Contributes to Neuronal Physiology
3.3. Autophagy and Neurodegenerative Diseases
3.4. Autophagy and Parkinson’s Disease
3.4.1. Role of Mitophagy in PD
3.4.2. Role of Macroautophagy in PD
3.4.3. Role of CMA in PD
3.4.4. Role of Lysosomes in PD
3.5. Is the Autophagy Machinery a Potential Target for Selective Intervention in PD?
4. Awaiting Satisfactory Answers—Future Research
5. General Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A
Appendix B
References
- Parkinson, J. An essay on the shaking palsy. 1817. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef]
- Del Rey, N.L.; Quiroga-Varela, A.; Garbayo, E.; Carballo-Carbajal, I.; Fernandez-Santiago, R.; Monje, M.H.G.; Trigo-Damas, I.; Blanco-Prieto, M.J.; Blesa, J. Advances in Parkinson’s disease: 200 years later. Front. Neuroanat. 2018, 12, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan-Or, Z.; Dion, P.A.; Rouleau, G.A. Genetic perspective on the role of the autophagy-lysosome pathway in Parkinson disease. Autophagy 2015, 11, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Smolders, S.; Van Broeckhoven, C. Genetic perspective on the synergistic connection between vesicular transport, lysosomal and mitochondrial pathways associated with Parkinson’s disease pathogenesis. Acta Neuropathol. Commun. 2020, 8, 63. [Google Scholar] [CrossRef]
- Beilina, A.; Cookson, M.R. Genes associated with Parkinson’s disease: Regulation of autophagy and beyond. J. Neurochem. 2016, 139, 91–107. [Google Scholar] [CrossRef]
- Gonzalez-Casacuberta, I.; Juarez-Flores, D.L.; Moren, C.; Garrabou, G. Bioenergetics and autophagic imbalance in patients-derived cell models of Parkinson disease supports systemic dysfunction in neurodegeneration. Front. Neurosci. 2019, 13, 894. [Google Scholar] [CrossRef]
- Van Veen, S.; Martin, S.; Van den Haute, C.; Benoy, V.; Lyons, J.; Vanhoutte, R.; Kahler, J.P.; Decuypere, J.-P.; Gelders, G.; Lambie, E.; et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature 2020, 578, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Smolders, S.; Van den Haute, C.; Heeman, B.; van Veen, S.; Crosiers, D.; Beletchi, I.; Verstraeten, A.; Gossye, H.; Gelders, G.; et al. Mutated ATP10B increases Parkinson’s disease risk by compromising lysosomal glucosylceramide export. Acta Neuropathol. 2020, 139, 1001–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos-Lobato, B.L.; Vidal, A.F.; Ribeiro-dos-Santos, Â. Regulatory miRNA–mRNA Networks in Parkinson’s Disease. Cells 2021, 10, 1410. [Google Scholar] [CrossRef]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Yavich, L.; Tanila, H.; Vepsalainen, S.; Jakala, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [Green Version]
- Todorova, A.; Jenner, P.; Ray Chaudhuri, K. Non-motor Parkinson’s: Integral to motor Parkinson’s, yet often neglected. Pract. Neurol. 2014, 14, 310–322. [Google Scholar] [CrossRef]
- Lin, K.J.; Lin, K.L.; Chen, S.D.; Liou, C.W.; Chuang, Y.C.; Lin, H.Y.; Lin, T.K. The overcrowded crossroads: Mitochondria, alpha-synuclein, and the endo-lysosomal system interaction in Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 5312. [Google Scholar] [CrossRef] [Green Version]
- Pieri, L.; Madiona, K.; Melki, R. Structural and functional properties of prefibrillar α-synuclein oligomers. Sci. Rep. 2016, 6, 24526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Balestrino, R.; Schapira, A.H.V. Parkinson disease. Eur. J. Neurol. 2020, 27, 27–42. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.F. Non-motor symptoms in Parkinson’s disease. Park. Relat. Disord. 2016, 22, S119–S122. [Google Scholar] [CrossRef]
- Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C.; Movement Disorder Society Evidence-Based Medicine, C. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 2018, 33, 1248–1266. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H.; Lang, A.E. Levodopa-related motor complications-phenomenology. Mov. Disord. 2008, 23, S509–S514. [Google Scholar] [CrossRef]
- Weintraub, D.; Claassen, D.O. Chapter 22—Impulse control and related disorders in Parkinson’s disease. In International Review of Neurobiology; Chaudhuri, K.R., Titova, N., Eds.; Academic Press: Cambridge, MA, USA, 2017; Volume 133, pp. 679–717. [Google Scholar]
- Garcia-Ruiz, P.J.; Martinez Castrillo, J.C.; Alonso-Canovas, A.; Herranz Barcenas, A.; Vela, L.; Sanchez Alonso, P.; Mata, M.; Olmedilla Gonzalez, N.; Mahillo Fernandez, I. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: A multicentre study. J. Neurol. Neurosurg. Psychiatry 2014, 85, 840–844. [Google Scholar] [CrossRef]
- Dijk, J.M.; Espay, A.J.; Katzenschlager, R.; de Bie, R.M.A. The choice between advanced therapies for Parkinson’s disease patients: Why, what, and when? J. Park. Dis. 2020, 10, S65–S73. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Seppi, K.; Ray Chaudhuri, K.; Coelho, M.; Fox, S.H.; Katzenschlager, R.; Perez Lloret, S.; Weintraub, D.; Sampaio, C.; the collaborators of the Parkinson’s Disease Update on Non-Motor Symptoms Study Group on behalf of the Movement Disorders Society Evidence-Based Medicine Committee. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov. Disord. 2019, 34, 180–198. [Google Scholar] [CrossRef] [Green Version]
- Schapira, A.H.; Bezard, E.; Brotchie, J.; Calon, F.; Collingridge, G.L.; Ferger, B.; Hengerer, B.; Hirsch, E.; Jenner, P.; Le Novere, N.; et al. Novel pharmacological targets for the treatment of Parkinson’s disease. Nat. Rev. Drug Discov. 2006, 5, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Elkouzi, A.; Vedam-Mai, V.; Eisinger, R.S.; Okun, M.S. Emerging therapies in Parkinson disease—Repurposed drugs and new approaches. Nat. Rev. Neurol. 2019, 15, 204–223. [Google Scholar] [CrossRef]
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. α-Synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013. [Google Scholar] [CrossRef] [Green Version]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamark, T.; Johansen, T. Aggrephagy: Selective disposal of protein aggregates by macroautophagy. Int. J. Cell Biol. 2012, 2012, 736905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzella, L.; Ahlberg, J.; Glaumann, H. Autophagy, heterophagy, microautophagy and crinophagy as the means for intracellular degradation. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1981, 36, 219–234. [Google Scholar] [CrossRef]
- Landstrom, A.S. A role for crinophagy in pancreatic islet B-cells. Minireview based on a doctoral thesis. Ups. J. Med. Sci. 1987, 92, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell. Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, Y.; Kikuchi, H.; Aizawa, S.; Furuta, A.; Hatanaka, Y.; Konya, C.; Uchida, K.; Wada, K.; Kabuta, T. Direct uptake and degradation of DNA by lysosomes. Autophagy 2013, 9, 1167–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chino, H.; Mizushima, N. ER-Phagy: Quality control and turnover of endoplasmic reticulum. Trends Cell Biol. 2020, 30, 384–398. [Google Scholar] [CrossRef]
- Mancias, J.D.; Vaites, L.P.; Nissim, S.; Biancur, D.E.; Kim, A.J.; Wang, X.; Liu, Y.; Goessling, W.; Kimmelman, A.C.; Harper, J.W. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. eLife 2015, 4, e10308. [Google Scholar] [CrossRef]
- Zhao, H.; Tang, M.; Liu, M.; Chen, L. Glycophagy: An emerging target in pathology. Clin. Chim. Acta 2018, 484, 298–303. [Google Scholar] [CrossRef]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Kounakis, K.; Chaniotakis, M.; Markaki, M.; Tavernarakis, N. Emerging roles of lipophagy in health and disease. Front. Cell Dev. Biol. 2019, 7, 185. [Google Scholar] [CrossRef]
- Otomo, T.; Yoshimori, T. Lysophagy: A method for monitoring lysosomal rupture followed by autophagy-dependent recovery. In Lysosomes; Springer: Berlin/Heidelberg, Germany, 2017; Volume 1594, pp. 141–149. [Google Scholar]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijaljica, D.; Devenish, R.J. Nucleophagy at a glance. J. Cell Sci. 2013, 126, 4325–4330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papandreou, M.E.; Tavernarakis, N. Nucleophagy: From homeostasis to disease. Cell Death Differ. 2019, 26, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.H.; Kim, Y.S.; Jo, D.S.; Choe, S.K.; Jo, E.K. Pexophagy: Molecular mechanisms and implications for health and diseases. Mol. Cells 2018, 41, 55–64. [Google Scholar]
- Tanaka, K.; Ichihara, A. Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem. Biophys. Res. Commun. 1989, 159, 1309–1315. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Palmer, A.; Rivett, A.J.; Knecht, E. Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem. 1995, 227, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Marshall, R.S.; Vierstra, R.D. Eat or be eaten: The autophagic plight of inactive 26S proteasomes. Autophagy 2015, 11, 1927–1928. [Google Scholar] [CrossRef] [Green Version]
