
Metabolic Reprogramming of Liver Fibrosis



Abstract
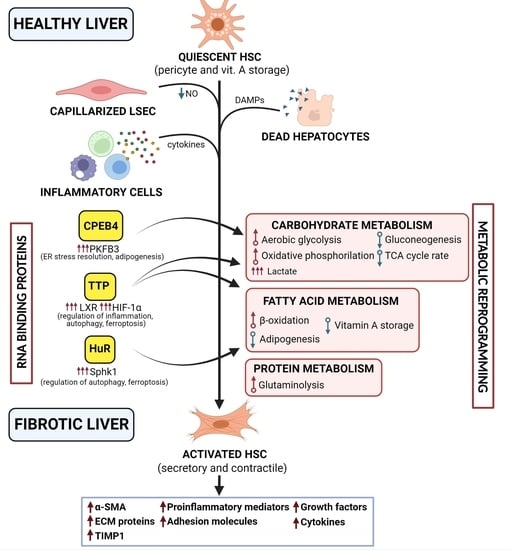
:
{kind=link}
{kind=link}
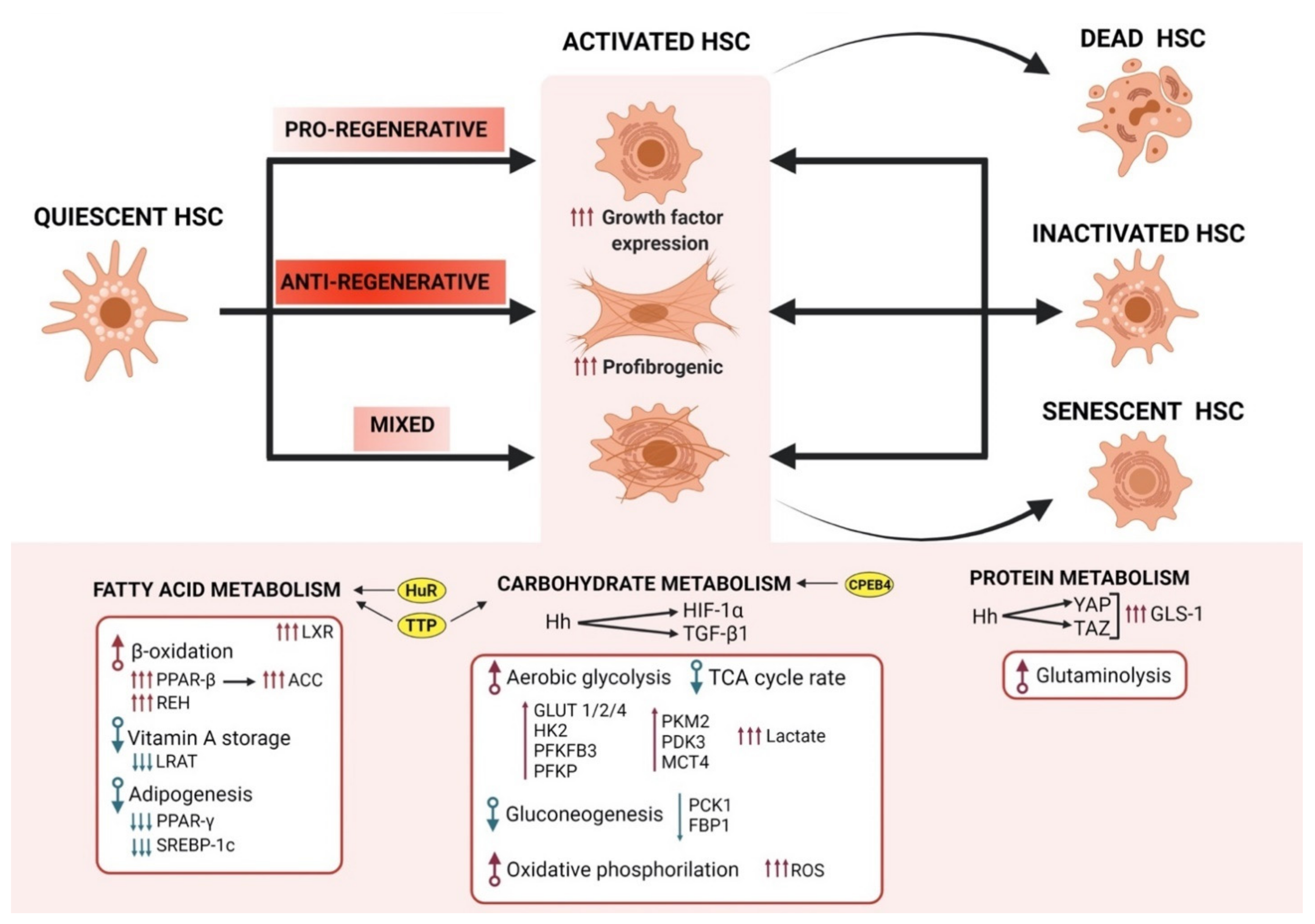{kind=link}
1. Introduction
2. Fibrogenesis and Chronic Liver Diseases
3. HSC-Dependent Molecular Mechanism of Liver Fibrosis
4. Metabolic Reprogramming of HSC in Fibrogenesis
5. RBPs Regulation of HSC Metabolic Reprogramming
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
 .