- Cebollero, E.; Reggiori, F.; Kraft, C. Reticulophagy and ribophagy: Regulated degradation of protein production factories. Int. J. Cell Biol. 2012, 2012, 182834. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, Y.; Furuta, A.; Kikuchi, H.; Aizawa, S.; Hatanaka, Y.; Konya, C.; Uchida, K.; Yoshimura, A.; Tamai, Y.; Wada, K.; et al. Discovery of a novel type of autophagy targeting RNA. Autophagy 2013, 9, 403–409. [Google Scholar] [CrossRef] [Green Version]
- Choi, I.; Zhang, Y.; Seegobin, S.P.; Pruvost, M.; Wang, Q.; Purtell, K.; Zhang, B.; Yue, Z. Microglia clear neuron-released alpha-synuclein via selective autophagy and prevent neurodegeneration. Nat. Commun. 2020, 11, 1386. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell. Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef]
- Grasso, D.; Ropolo, A.; Lo Re, A.; Boggio, V.; Molejon, M.I.; Iovanna, J.L.; Gonzalez, C.D.; Urrutia, R.; Vaccaro, M.I. Zymophagy, a novel selective autophagy pathway mediated by VMP1-USP9x-p62, prevents pancreatic cell death. J. Biol. Chem. 2011, 286, 8308–8324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.; Menzies, F.M.; Narayanan, U.; Renna, M. Mammalian macroautophagy at a glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itakura, E.; Mizushima, N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy 2010, 6, 764–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biazik, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.-L. Ultrastructural relationship of the phagophore with surrounding organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, B.; Kroemer, G. Autophagy in the pathogenesis of disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Noda, N.N.; Ohsumi, Y.; Inagaki, F. ATG systems from the protein structural point of view. Chem. Rev. 2009, 109, 1587–1598. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef] [PubMed]
- Durgan, J.; Lystad, A.H.; Sloan, K.; Carlsson, S.R.; Wilson, M.I.; Marcassa, E.; Ulferts, R.; Webster, J.; Lopez-Clavijo, A.F.; Wakelam, M.J.; et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 2021, 81, 2031–2040.e8. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; Leidal, A.M.; Debnath, J.; Hansen, M. Beyond autophagy: The expanding roles of ATG8 proteins. Trends Biochem. Sci. 2021, 46, 673–686. [Google Scholar] [CrossRef]
- Fletcher, K.; Ulferts, R.; Jacquin, E.; Veith, T.; Gammoh, N.; Arasteh, J.M.; Mayer, U.; Carding, S.R.; Wileman, T.; Beale, R.; et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 2018, 37, e97840. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy in neurons: It is not all about food. Trends Mol. Med. 2006, 12, 461–464. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Autophagy in neurons. Annu. Rev. Cell. Dev. Biol. 2019, 35, 477–500. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Quality control in neurons: Mitophagy and other selective autophagy mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef] [PubMed]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Azarnia Tehran, D.; Kuijpers, M.; Haucke, V. Presynaptic endocytic factors in autophagy and neurodegeneration. Curr. Opin. Neurobiol. 2018, 48, 153–159. [Google Scholar] [CrossRef]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Ohsumi, Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Bonam, S.R.; Bayry, J.; Tschan, M.P.; Muller, S. Progress and challenges in the use of MAP1LC3 as a legitimate marker for measuring dynamic autophagy in vivo. Cells 2020, 9, 1321. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy 2021, 17, 1–382. [Google Scholar] [PubMed]
- Yue, Z.; Friedman, L.; Komatsu, M.; Tanaka, K. The cellular pathways of neuronal autophagy and their implication in neurodegenerative diseases. Biochim. Biophys. Acta 2009, 1793, 1496–1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavoe, A.K.H.; Gopal, P.P.; Gubas, A.; Tooze, S.A.; Holzbaur, E.L.F. Expression of WIPI2B counteracts age-related decline in autophagosome biogenesis in neurons. eLife 2019, 8, e44219. [Google Scholar] [CrossRef]
- Kim, S.; Nam, Y.; Kim, C.; Lee, H.; Hong, S.; Kim, H.S.; Shin, S.J.; Park, Y.H.; Mai, H.N.; Oh, S.M.; et al. Neuroprotective and anti-inflammatory effects of low-moderate dose ionizing radiation in models of Alzheimer’s disease. Int. J. Mol. Sci. 2020, 21, 3678. [Google Scholar] [CrossRef]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune connections in aging and neurodegenerative diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Dadar, M.; Munjal, A.; Dhama, K.; Karthik, K.; Tiwari, R.; Yatoo, M.; Iqbal, H.; Singh, K.P.; Joshi, S.K. A comprehensive review of autophagy and its various roles in infectious, non-infectious, and lifestyle diseases: Current knowledge and prospects for disease prevention, novel drug design, and therapy. Cells 2019, 8, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonam, S.R.; Muller, S. Parkinson’s disease is an autoimmune disease: A reappraisal. Autoimmun. Rev. 2020, 19, 102684. [Google Scholar] [CrossRef]
- El Haddad, S.; Serrano, A.; Moal, F.; Normand, T.; Robin, C.; Charpentier, S.; Valery, A.; Brulé-Morabito, F.; Auzou, P.; Mollet, L.; et al. Disturbed expression of autophagy genes in blood of Parkinson’s disease patients. Gene 2020, 738, 144454. [Google Scholar] [CrossRef]
- Rott, R.; Szargel, R.; Shani, V.; Bisharat, S.; Engelender, S. Alpha-synuclein ubiquitination and novel therapeutic targets for Parkinson’s disease. CNS Neurol. Disord. Drug Targets 2014, 13, 630–637. [Google Scholar] [CrossRef]
- Popova, B.; Kleinknecht, A.; Braus, G.H. Posttranslational modifications and clearing of α-synuclein aggregates in yeast. Biomolecules 2015, 5, 617–634. [Google Scholar] [CrossRef] [Green Version]
- Parker, W.D., Jr.; Boyson, S.J.; Parks, J.K. Abnormalities of the electron transport chain in idiopathic Parkinson’s disease. Ann. Neurol. 1989, 26, 719–723. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Deus, C.M.; Yambire, K.F.; Oliveira, P.J.; Raimundo, N. Mitochondria-lysosome crosstalk: From physiology to neurodegeneration. Trends Mol. Med. 2020, 26, 71–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Qi, H.; Tang, Y.; Shen, H.M. Post-translational modifications of key machinery in the control of mitophagy. Trends Biochem. Sci. 2020, 45, 58–75. [Google Scholar] [CrossRef]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef]
- Poole, A.C.; Thomas, R.E.; Andrews, L.A.; McBride, H.M.; Whitworth, A.J.; Pallanck, L.J. The PINK1/Parkin pathway regulates mitochondrial morphology. Proc. Natl Acad. Sci. USA 2008, 105, 1638–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Dernie, F. Mitophagy in Parkinson’s disease: From pathogenesis to treatment target. Neurochem. Int. 2020, 138, 104756. [Google Scholar] [CrossRef]
- Leiva-Rodríguez, T.; Romeo-Guitart, D.; Marmolejo-Martínez-Artesero, S.; Herrando-Grabulosa, M.; Bosch, A.; Forés, J.; Casas, C. ATG5 overexpression is neuroprotective and attenuates cytoskeletal and vesicle-trafficking alterations in axotomized motoneurons. Cell Death Dis. 2018, 9, 626. [Google Scholar] [CrossRef]
- Hu, Z.-Y.; Chen, B.; Zhang, J.-P.; Ma, Y.-Y. Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J. Biol. Chem. 2017, 292, 18062–18074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Chen, D.; Pang, S.; Feng, X.; Huang, W.; Hawley, R.G.; Yan, B. Genetic analysis of the ATG7 gene promoter in sporadic Parkinson’s disease. Neurosci. Lett. 2013, 534, 193–198. [Google Scholar] [CrossRef]
- Moors, T.E.; Hoozemans, J.J.; Ingrassia, A.; Beccari, T.; Parnetti, L.; Chartier-Harlin, M.C.; van de Berg, W.D. Therapeutic potential of autophagy-enhancing agents in Parkinson’s disease. Mol. Neurodegener. 2017, 12, 11. [Google Scholar] [CrossRef] [Green Version]
- Korvatska, O.; Strand, N.S.; Berndt, J.D.; Strovas, T.; Chen, D.H.; Leverenz, J.B.; Kiianitsa, K.; Mata, I.F.; Karakoc, E.; Greenup, J.L.; et al. Altered splicing of ATP6AP2 causes X-linked parkinsonism with spasticity (XPDS). Hum. Mol. Genet. 2013, 22, 3259–3268. [Google Scholar] [CrossRef] [Green Version]
- Rujano, M.A.; Cannata Serio, M.; Panasyuk, G.; Peanne, R.; Reunert, J.; Rymen, D.; Hauser, V.; Park, J.H.; Freisinger, P.; Souche, E.; et al. Mutations in the X-linked ATP6AP2 cause a glycosylation disorder with autophagic defects. J. Exp. Med. 2017, 214, 3707–3729. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Crosstalk between lysosomes and mitochondria in Parkinson’s disease. Front. Cell Dev. Biol. 2017, 5, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, L.Y.; Giasson, B.I.; Bonini, N.M. DJ-1 is critical for mitochondrial function and rescues PINK1 loss of function. Proc. Natl. Acad. Sci. USA 2010, 107, 9747–9752. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Cookson, M.R. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy 2011, 7, 531–532. [Google Scholar] [CrossRef] [Green Version]
- Burchell, V.S.; Nelson, D.E.; Sanchez-Martinez, A.; Delgado-Camprubi, M.; Ivatt, R.M.; Pogson, J.H.; Randle, S.J.; Wray, S.; Lewis, P.A.; Houlden, H.; et al. The Parkinson’s disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat. Neurosci. 2013, 16, 1257–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H. Glucocerebrosidase and Parkinson disease: Recent advances. Mol. Cell. Neurosci. 2015, 66, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Aflaki, E.; Westbroek, W.; Sidransky, E. The complicated relationship between gaucher disease and parkinsonism: Insights from a rare disease. Neuron 2017, 93, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, K.Y.; Fernandez-Hernandez, H.; Hu, J.; Schaffner, A.; Pankratz, N.; Hsu, N.Y.; Chuang, L.S.; Carmi, S.; Villaverde, N.; Li, X.; et al. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaai7795. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Mira, M.T.; Alcais, A.; Nguyen, V.T.; Moraes, M.O.; Di Flumeri, C.; Vu, H.T.; Mai, C.P.; Nguyen, T.H.; Nguyen, N.B.; Pham, X.K.; et al. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 2004, 427, 636–640. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinn, S.; Drolet, R.E.; Cramer, P.E.; Wong, A.H.; Toolan, D.M.; Gretzula, C.A.; Voleti, B.; Vassileva, G.; Disa, J.; Tadin-Strapps, M.; et al. TMEM175 deficiency impairs lysosomal and mitochondrial function and increases alpha-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 2389–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Deng, H.X.; Wong, Y.C.; Siddique, T.; Krainc, D. The Parkinson’s disease-linked protein TMEM230 is required for Rab8a-mediated secretory vesicle trafficking and retromer trafficking. Hum. Mol. Genet. 2017, 26, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.-F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [Green Version]
- Narendra, D.; Kane, L.A.; Hauser, D.N.; Fearnley, I.M.; Youle, R.J. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 2010, 6, 1090–1106. [Google Scholar] [CrossRef]
- Heo, J.M.; Ordureau, A.; Paulo, J.A.; Rinehart, J.; Harper, J.W. The PINK1-PARKIN mitochondrial ubiquitylation pathway drives a program of OPTN/NDP52 recruitment and TBK1 activation to promote mitophagy. Mol. Cell 2015, 60, 7–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M.; Sliter, D.A.; Kane, L.A.; Sarraf, S.A.; Wang, C.; Burman, J.L.; Sideris, D.P.; Fogel, A.I.; Youle, R.J. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 2015, 524, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutagalung, A.H.; Novick, P.J. Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev. 2011, 91, 119–149. [Google Scholar] [CrossRef] [Green Version]
- Song, P.; Trajkovic, K.; Tsunemi, T.; Krainc, D. Parkin modulates endosomal organization and function of the endo-lysosomal pathway. J. Neurosci. 2016, 36, 2425–2437. [Google Scholar] [CrossRef] [Green Version]
- Yamano, K.; Fogel, A.I.; Wang, C.; van der Bliek, A.M.; Youle, R.J. Mitochondrial Rab GAPs govern autophagosome biogenesis during mitophagy. eLife 2014, 3, e01612. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple roles of the small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Demers-Lamarche, J.; Guillebaud, G.; Tlili, M.; Todkar, K.; Belanger, N.; Grondin, M.; Nguyen, A.P.; Michel, J.; Germain, M. Loss of mitochondrial function impairs lysosomes. J. Biol. Chem. 2016, 291, 10263–10276. [Google Scholar] [CrossRef] [Green Version]
- Sterky, F.H.; Lee, S.; Wibom, R.; Olson, L.; Larsson, N.G. Impaired mitochondrial transport and Parkin-independent degeneration of respiratory chain-deficient dopamine neurons in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12937–12942. [Google Scholar] [CrossRef] [Green Version]
- Bingol, B.; Tea, J.S.; Phu, L.; Reichelt, M.; Bakalarski, C.E.; Song, Q.; Foreman, O.; Kirkpatrick, D.S.; Sheng, M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 2014, 510, 370–375. [Google Scholar] [CrossRef]
- Luo, H.; Krigman, J.; Zhang, R.; Yang, M.; Sun, N. Pharmacological inhibition of USP30 activates tissue-specific mitophagy. Acta Physiol. 2021, 232, e13666. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [Green Version]
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ren, Y.; Gui, C.; Zhao, M.; Wu, X.; Mao, K.; Li, W.; Zou, F. Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T alpha-synuclein model of Parkinson’s disease. Cell Death Dis. 2018, 9, 700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Gao, L.; Lu, F.; Wang, B.; Gao, F.; Zhu, G.; Cai, Z.; Lai, J.; Yang, Q. Transcription factor myocyte enhancer factor 2D regulates interleukin-10 production in microglia to protect neuronal cells from inflammation-induced death. J. Neuroinflamm. 2015, 12, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- She, H.; Yang, Q.; Shepherd, K.; Smith, Y.; Miller, G.; Testa, C.; Mao, Z. Direct regulation of complex I by mitochondrial MEF2D is disrupted in a mouse model of Parkinson disease and in human patients. J. Clin. Investig. 2011, 121, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Acin-Perez, R.; Villarroya-Beltri, C.; Mazzeo, C.; Nunez-Andrade, N.; Gabande-Rodriguez, E.; Ledesma, M.D.; Blazquez, A.; Martin, M.A.; Falcon-Perez, J.M.; et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015, 22, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Palikaras, K.; Daskalaki, I.; Markaki, M.; Tavernarakis, N. Mitophagy and age-related pathologies: Development of new therapeutics by targeting mitochondrial turnover. Pharmacol. Ther. 2017, 178, 157–174. [Google Scholar] [CrossRef]
- Villa, E.; Marchetti, S.; Ricci, J.E. No Parkin zone: Mitophagy without Parkin. Trends Cell Biol. 2018, 28, 882–895. [Google Scholar] [CrossRef]
- Winslow, A.R.; Rubinsztein, D.C. The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy 2011, 7, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nat. Cell Biol. 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Arotcarena, M.-L.; Teil, M.; Dehay, B. Autophagy in synucleinopathy: The overwhelmed and defective machinery. Cells 2019, 8, 565. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Rubinsztein, D.C. Autophagy in neuronal development and plasticity. Trends Neurosci. 2020, 43, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pages, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Eleuteri, S.; Albanese, A. VPS35-based approach: A potential innovative treatment in Parkinson’s disease. Front. Neurol. 2019, 10, 1272. [Google Scholar] [CrossRef]
- Miura, E.; Hasegawa, T.; Konno, M.; Suzuki, M.; Sugeno, N.; Fujikake, N.; Geisler, S.; Tabuchi, M.; Oshima, R.; Kikuchi, A.; et al. VPS35 dysfunction impairs lysosomal degradation of alpha-synuclein and exacerbates neurotoxicity in a Drosophila model of Parkinson’s disease. Neurobiol. Dis. 2014, 71, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zavodszky, E.; Seaman, M.N.J.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.L.; Liu, W.; Hu, J.X.; Erion, J.R.; Ye, J.; Mei, L.; Xiong, W.C. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015, 12, 1631–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, A.A.; Soto-Avellaneda, A.; Yong Jin, H.; Stojkovska, I.; Lai, N.K.; Albright, J.E.; Webb, A.R.; Oe, E.; Valarde, J.P.; Oxford, A.E.; et al. Enhanced hyaluronan signaling and autophagy dysfunction by VPS35 D620N. Neuroscience 2020, 441, 33–45. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef]
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuervo, A.M.; Wong, E. Chaperone-mediated autophagy: Roles in disease and aging. Cell Res. 2014, 24, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone mediated autophagy in the crosstalk of neurodegenerative diseases and metabolic disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472. [Google Scholar] [CrossRef] [Green Version]
- Orenstein, S.J.; Kuo, S.-H.; Tasset, I.; Arias, E.; Koga, H.; Fernandez-Carasa, I.; Cortes, E.; Honig, L.S.; Dauer, W.; Consiglio, A.; et al. Interplay of LRRK2 with chaperone-mediated autophagy. Nat. Neurosci. 2013, 16, 394–406. [Google Scholar] [CrossRef] [Green Version]
- Xilouri, M.; Vogiatzi, T.; Vekrellis, K.; Park, D.; Stefanis, L. Abberant alpha-synuclein confers toxicity to neurons in part through inhibition of chaperone-mediated autophagy. PLoS ONE 2009, 4, e5515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, Y.; Dodiya, H.; Aebischer, P.; Olanow, C.W.; Kordower, J.H. Alterations in lysosomal and proteasomal markers in Parkinson’s disease: Relationship to alpha-synuclein inclusions. Neurobiol. Dis. 2009, 35, 385–398. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647. [Google Scholar] [CrossRef]
- Gao, L.; She, H.; Li, W.; Zeng, J.; Zhu, J.; Jones, D.P.; Mao, Z.; Gao, G.; Yang, Q. Oxidation of survival factor MEF2D in neuronal death and Parkinson’s disease. Antioxid. Redox Signal. 2014, 20, 2936–2948. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Wang, X.; Feng, X.; Zhang, A.; Li, J.; Gu, K.; Huang, J.; Pang, S.; Dong, H.; Gao, H.; et al. Altered expression of autophagic genes in the peripheral leukocytes of patients with sporadic Parkinson’s disease. Brain Res. 2011, 1394, 105–111. [Google Scholar] [CrossRef]
- Lim, H.; Lim, Y.-M.; Kim, K.H.; Jeon, Y.E.; Park, K.; Kim, J.; Hwang, H.-Y.; Lee, D.J.; Pagire, H.; Kwon, H.J.; et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum–mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef]
- Xu, C.Y.; Kang, W.Y.; Chen, Y.M.; Jiang, T.F.; Zhang, J.; Zhang, L.N.; Ding, J.Q.; Liu, J.; Chen, S.D. DJ-1 Inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front. Aging Neurosci. 2017, 9, 308. [Google Scholar] [CrossRef] [Green Version]
- Brekk, O.R.; Makridakis, M.; Mavroeidi, P.; Vlahou, A.; Xilouri, M.; Stefanis, L. Impairment of chaperone-mediated autophagy affects neuronal homeostasis through altered expression of DJ-1 and CRMP-2 proteins. Mol. Cell. Neurosci. 2019, 95, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztein, D.C.; Mariño, G.; Kroemer, G. Autophagy and Aging. Cell 2011, 146, 682–695. [Google Scholar] [CrossRef] [Green Version]
- Metaxakis, A.; Ploumi, C.; Tavernarakis, N. Autophagy in age-associated neurodegeneration. Cells 2018, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, M.; Bonam, S.R.; Schall, N.; Bendorius, M.; Korganow, A.-S.; Lumbroso, C.; Muller, S. Implication of a lysosomal antigen in the pathogenesis of lupus erythematosus. J. Autoimmun. 2021, 120, 102633. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Hunt, C.P.; Thacker, E.A.; Toste, C.M.; Boularand, S.; Deprets, S.; Dubois, L.; Sanders, L.H. Mitochondrial DNA damage as a potential biomarker of LRRK2 kinase activity in LRRK2 Parkinson’s disease. Sci. Rep. 2020, 10, 17293. [Google Scholar] [CrossRef] [PubMed]
- Balducci, C.; Pierguidi, L.; Persichetti, E.; Parnetti, L.; Sbaragli, M.; Tassi, C.; Orlacchio, A.; Calabresi, P.; Beccari, T.; Rossi, A. Lysosomal hydrolases in cerebrospinal fluid from subjects with Parkinson’s disease. Mov. Disord. 2007, 22, 1481–1484. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Murphy, K.; Gysbers, A.; Abbott, S.; Tayebi, N.; Kim, W.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G. Reduced glucocerebrosidase is associated with increased α-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [Green Version]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.E.; Cottle, L.; Gysbers, A.M.; Cooper, A.A.; Halliday, G.M. ATP13A2 (PARK9) protein levels are reduced in brain tissue of cases with Lewy bodies. Acta Neuropathol. Commun. 2013, 1, 11. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Huang, J.; Feng, X.; Zhang, A.; Li, J.; Pang, S.; Gu, K.; Dong, H.; Zhang, J.; Gao, H.; et al. Decreased expression of lysosomal alpha-galactosiase a gene in sporadic Parkinson’s disease. Neurochem. Res. 2011, 36, 1939–1944. [Google Scholar] [CrossRef]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marquez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Decressac, M.; Mattsson, B.; Weikop, P.; Lundblad, M.; Jakobsson, J.; Bjorklund, A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc. Natl. Acad. Sci. USA 2013, 110, E1817–E1826. [Google Scholar] [CrossRef] [Green Version]
- Dehay, B.; Bové, J.; Rodríguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.-X.; Wang, S.-F.; Tan, Y.; Song, J.-X.; Zhu, Z.; Wang, Z.-Y.; Wu, M.-Y.; Cai, C.-Z.; Huang, Z.-J.; Tan, J.-Q.; et al. Pharmacological enhancement of TFEB-mediated autophagy alleviated neuronal death in oxidative stress-induced Parkinson’s disease models. Cell Death Dis. 2020, 11, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Dai, Y.; Liu, S.; Fan, Y.; Ding, Z.; Li, D. TFEB biology and agonists at a glance. Cells 2021, 10, 333. [Google Scholar] [CrossRef]
- Orrù, V.; Steri, M.; Sidore, C.; Marongiu, M.; Serra, V.; Olla, S.; Sole, G.; Lai, S.; Dei, M.; Mulas, A.; et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 2020, 52, 1036–1045. [Google Scholar] [CrossRef] [PubMed]
- Zerr, I. RT-QuIC for detection of prodromal α-synucleinopathies. Lancet Neurol. 2021, 20, 165–166. [Google Scholar] [CrossRef]
- Curtis, L.; Lees, A.J.; Stern, G.M.; Marmot, M.G. Effect of L-dopa on course of Parkinson’s disease. Lancet 1984, 2, 211–212. [Google Scholar] [CrossRef]
- Pajares, M.; Rojo, A.; Manda, G.; Boscá, L.; Cuadrado, A. Inflammation in Parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9, 1687. [Google Scholar] [CrossRef] [PubMed]
- Parmar, M.; Grealish, S.; Henchcliffe, C. The future of stem cell therapies for Parkinson disease. Nat. Rev. Neurosci. 2020, 21, 103–115. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-derived dopamine progenitor cells for Parkinson’s disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef] [PubMed]
- Dawson, V.L.; Dawson, T.M. Promising disease-modifying therapies for Parkinson’s disease. Sci. Transl. Med. 2019, 11, eaba1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujols, J.; Peña-Díaz, S.; Pallarès, I.; Ventura, S. Chemical chaperones as novel drugs for Parkinson’s disease. Trends Mol. Med. 2020, 26, 408–421. [Google Scholar] [CrossRef]
- Robinson, E.J.; Aguiar, S.; Smidt, M.P.; van der Heide, L.P. MCL1 as a therapeutic target in Parkinson’s disease? Trends Mol. Med. 2019, 25, 1056–1065. [Google Scholar] [CrossRef]
- Harris, J.P.; Burrell, J.C.; Struzyna, L.A.; Chen, H.I.; Serruya, M.D.; Wolf, J.A.; Duda, J.E.; Cullen, D.K. Emerging regenerative medicine and tissue engineering strategies for Parkinson’s disease. NPJ Parkinson’s Dis. 2020, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Marin-Aguilar, F.; Castejon-Vega, B.; Alcocer-Gomez, E.; Lendines-Cordero, D.; Cooper, M.A.; de la Cruz, P.; Andujar-Pulido, E.; Perez-Alegre, M.; Muntane, J.; Perez-Pulido, A.J.; et al. NLRP3 inflammasome inhibition by MCC950 in aged mice improves health via enhanced autophagy and PPARalpha activity. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1457–1464. [Google Scholar] [CrossRef]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.-I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef]
- Park, H.; Kim, J.; Shin, C.; Lee, S. Intersection between redox homeostasis and autophagy: Valuable insights into neurodegeneration. Antioxidants 2021, 10, 694. [Google Scholar] [CrossRef] [PubMed]
- Georgakopoulos, N.D.; Wells, G.; Campanella, M. The pharmacological regulation of cellular mitophagy. Nat. Chem. Biol. 2017, 13, 136–146. [Google Scholar] [CrossRef]
- Koentjoro, B.; Park, J.-S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef]
- Yi, W.; MacDougall, E.J.; Tang, M.Y.; Krahn, A.I.; Gan-Or, Z.; Trempe, J.-F.; Fon, E.A. The landscape of Parkin variants reveals pathogenic mechanisms and therapeutic targets in Parkinson’s disease. Hum. Mol. Genet. 2019, 28, 2811–2825. [Google Scholar] [CrossRef]
- Kluge, A.F.; Lagu, B.R.; Maiti, P.; Jaleel, M.; Webb, M.; Malhotra, J.; Mallat, A.; Srinivas, P.A.; Thompson, J.E. Novel highly selective inhibitors of ubiquitin specific protease 30 (USP30) accelerate mitophagy. Bioorg. Med. Chem. Lett. 2018, 28, 2655–2659. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Banerjee, R.; Raju, A.; Ngima Nthenge-Ngumbau, D.; Singh, R.; Jaisankar, P.; Mohanakumar, K.P.; Biswas, S.C. Tetrahydroisoquinoline molecule of Indian ayurveda medicine: Therapeutic potential in Parkinson’s disease. Park. Relat. Disord. 2018, 46, e85. [Google Scholar] [CrossRef]
- Suresh, S.N.; Manjithaya, R. A small molecule autophagy inducer exerts cytoprotection against α-synuclein toxicity. Eur. J. Pharmacol. 2019, 862, 172635. [Google Scholar] [CrossRef]
- Kovács, T.; Billes, V.; Komlós, M.; Hotzi, B.; Manzéger, A.; Tarnóci, A.; Papp, D.; Szikszai, F.; Szinyákovics, J.; Rácz, Á.; et al. The small molecule AUTEN-99 (autophagy enhancer-99) prevents the progression of neurodegenerative symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Jiang, C.; Chen, W.; Zhang, G.; Luo, D.; Cao, Y.; Wu, J.; Ding, Y.; Liu, B. Baicalein induces apoptosis and autophagy via endoplasmic reticulum stress in hepatocellular carcinoma cells. Biomed. Res. Int. 2014, 2014, 732516. [Google Scholar] [CrossRef] [Green Version]
- Yan, W.; Ma, X.; Zhao, X.; Zhang, S. Baicalein induces apoptosis and autophagy of breast cancer cells via inhibiting PI3K/AKT pathway in vivo and vitro. Drug Des. Dev. Ther. 2018, 12, 3961–3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, X.; Di, C.; Wang, Z.; Mi, X.; Liu, Y.; Zhao, Q.; Mao, A.; Chen, W.; Gan, L.; et al. MitoQ regulates autophagy by inducing a pseudo-mitochondrial membrane potential. Autophagy 2017, 13, 730–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.C.; Huang, H.J.; Wang, Y.T.; Lin, A.M. Baicalein attenuates alpha-synuclein aggregation, inflammasome activation and autophagy in the MPP(+)-treated nigrostriatal dopaminergic system in vivo. J. Ethnopharmacol. 2016, 194, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Uversky, V.N.; Huang, M.; Kang, H.; Xu, F.; Liu, X.; Lian, L.; Liang, Q.; Jiang, H.; Liu, A.; et al. Baicalein inhibits alpha-synuclein oligomer formation and prevents progression of alpha-synuclein accumulation in a rotenone mouse model of Parkinson’s disease. Biochim. Biophys. Acta 2016, 1862, 1883–1890. [Google Scholar] [CrossRef]
- Li, Y.; Lin, S.; Xu, C.; Zhang, P.; Mei, X. Triggering of autophagy by baicalein in response to apoptosis after spinal cord injury: Possible involvement of the PI3K activation. Biol. Pharm. Bull. 2018, 41, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, S.N.; Chavalmane, A.K.; Dj, V.; Yarreiphang, H.; Rai, S.; Paul, A.; Clement, J.P.; Alladi, P.A.; Manjithaya, R. A novel autophagy modulator 6-Bio ameliorates SNCA/α-synuclein toxicity. Autophagy 2017, 13, 1221–1234. [Google Scholar] [CrossRef]
- Deng, Y.N.; Shi, J.; Liu, J.; Qu, Q.M. Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem. Int. 2013, 63, 1–9. [Google Scholar] [CrossRef]
- Spinelli, K.J.; Osterberg, V.R.; Meshul, C.K.; Soumyanath, A.; Unni, V.K. Curcumin treatment improves motor behavior in α-synuclein transgenic mice. PLoS ONE 2015, 10, e0128510. [Google Scholar]
- Jiang, T.-F.; Zhang, Y.-J.; Zhou, H.-Y.; Wang, H.-M.; Tian, L.-P.; Liu, J.; Ding, J.-Q.; Chen, S.-D. Curcumin ameliorates the neurodegenerative pathology in A53T α-synuclein cell model of Parkinson’s disease through the downregulation of mTOR/p70S6K signaling and the recovery of macroautophagy. J. Neuroimmune Pharmacol. 2013, 8, 356–369. [Google Scholar] [CrossRef]
- Song, J.-X.; Sun, Y.-R.; Peluso, I.; Zeng, Y.; Yu, X.; Lu, J.-H.; Xu, Z.; Wang, M.-Z.; Liu, L.-F.; Huang, Y.-Y.; et al. A novel curcumin analog binds to and activates TFEB in vitro and in vivo independent of MTOR inhibition. Autophagy 2016, 12, 1372–1389. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Garcia-Yague, A.J.; Scannevin, R.H.; Casarejos, M.J.; Kugler, S.; Rabano, A.; Cuadrado, A. Repurposing the NRF2 activator dimethyl fumarate as therapy against synucleinopathy in Parkinson’s disease. Antioxid. Redox Signal. 2016, 25, 61–77. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Moon, J.H.; Kim, S.W.; Jeong, J.K.; Nazim, U.M.D.; Lee, Y.J.; Seol, J.W.; Park, S.Y. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 2015, 6, 9701–9717. [Google Scholar] [CrossRef] [Green Version]
- Ng, C.H.; Guan, M.S.; Koh, C.; Ouyang, X.; Yu, F.; Tan, E.K.; O’Neill, S.P.; Zhang, X.; Chung, J.; Lim, K.L. AMP kinase activation mitigates dopaminergic dysfunction and mitochondrial abnormalities in Drosophila models of Parkinson’s disease. J. Neurosci. 2012, 32, 14311–14317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, K.; Zeng, Y.; Hancock, T.; Segatori, L. Genetic and chemical activation of TFEB mediates clearance of aggregated α-synuclein. PLoS ONE 2015, 10, e0120819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of acid β-glucosidase with pharmacological chaperone provides insight into Gaucher disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar]
- Lu, J.-H.; Tan, J.-Q.; Durairajan, S.S.K.; Liu, L.-F.; Zhang, Z.-H.; Ma, L.; Shen, H.-M.; Chan, H.Y.E.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, S.; Mak, S.; Zuo, X.; Li, H.; Wang, Y.; Han, Y. Neuroprotection against MPP(+)-induced cytotoxicity through the activation of PI3-K/Akt/GSK3beta/MEF2D signaling pathway by rhynchophylline, the major tetracyclic oxindole alkaloid isolated from uncaria rhynchophylla. Front. Pharmacol. 2018, 9, 768. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Graziani, I.; Piccirillo, S.; Rotilio, G.; Ciriolo, M.R. Carcinoma cells activate AMP-activated protein kinase-dependent autophagy as survival response to kaempferol-mediated energetic impairment. Autophagy 2010, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; Graziani, I.; De Zio, D.; Dini, L.; Centonze, D.; Rotilio, G.; Ciriolo, M.R. Neuroprotection of kaempferol by autophagy in models of rotenone-mediated acute toxicity: Possible implications for Parkinson’s disease. Neurobiol. Aging 2012, 33, 767–785. [Google Scholar] [CrossRef]
- Myöhänen, T.; Hannula, M.; Van Elzen, R.; Gerard, M.; Van Der Veken, P.; García-Horsman, J.; Baekelandt, V.; Männistö, P.; Lambeir, A. A prolyl oligopeptidase inhibitor, KYP-2047, reduces α-synuclein protein levels and aggregates in cellular and animal models of Parkinson’s disease. Br. J. Pharmacol. 2012, 166, 1097–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savolainen, M.H.; Richie, C.T.; Harvey, B.K.; Männistö, P.T.; Maguire-Zeiss, K.A.; Myöhänen, T.T. The beneficial effect of a prolyl oligopeptidase inhibitor, KYP-2047, on alpha-synuclein clearance and autophagy in A30P transgenic mouse. Neurobiol. Dis. 2014, 68, 1–15. [Google Scholar] [CrossRef]
- Kilpeläinen, T.P.; Hellinen, L.; Vrijdag, J.; Yan, X.; Svarcbahs, R.; Vellonen, K.-S.; Lambeir, A.-M.; Huttunen, H.; Urtti, A.; Wallen, E.A.A.; et al. The effect of prolyl oligopeptidase inhibitors on alpha-synuclein aggregation and autophagy cannot be predicted by their inhibitory efficacy. Biomed. Pharmacother. 2020, 128, 110253. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Ju, S.; Lachenmayer, M.L.; Liken, J.; Stock, A.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine stimulates autophagy and reduces accumulation of α-synuclein in cells and in mouse brain. Mol. Psychiatry 2013, 18, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.; Floto, R.A.; Berger, Z.; Imarisio, S.; Cordenier, A.; Pasco, M.; Cook, L.J.; Rubinsztein, D.C. Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 2005, 170, 1101–1111. [Google Scholar] [CrossRef] [PubMed]
- Forlenza, O.V.; De-Paula, V.J.R.; Diniz, B.S.O. Neuroprotective effects of lithium: Implications for the treatment of Alzheimer’s disease and related neurodegenerative disorders. ACS Chem. Neurosci. 2014, 5, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Motoi, Y.; Shimada, K.; Ishiguro, K.; Hattori, N. Lithium and autophagy. ACS Chem. Neurosci. 2014, 5, 434–442. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Xiong, N.; Liu, L.; Huang, J.; Han, C.; Zhang, G.; Li, J.; Xu, X.; Lin, Z.; Wang, T. Lithium protects dopaminergic cells from rotenone toxicity via autophagy enhancement. BMC Neurosci. 2015, 16, 82. [Google Scholar] [CrossRef] [Green Version]
- Lazzara, C.A.; Kim, Y.-H. Potential application of lithium in Parkinson’s and other neurodegenerative diseases. Front. Neurosci. 2015, 9, 403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef] [Green Version]
- Saber, S.; El-Kader, E.M.A. Novel complementary coloprotective effects of metformin and MCC950 by modulating HSP90/NLRP3 interaction and inducing autophagy in rats. Inflammopharmacology 2021, 29, 237–251. [Google Scholar] [CrossRef]
- Corcoran, S.E.; Reena, H.; Matthew, A.C. Pharmacological Inhibition of the Nod-Like Receptor Family Pyrin Domain Containing 3 Inflammasome with MCC950. Pharmacol. Rev. 2021, 73, 968–1000. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulovic, M.; Jovanovic, M.; Xilouri, M.; Stefanis, L.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Paunovic, V.; Ardah, M.T.; El-Agnaf, O.M.; Kostic, V.; et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Perez-Revuelta, B.I.; Hettich, M.M.; Ciociaro, A.; Rotermund, C.; Kahle, P.J.; Krauss, S.; Di Monte, D.A. Metformin lowers Ser-129 phosphorylated alpha-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014, 5, e1209. [Google Scholar] [CrossRef] [Green Version]
- Jovanovic-Tucovic, M.; Harhaji-Trajkovic, L.; Dulovic, M.; Tovilovic-Kovacevic, G.; Zogovic, N.; Jeremic, M.; Mandic, M.; Kostic, V.; Trajkovic, V.; Markovic, I. AMP-activated protein kinase inhibits MPP+-induced oxidative stress and apoptotic death of SH-SY5Y cells through sequential stimulation of Akt and autophagy. Eur. J. Pharmacol. 2019, 863, 172677. [Google Scholar] [CrossRef]
- Patil, S.P.; Jain, P.D.; Ghumatkar, P.J.; Tambe, R.; Sathaye, S. Neuroprotective effect of metformin in MPTP-induced Parkinson’s disease in mice. Neuroscience 2014, 277, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.-G.; Wong, V.K.-W.; Xu, S.-W.; Chan, W.-K.; Ng, C.-I.; Liu, L.; Law, B.Y.-K. Onjisaponin B derived from radix polygalae enhances autophagy and accelerates the degradation of mutant α-synuclein and huntingtin in pc-12 cells. Int. J. Mol. Sci. 2013, 14, 22618–22641. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhai, Y.; Yuan, J.; Hu, Y. New insights into Paeoniaceae used as medicinal plants in China. Sci. Rep. 2019, 9, 18469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Cao, Y.B.; Hu, L.F.; Yang, Y.P.; Li, J.; Wang, F.; Liu, C.F. ASICs mediate the modulatory effect by paeoniflorin on alpha-synuclein autophagic degradation. Brain Res. 2011, 1396, 77–87. [Google Scholar] [CrossRef]
- Yang, L.-Y.; Greig, N.H.; Tweedie, D.; Jung, Y.J.; Chiang, Y.-H.; Hoffer, B.J.; Miller, J.P.; Chang, K.-H.; Wang, J.-Y. The p53 inactivators pifithrin-μ and pifithrin-α mitigate TBI-induced neuronal damage through regulation of oxidative stress, neuroinflammation, autophagy and mitophagy. Exp. Neurol. 2020, 324, 113135. [Google Scholar] [CrossRef]
- Duan, W.; Zhu, X.; Ladenheim, B.; Yu, Q.-S.; Guo, Z.; Oyler, J.; Cutler, R.G.; Cadet, J.L.; Greig, N.H.; Mattson, M.P. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol. 2002, 52, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.-J.; Nam, Y.; Lee, J.W.; Nguyen, P.-K.T.; Yoo, J.E.; Tran, T.-V.; Jeong, J.H.; Jang, C.-G.; Oh, Y.J.; Youdim, M.B.H.; et al. N-Methyl, N-propynyl-2-phenylethylamine (MPPE), a selegiline analog, attenuates MPTP-induced dopaminergic toxicity with guaranteed behavioral safety: Involvement of inhibitions of mitochondrial oxidative burdens and p53 gene-elicited pro-apoptotic change. Mol. Neurobiol. 2016, 53, 6251–6269. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Ariyoshi, M.; Okawa, Y.; Kaimoto, S.; Uchihashi, M.; Fukai, K.; Iwai-Kanai, E.; Ikeda, K.; Ueyama, T.; Ogata, T.; et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic beta-cell function in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 3116–3121. [Google Scholar] [CrossRef] [Green Version]
- Santini, E.; Heiman, M.; Greengard, P.; Valjent, E.; Fisone, G. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci. Signal. 2009, 2, ra36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T.; Rawal, P.; Wu, Y.; Xie, W.; Jankovic, J.; Le, W. Rapamycin protects against rotenone-induced apoptosis through autophagy induction. Neuroscience 2009, 164, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Tain, L.S.; Mortiboys, H.; Tao, R.N.; Ziviani, E.; Bandmann, O.; Whitworth, A.J. Rapamycin activation of 4E-BP prevents parkinsonian dopaminergic neuron loss. Nat. Neurosci. 2009, 12, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Malagelada, C.; Jin, Z.H.; Jackson-Lewis, V.; Przedborski, S.; Greene, L.A. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson’s disease. J. Neurosci. 2010, 30, 1166–1175. [Google Scholar] [CrossRef]
- Maiese, K.; Chong, Z.Z.; Shang, Y.C.; Wang, S. mTOR: On target for novel therapeutic strategies in the nervous system. Trends Mol. Med. 2013, 19, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Bai, X.; Wey, M.C.; Fernandez, E.; Hart, M.J.; Gelfond, J.; Bokov, A.F.; Rani, S.; Strong, R. Rapamycin improves motor function, reduces 4-hydroxynonenal adducted protein in brain, and attenuates synaptic injury in a mouse model of synucleinopathy. Pathobiol. Aging Age Relat. Dis. 2015, 5, 28743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.-z.; Chen, X.-p.; Zhao, K.; Bai, L.-m.; Zhang, H.; Zhou, X.-p. Therapeutic effects of valproate combined with lithium carbonate on MPTP-induced Parkinsonism in mice: Possible mediation through enhanced autophagy. Int. J. Neurosci. 2012, 123, 73–79. [Google Scholar] [CrossRef]
- Price, P.A.; Parkes, J.D.; Marsden, C.D. Sodium valproate in the treatment of levodopa-induced dyskinesia. J. Neurol. Neurosurg. Psychiatry 1978, 41, 702–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nutt, J.G.; Williams, A.C.; Calne, D.B. Effect of sodium valproate on parkinsonism and L-DOPA induced dyskinesia. Brain Res. Bull. 1980, 5, 589–593. [Google Scholar] [CrossRef]
- Ristic, A.J.; Vojvodic, N.; Jankovic, S.; Sindelic, A.; Sokic, D. The frequency of reversible parkinsonism and cognitive decline associated with valproate treatment: A study of 364 patients with different types of epilepsy. Epilepsia 2006, 47, 2183–2185. [Google Scholar] [CrossRef] [PubMed]
- Silver, M.; Factor, S.A. Valproic acid-induced parkinsonism: Levodopa responsiveness with dyskinesia. Park. Relat. Disord. 2013, 19, 758–760. [Google Scholar] [CrossRef]
- Büttner, S.; Broeskamp, F.; Sommer, C.; Markaki, M.; Habernig, L.; Alavian-Ghavanini, A.; Carmona-Gutierrez, D.; Eisenberg, T.; Michael, E.; Kroemer, G.; et al. Spermidine protects against α-synuclein neurotoxicity. Cell Cycle 2014, 13, 3903–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saiki, S.; Sasazawa, Y.; Fujimaki, M.; Kamagata, K.; Kaga, N.; Taka, H.; Li, Y.; Souma, S.; Hatano, T.; Imamichi, Y.; et al. A metabolic profile of polyamines in parkinson disease: A promising biomarker. Ann. Neurol. 2019, 86, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Zhao, K.; Calingasan, N.Y.; Luo, G.; Szeto, H.H.; Beal, M.F. Mitochondria targeted peptides protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity. Antioxid. Redox Signal. 2009, 11, 2095–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Anantharam, V.; Kalyanaraman, B.; Kanthasamy, A.G. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: Preclinical and clinical outcomes. Biochim. Biophys. Acta 2014, 1842, 1282–1294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escribano-López, I.; de Marañon, A.M.; Iannantuoni, F.; López-Domènech, S.; Abad-Jiménez, Z.; Díaz, P.; Solá, E.; Apostolova, N.; Rocha, M.; Víctor, V.M. The mitochondrial antioxidant SS-31 modulates oxidative stress, endoplasmic reticulum stress, and autophagy in type 2 diabetes. J. Clin. Med. 2019, 8, 1322. [Google Scholar] [CrossRef] [Green Version]
- Muller, S. Excipients: Not so inert? when the excipient plays the role of an active substance, as exemplified by systemic lupus. Swiss Med. Wkly. 2018, 148, w14631. [Google Scholar] [CrossRef] [Green Version]
- DeBosch, B.J.; Heitmeier, M.R.; Mayer, A.L.; Higgins, C.B.; Crowley, J.R.; Kraft, T.E.; Chi, M.; Newberry, E.P.; Chen, Z.; Finck, B.N.; et al. Trehalose inhibits solute carrier 2A (SLC2A) proteins to induce autophagy and prevent hepatic steatosis. Sci. Signal. 2016, 9, ra21. [Google Scholar] [CrossRef] [Green Version]
- Sarkar, S.; Davies, J.E.; Huang, Z.; Tunnacliffe, A.; Rubinsztein, D.C. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J. Biol. Chem. 2007, 282, 5641–5652. [Google Scholar] [CrossRef] [Green Version]
- Rusilowicz-Jones, E.V.; Barone, F.G.; Lopes, F.M.; Stephen, E.; Mortiboys, H.; Urbé, S.; Clague, M.J. Benchmarking a highly selective USP30 inhibitor for enhancement of mitophagy and pexophagy. bioRxiv 2021. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.N.; Chavalmane, A.K.; Pillai, M.; Ammanathan, V.; Vidyadhara, D.J.; Yarreiphang, H.; Rai, S.; Paul, A.; Clement, J.P.; Alladi, P.A.; et al. Modulation of autophagy by a small molecule inverse agonist of ERRα Is neuroprotective. Front. Mol. Neurosci. 2018, 11, 109. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.; Noda, T.; Yoshimori, T.; Rubinsztein, D. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat. Chem. Biol. 2011, 7, 9–17. [Google Scholar] [CrossRef]
- Javed, H.; Nagoor Meeran, M.F.; Azimullah, S.; Adem, A.; Sadek, B.; Ojha, S.K. Plant extracts and phytochemicals targeting alpha-synuclein aggregation in Parkinson’s disease models. Front. Pharmacol. 2018, 9, 1555. [Google Scholar]
- Vucicevic, L.; Misirkic-Marjanovic, M.; Harhaji-Trajkovic, L.; Maric, N.; Trajkovic, V. Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs. Cell Stress 2018, 2, 282–291. [Google Scholar] [CrossRef]
- Thellung, S.; Corsaro, A.; Nizzari, M.; Barbieri, F.; Florio, T. Autophagy activator drugs: A new opportunity in neuroprotection from misfolded protein toxicity. Int. J. Mol. Sci. 2019, 20, 901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, B.; Potkar, R.; Trejo, M.; Rockenstein, E.; Patrick, C.; Gindi, R.; Adame, A.; Wyss-Coray, T.; Masliah, E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J. Neurosci. 2009, 29, 13578–13588. [Google Scholar] [CrossRef] [Green Version]
- Perrone, L.; Squillaro, T.; Napolitano, F.; Terracciano, C.; Sampaolo, S.; Melone, M.A.B. The autophagy signaling pathway: A potential multifunctional therapeutic target of curcumin in neurological and neuromuscular diseases. Nutrients 2019, 11, 1881. [Google Scholar] [CrossRef] [Green Version]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosi, G.; Ghezzi, C.; Zangaglia, R.; Levandis, G.; Pacchetti, C.; Blandini, F. Ambroxol-induced rescue of defective glucocerebrosidase is associated with increased LIMP-2 and saposin C levels in GBA1 mutant Parkinson’s disease cells. Neurobiol. Dis. 2015, 82, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Migdalska-Richards, A.; Daly, L.; Bezard, E.; Schapira, A.H.V. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 2016, 80, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Magalhaes, J.; Gegg, M.E.; Migdalska-Richards, A.; Schapira, A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018, 8, 1385. [Google Scholar] [CrossRef]
- Mullin, S.; Smith, L.; Lee, K.; D’Souza, G.; Woodgate, P.; Elflein, J.; Hällqvist, J.; Toffoli, M.; Streeter, A.; Hosking, J.; et al. Ambroxol for the treatment of patients with Parkinson disease with and without glucocerebrosidase gene mutations: A nonrandomized, noncontrolled trial. JAMA Neurol. 2020, 77, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Wu, Y.; Li, X.; Zhu, J.X.; Xie, W.; Le, W.; Fan, Z.; Jankovic, J.; Pan, T. Resveratrol-activated AMPK/SIRT1/autophagy in cellular models of Parkinson’s disease. Neurosignals 2011, 19, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Hernández, Á.; Cordero, M.D.; Salviati, L.; Artuch, R.; Pineda, M.; Briones, P.; Gómez Izquierdo, L.; Cotán, D.; Navas, P.; Sánchez-Alcázar, J.A. Coenzyme Q deficiency triggers mitochondria degradation by mitophagy. Autophagy 2009, 5, 19–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.Z.; Chen, Y.Z.; Su, M.; Zheng, H.F.; Yang, Y.P.; Chen, J.; Liu, C.F. dl-3-n-Butylphthalide prevents oxidative damage and reduces mitochondrial dysfunction in an MPP(+)-induced cellular model of Parkinson’s disease. Neurosci. Lett. 2010, 475, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ye, M.; Xu, W.; Yu, M.; Liu, X.; Chen, Y. DL-3-n-butylphthalide therapy for Parkinson’s disease: A randomized controlled trial. Exp. Ther. Med. 2019, 17, 3800–3806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Ma, F.; Huang, L.; Zhang, Y.; Peng, Y.; Xing, C.; Feng, Y.; Wang, X.; Peng, Y. Dl-3-n-Butylphthalide (NBP): A promising therapeutic agent for ischemic stroke. CNS Neurol. Disord. Drug Targets 2018, 17, 338–347. [Google Scholar] [CrossRef]
- Xiong, N.; Huang, J.; Chen, C.; Zhao, Y.; Zhang, Z.; Jia, M.; Zhang, Z.; Hou, L.; Yang, H.; Cao, X.; et al. Dl-3-n-butylphthalide, a natural antioxidant, protects dopamine neurons in rotenone models for Parkinson’s disease. Neurobiol. Aging 2012, 33, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Tian, A.; Ma, X.; Li, H.; Zhang, R. Dl-3n-butylphthalide improves spatial learning and memory in rats with vascular dementia by reducing autophagy via regulation of the mTOR signaling pathway. Exp. Ther. Med. 2020, 19, 1940–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, D.; Zhang, Y.; Peng, T. Abstract 20228: Nicotinamide riboside improves autophagic flux and prevents doxorubicin-induced cardiac injury. Circulation 2017, 136, A20228. [Google Scholar]
- Maiese, K. New insights for nicotinamide: Metabolic disease, autophagy, and mTOR. Front. Biosci. 2020, 25, 1925–1973. [Google Scholar] [CrossRef] [PubMed]
- Hebron, M.L.; Lonskaya, I.; Moussa, C.E.-H. Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum. Mol. Genet. 2013, 22, 3315–3328. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Abraham, N.; Gao, G.; Yang, Q. Dysregulation of autophagy and mitochondrial function in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- Issa, A.-R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Bonam, S.R.; Ruff, M.; Muller, S. HSPA8/HSC70 in immune disorders: A molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells 2019, 8, 849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renna, M.; Jimenez-Sanchez, M.; Sarkar, S.; Rubinsztein, D.C. Chemical inducers of autophagy that enhance the clearance of mutant proteins in neurodegenerative diseases. J. Biol. Chem. 2010, 285, 11061–11067. [Google Scholar] [CrossRef] [Green Version]
- Rubinsztein, D.C.; Codogno, P.; Levine, B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 2012, 11, 709–730. [Google Scholar] [CrossRef] [Green Version]
- Gros, F.; Muller, S. Pharmacological regulators of autophagy and their link with modulators of lupus disease. Br. J. Pharmacol. 2014, 171, 4337–4359. [Google Scholar] [CrossRef] [Green Version]
- Levine, B.; Packer, M.; Codogno, P. Development of autophagy inducers in clinical medicine. J. Clin. Investig. 2015, 125, 14–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardones, P.; Rubinsztein, D.C.; Hetz, C. Mystery solved: Trehalose kickstarts autophagy by blocking glucose transport. Sci. Signal. 2016, 9, fs2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, S.E.; Colon-Ramos, D.A. The journey of the synaptic autophagosome: A cell biological perspective. Neuron 2020, 105, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Wang, F.; Schall, N.; Muller, S. Rescue of autophagy and lysosome defects in salivary glands of MRL/lpr mice by a therapeutic phosphopeptide. J. Autoimmun. 2018, 90, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Brun, S.; Schall, N.; Bonam, S.R.; Bigaut, K.; Mensah-Nyagan, A.-G.; de Sèze, J.; Muller, S. An autophagy-targeting peptide to treat chronic inflammatory demyelinating polyneuropathies. J. Autoimmun. 2018, 92, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Daubeuf, F.; Schall, N.; Petit-Demoulière, N.; Frossard, N.; Muller, S. An autophagy modulator peptide prevents lung function decrease and corrects established inflammation in murine models of airway allergy. Cells 2021, 10, 2468. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, R.; Poornima, P.; Priya, L.B.; Huang, C.Y.; Padma, V.V. Neferine prevents autophagy induced by hypoxia through activation of Akt/mTOR pathway and Nrf2 in muscle cells. Biomed. Pharmacother. 2016, 83, 1407–1413. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.K.; Wu, A.G.; Wang, J.R.; Liu, L.; Law, B.Y. Neferine attenuates the protein level and toxicity of mutant huntingtin in PC-12 cells via induction of autophagy. Molecules 2015, 20, 3496–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, P.K.; Fahrner, A.; Vats, S.; Seranova, E.; Sharma, V.; Chipara, M.; Desai, P.; Torresi, J.; Rosenstock, T.; Kumar, D.; et al. Chemical screening approaches enabling drug discovery of autophagy modulators for biomedical applications in human diseases. Front. Cell Dev. Biol. 2019, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Suresh, S.N.; Chakravorty, A.; Giridharan, M.; Garimella, L.; Manjithaya, R. Pharmacological tools to modulate autophagy in neurodegenerative diseases. J. Mol. Biol. 2020, 432, 2822–2842. [Google Scholar] [CrossRef]
- Hatstat, A.K.; Ahrendt, H.D.; Foster, M.W.; Mayne, L.; Moseley, M.A.; Englander, S.W.; McCafferty, D.G. Characterization of small-molecule-induced changes in Parkinson’s-related trafficking via the nedd4 ubiquitin signaling cascade. Cell Chem. Biol. 2021, 28, 14.e19–25.e19. [Google Scholar] [CrossRef]
- Zhu, Y.; Shan, X.; Safarpour, F.; Erro Go, N.; Li, N.; Shan, A.; Huang, M.C.; Deen, M.; Holicek, V.; Ashmus, R.; et al. Pharmacological inhibition of O-GlcNAcase enhances autophagy in brain through an mTOR-independent pathway. ACS Chem. Neurosci. 2018, 9, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.; Xu, M.; Davey, A.K.; Danon, J.J.; Mellick, G.D.; Kassiou, M.; Rudrawar, S. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem. Neurosci. 2019, 10, 2209–2221. [Google Scholar] [CrossRef]
- Whitworth, A.J.; Wes, P.D.; Pallanck, L.J. Drosophila models pioneer a new approach to drug discovery for Parkinson’s disease. Drug Discov. Today 2006, 11, 119–126. [Google Scholar] [CrossRef]
- Chen-Plotkin, A.S.; Albin, R.; Alcalay, R.; Babcock, D.; Bajaj, V.; Bowman, D.; Buko, A.; Cedarbaum, J.; Chelsky, D.; Cookson, M.R.; et al. Finding useful biomarkers for Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaam6003. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.M.C.; Bannerman, D.M. Mouse models of neurodegeneration: Know your question, know your mouse. Sci. Transl. Med. 2019, 11, eaaq1818. [Google Scholar] [CrossRef] [PubMed]





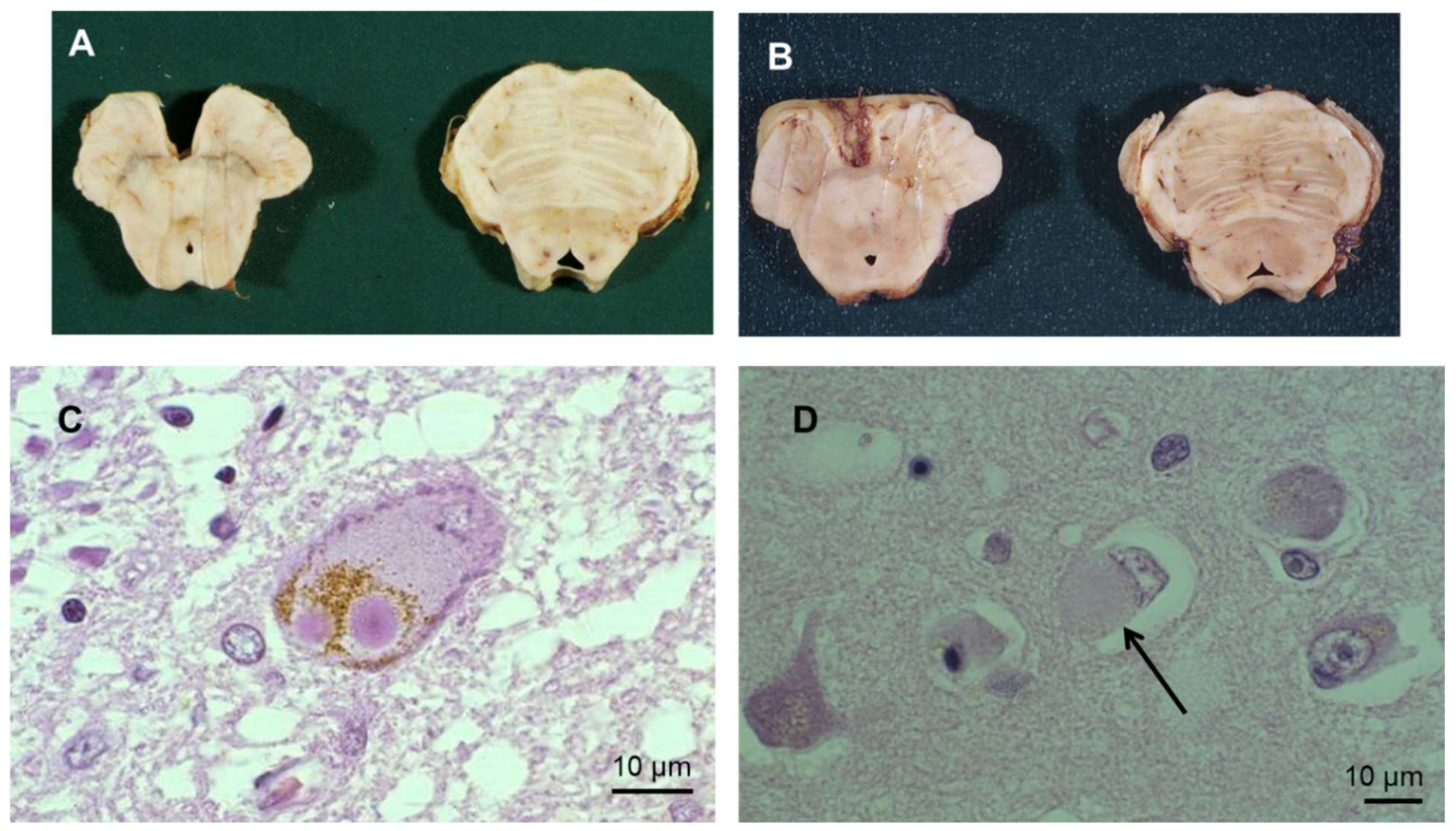{kind=link}
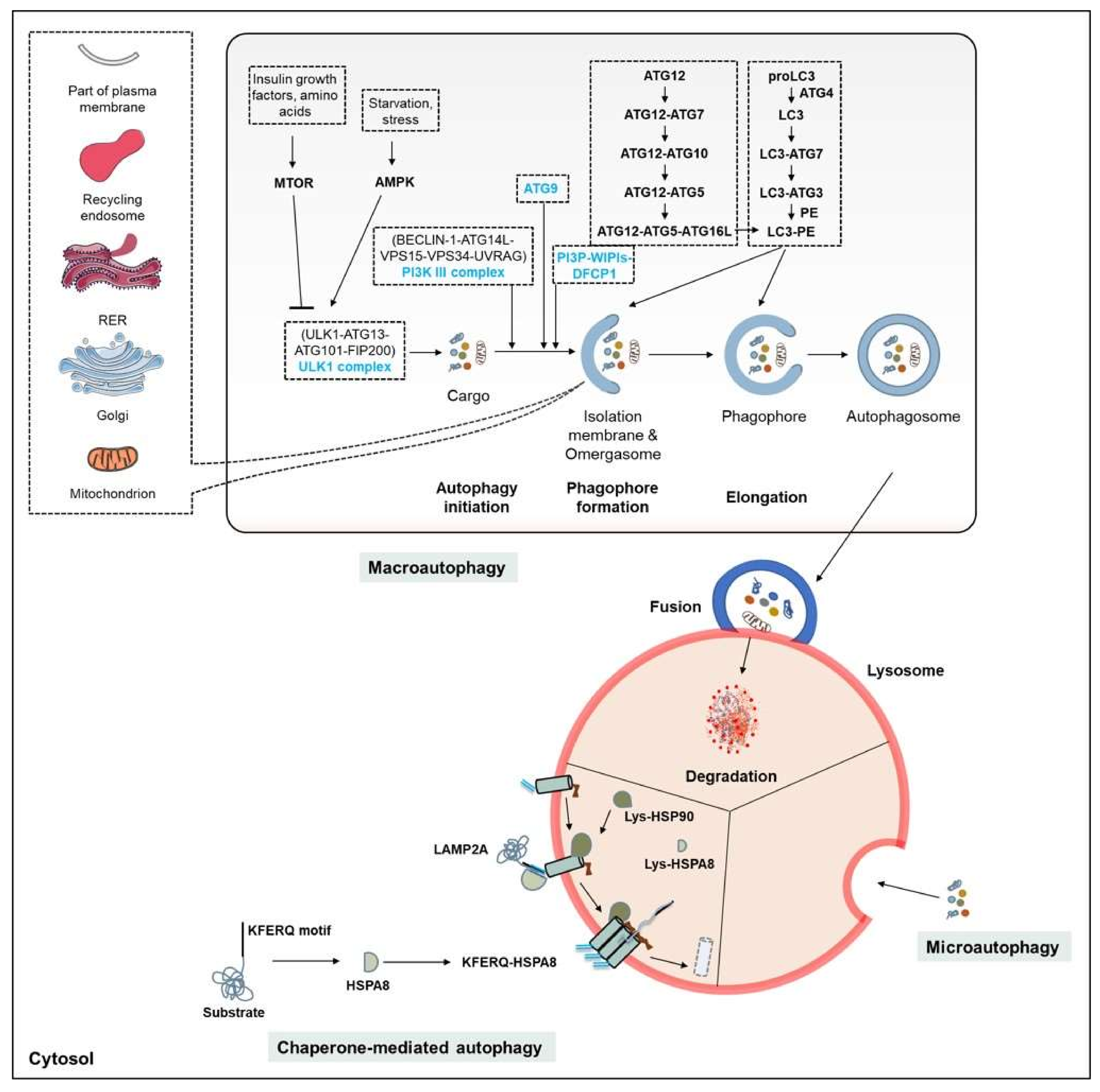{kind=link}
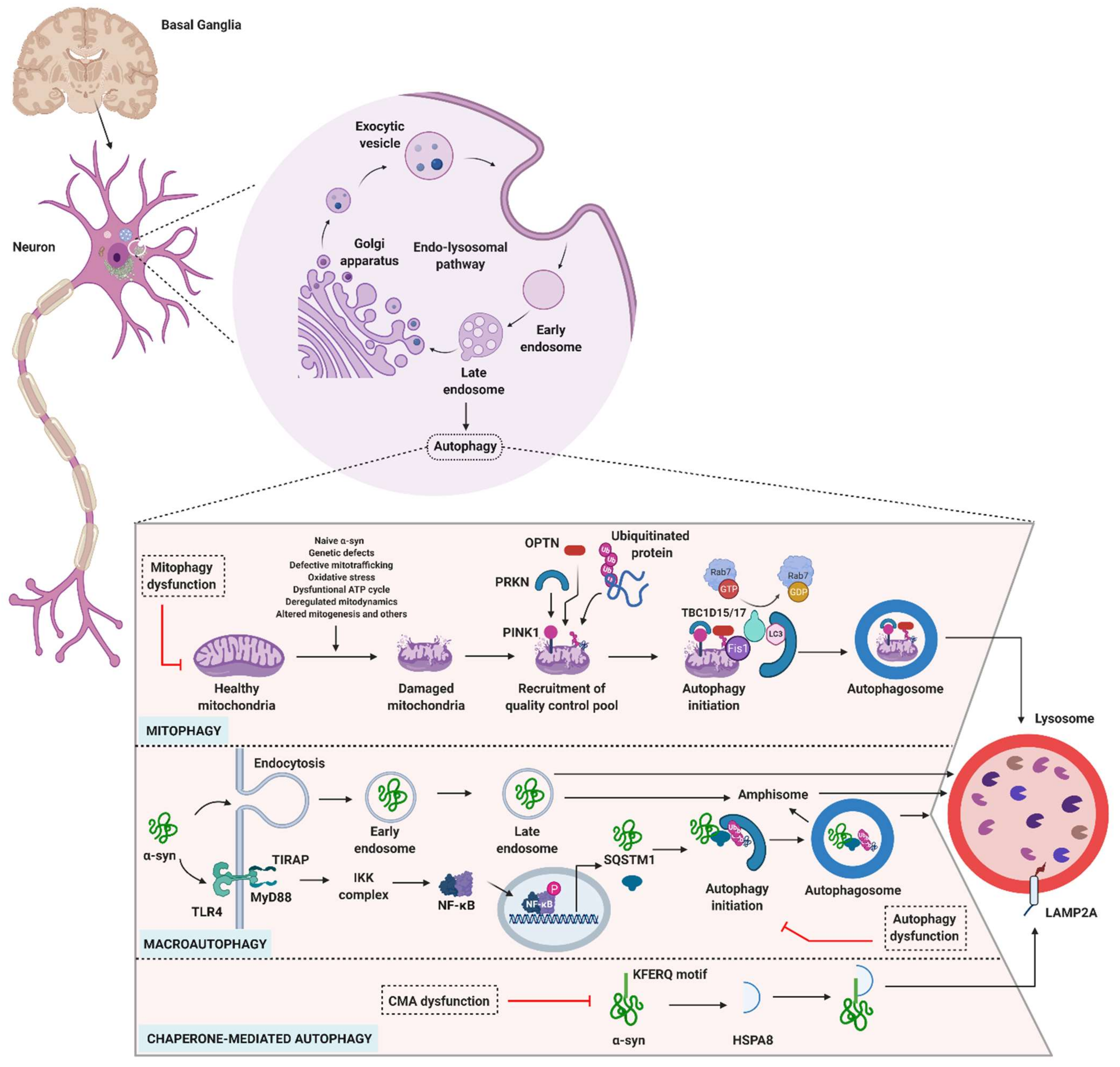{kind=link}
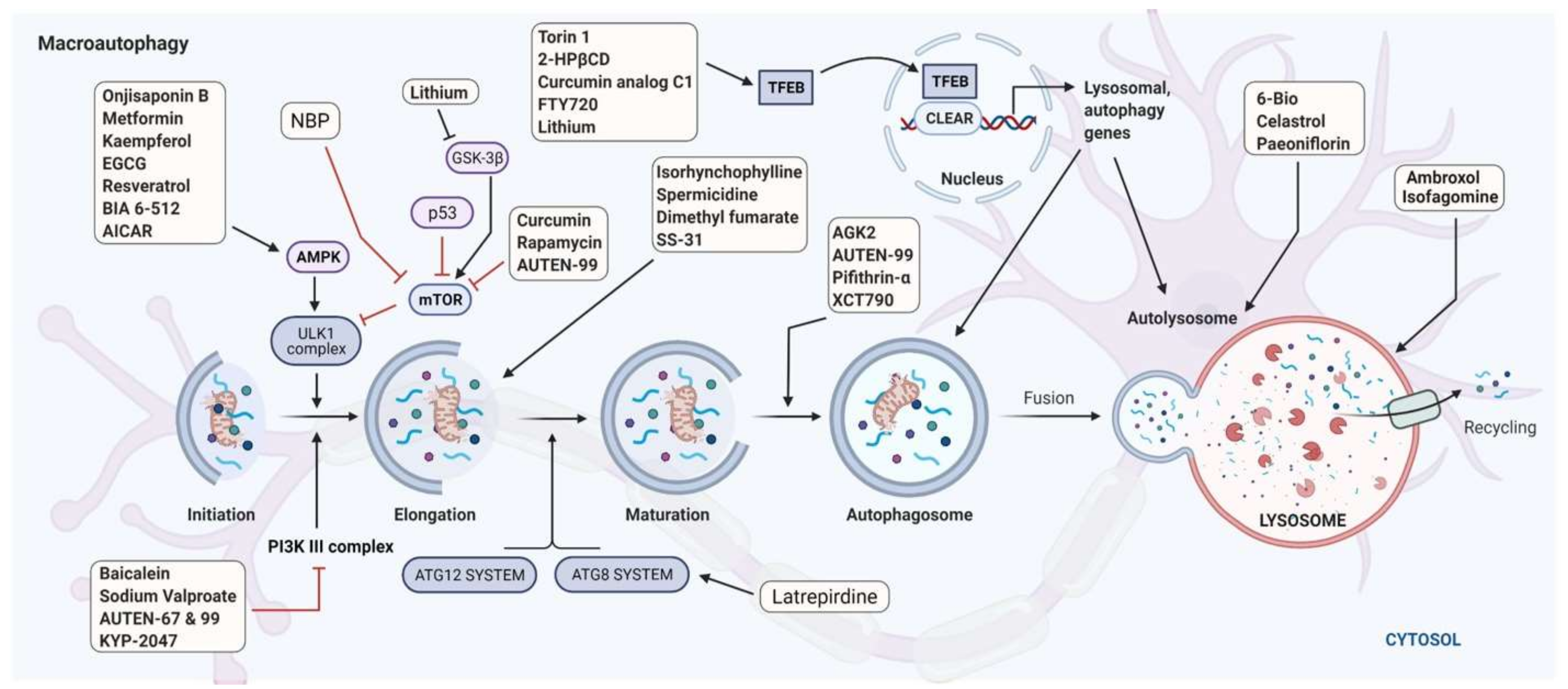{kind=link}
| Processes | Organelles and Substrates | Selective Markers | Functions |
|---|---|---|---|
| Aggrephagy | Protein aggregates | MAP1LC3, Alfy |
|
| Crinophagy | Secretory vesicles or granules | HOPS |
|
| CMA | Proteins that contain KFERQ-motif | LAMP2A, HSPA8 |
|
| DNautophagy | DNA | LAMP2C |
|
| ER-phagy (also referred to as reticulophagy) | Endoplasmic reticulum | Syntaxin 17, DFCP1 |
|
| Ferritinophagy | Ferritin | NCOA4 |
|
| Glycophagy | Glycogen | STBD1/Genethonin-1 |
|
| LAP | Bacteria, living and dead cells | Lipidated LC3 |
|
| Lipophagy | Lipid droplets | LIPA |
|
| Lysophagy | Lysosomes | LAMP2A | |
| Mitophagy | Mitochondria | PINK1, RKN |
|
| Nucleophagy | Nucleus | ATG39 * |
|
| Pexophagy | Peroxisomes | ATG36 |
|
| Proteaphagy | Proteasomes | Proteasome (depends on the size of the proteasome) |
|
| Ribophagy | Ribosomes | Ubp3, Bre5, Rsp5 |
|
| RNautophagy | RNA | LAMP2C |
|
| Synucleinphagy | α-syn | SQSTM1 |
|
| Xenophagy | Invading microbes | CALCOCO2/NDP52 |
|
| Zymophagy | Pancreatic zymogen granules | VMP1 |
|
| Genes | Functions | Link to PD |
|---|---|---|
| ATG5 | ATG5-dependent autophagy protects various cells, including neurons, from apoptosis [99]. |
|
| ATG7 | ATG7 is a vital part of the ATG8 and ATG12 conjugating systems of autophagy, and plays an essential role in neuronal development [101]. |
|
| ATG12 | In conjunction with other ATGs, it forms an ATG12 system, which is essential for autophagosome formation. |
|
| ATP6AP2 | Vacuolar ATPase localized on the lysosomal membrane that regulates pH. | |
| ATP10B | P4-type ATPase present on the late endo/lysosomal compartment. In addition to its lipid flippase function (transfer of lipids from the exoplasmic to the cytoplasmic membrane leaflet), it also transports cell cycle regulator proteins from the ER to endosomes and lysosomes [9,13]. |
|
| ATP13A2/PARK9 | P5-type ATPase localized on vesicular structures, particularly lysosomes; functions as a cation transporter [10]. |
|
| DJ-1/PARK7 | Functions as an antioxidant and chaperone. Through its diversified functions, it maintains mitochondrial homeostasis [106]. |
|
| DNAJC13/PARK21 | Retromer-mediated endosomal protein sorting [106]. |
|
| FBOX7/PARK15 | In association with PINK1 and PRKN, regulates mitophagy [109]. |
|
| GBA1 | GBA is a lysosomal enzyme, which degrades cell membrane glycolipids, i.e., glycosylceramide [48]. |
|
| HSPA8 | Acts as a substrate carrier protein in CMA. |
|
| LRRK2 | LRRK2 is known for sorting various vesicles, based on its interactions with Rab5b, Rab7, Rab7L1, Hsc70, and others [10]. |
|
| MAP1LC3 | MAP1LC3B and its subfamily members are crucial to autophagosome formation, elongation and maturation [88]. |
|
| PRKN/PARK2 | Gene that encodes PRKN (E3 ligase), which plays an essential role in mitophagy in parallel with PINK1. |
|
| PINK1/PARK6 | Member of the serine-threonine kinases class, plays a crucial role in mitochondria quality control by inducing mitophagy. |
|
| TMEM175 | Gene that encodes endo/lysosomal K+ channel [117]. |
|
| TMEM230 | Gene that encodes transmembrane protein involved in retromer and secretory vesicle trafficking functions. |
|
| VPS13C/PARK23 | Mitophagy development. |
|
| VPS35/PARK17 | As a part of the retromer complex, it performs protein trafficking functions [106]. |
|
| Candidate | Mechanism of Action | Comments |
|---|---|---|
| AGK2 | Enhances autolysosome formation |
|
| AUTEN-99 (autophagy enhancer-99) | Inhibits the phosphatase activity of MTMR14 (Jumpy), a negative regulator of autophagy | |
| Baicalein | Induces PI3K-mediated autophagy |
|
| 6-Bio | Enhances autolysosome formation in DA neurons |
|
| Celastrol | Enhances autolysosome formation |
|
| Curcumin | Inhibits MTOR/p70S6K |
|
| Curcumin analogue C1 | MTOR-independent TFEB enhancer. Selectively binds to the N-terminus of TFEB and promotes its nuclear translocation | |
| Dimethyl fumarate | Modulator of SQSTM1-dependent autophagy |
|
| Epigallocatechin gallate (EGCG) | Activates AMPK | |
| 2-HPβCD | Activates TFEB |
|
| Isofagomine | GCase |
|
| (Iso)rhynchophylline | Activation of BECN-mediated autophagy |
|
| Kaempferol | AMPK/MTOR-mediated pathway | |
| KYP-2047 | Prolyl endopeptidase inhibitor III | |
| Latrepirdine (Dimebon; dimebolin) | Enhances ATG8-dependent autophagy | |
| Lithium | Reduces IP3 production by inhibiting inositol monophosphatase |
|
| MCC950 | Potent, selective inhibitor of NLRP3 with IC50 of 7.5 nM in BMDMs | |
| Metformin | Antihyperglycemic agent of the biguanide class. Acts via both AMPK-dependent and AMPK-independent mechanisms |
|
| Onjisaponin B | Triggers autophagy via the AMPK/MTOR-mediated signaling pathway |
|
| Paeoniflorin | Enhances autolysosome formation | |
| Pifithrin (PFT)-α | Specific inhibitor of p53 transcription activity. Displays potent p53-independent activity in cells. |
|
| Rapamycin and its derivatives (CCI-779, RAD001 and AP23573) | Inhibitors of MTOR | |
| Sodium valproate | Synergistic to lithium-induced autophagy |
|
| Spermidine | Autophagy inducer that maintains cellular and neuronal homeostasis. May achieve effects via BECN1 and TFEB-mediated pathways |
|
| SS-31 (D-Arg-2′6′-dimethylTyr-Lys-Phe-NH2) | BECN1-mediated autophagy | |
| Torin1 | MTOR-dependent TFEB enhancer | |
| Trehalose | Induces autophagy via lysosome-mediated TFEB activation | |
| MF-094 | Selective USP30 inhibitor | |
| XCT790 | Modulates autophagosome formation by estrogen-related receptor α-dependent manner |
|
| Drugs | Characteristics and Mechanism of Action | Stage of Development/Sponsors | Identifiers |
|---|---|---|---|
| Ambroxol | Well-known expectorant and mucolytic agent. In addition to its diverse functions, also acts as a potent chaperone for GCase [277]. In Vitro and in vivo studies in wild-type and transgenic models indicate that its therapeutic effects in the context of PD involve modulating the autophagy process [221,278,279]. These effects are mediated by the TFEB pathway and through increased exocytosis [280]. A recent clinical study in PD patients with and without GBA1 mutations provided positive preliminary data for large cohort studies [281]. | Phase II/University College, London, UK (NCT02941822), Lawson Health Research Institute (NCT02914366) | NCT02941822; NCT02914366 |
| BIA 6-512 (trans-resveratrol) | An analogue of a natural compound, resveratrol, which has a wide pharmacological action, especially in PD [282,283]. Potential antioxidant; it also initiates Sirtuin 1 (SIRT1)-dependent AMPK activation [103,283]. | Phase I/Bial—Portela C S.A. | NCT03095092 |
| Coenzyme Q10 (CoQ) | Free radical scavenger used as a supplement in PD. Reduced CoQ in cells triggers mitophagy [284]. | Phase III/Weill Medical College of Cornell University (NCT00740714), Technische Universität Dresden, Germany (NCT00180037) | NCT00740714; NCT00180037 |
| DL-3-n-butylphthalide (NBP) | An active component isolated from Apium graveolens (Apiaceae) [285]. Its antioxidant function, and capacity to correct mitochondrial dysfunction is widely documented [286]. Approved by the Chinese FDA for the treatment of ischemic stroke [287]. Various preclinical models of PD have confirmed its efficacy in alleviating neuronal toxicity via autophagic mechanisms; indirectly inhibits MTOR-mediated autophagy [286,288,289]. | Phase II/First Affiliated Hospital of Bengbu Medical College (Bengbu, China) | ChiCTR1800018892 |
| MitoQ (mitoquinone) | A conjugate of lipophilic triphenylphosphonium cation and coenzyme Q10. Powerful antioxidant. Studies with MitoQ on leukemia cells (HL-60) and hepatocellular carcinoma cells (HepG2) confirmed the induction of autophagy following activation of AMPK and inhibition of MTOR [207]. | Phase II/Antipodean Pharmaceuticals, Inc. | NCT00329056 |
| Nicotinamide riboside | Used as a supplement. Nicotinamide (in various forms) induces autophagy by acting on SIRT1-dependent and MTOR-mediated mechanisms [290,291]. | ND/Haukeland University Hospital, Norway | NCT03816020 |
| Nilotinib | An Abelson kinase/tyrosine kinase inhibitor. Approved by the US FDA (2007) for the treatment of chronic myelogenous leukemia. Preclinical studies on transgenic mice models (A53T mutation of human α-syn) have shown that it increases α-syn clearance via BECN1-mediated autophagy [292]. Reduces cell death and improves motor function. Under evaluation for treatment of other neurodegenerative diseases (AD, NCT02947893; HD, NCT03764215). | Phase II/Georgetown University, USA | NCT02954978 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonam, S.R.; Tranchant, C.; Muller, S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells 2021, 10, 3547. https://doi.org/10.3390/cells10123547
Bonam SR, Tranchant C, Muller S. Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells. 2021; 10(12):3547. https://doi.org/10.3390/cells10123547
Chicago/Turabian StyleBonam, Srinivasa Reddy, Christine Tranchant, and Sylviane Muller. 2021. "Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease" Cells 10, no. 12: 3547. https://doi.org/10.3390/cells10123547
APA StyleBonam, S. R., Tranchant, C., & Muller, S. (2021). Autophagy-Lysosomal Pathway as Potential Therapeutic Target in Parkinson’s Disease. Cells, 10(12), 3547. https://doi.org/10.3390/cells10123547