.Conflicts of Interest
Abbreviations
References
- Roth, G.A.; Abate, D.; Abate, K.H.; Abay, S.M.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; Abdela, J.; Abdelalim, A.; et al. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: A systematic analysis for the Global Burden of Disease Study. Lancet 2018, 392, 1736–1788. [Google Scholar] [CrossRef] [Green Version]
- International Agency for Research in Cancer; World Health Organization. Global Cancer Observatory. Available online: https://gco.iarc.fr/ (accessed on 8 November 2021).
- Sepanlou, S.G.; Safiri, S.; Bisignano, C.; Ikuta, K.S.; Merat, S.; Saberifiroozi, M.; Poustchi, H.; Tsoi, D.; Colombara, D.V.; Abdoli, A.; et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet Gastroenterol. Hepatol. 2020, 5, 245–266. [Google Scholar] [CrossRef] [Green Version]
- Eurotransplant Liver-Eurotransplant Annual Report. Available online: https://www.eurotransplant.org/organs/liver/ (accessed on 8 November 2021).
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef]
- Affo, S.; Yu, L.-X.; Schwabe, R.F. The Role of Cancer-Associated Fibroblasts and Fibrosis in Liver Cancer. Annu. Rev. Pathol. 2017, 12, 153–186. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Friedman, S.L. Hepatic fibrosis: A convergent response to liver injury that is reversible. J. Hepatol. 2020, 73, 210–211. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.T.; Wang, N.; Tan, H.Y.; Li, S.; Feng, Y. Targeting Hepatic Stellate Cells for the Treatment of Liver Fibrosis by Natural Products: Is It the Dawning of a New Era? Front. Pharmacol. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, P.; Wang, S.; Friedman, S.L. The Power of Plasticity—Metabolic Regulation of Hepatic Stellate Cells. Cell Metab. 2021, 33, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis–from bench to bedside. J. Hepatol. 2003, 38, 38–53. [Google Scholar] [CrossRef]
- Afratis, N.A.; Selman, M.; Pardo, A.; Sagi, I. Emerging insights into the role of matrix metalloproteases as therapeutic targets in fibrosis. Matrix Biol. 2018, 68–69, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Roehlen, N.; Crouchet, E.; Baumert, T.F. Liver Fibrosis: Mechanistic Concepts and Therapeutic Perspectives. Cells 2020, 9, 875. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, S.H.; Hirsova, P.; Gores, G.J. Non-alcoholic steatohepatitis pathogenesis: Sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018, 67, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global epidemiology of NAFLD-related HCC: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Eslam, M.; Kawaguchi, T.; Tsutsumi, T.; Nakano, D.; Yoshinaga, S.; Takahashi, H.; Anzai, K.; George, J.; Torimura, T. MAFLD identifies patients with significant hepatic fibrosis better than NAFLD. Liver Int. 2020, 40, 3018–3030. [Google Scholar] [CrossRef]
- Lee, Y.A.; Friedman, S.L. Stellate Cells and Fibrosis. In The Liver: Biology and Pathobiology, Sixth Edition; John Wiley & Sons Ltd: New York City, NY, USA, 2020; pp. 444–454. [Google Scholar]
- Gomes, R.N.; Manuel, F.; Nascimento, D.S. The bright side of fibroblasts: Molecular signature and regenerative cues in major organs. npj Regen. Med. 2021, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Friedman, S.L. Fibrosis Regression After Eradication of Hepatitis C Virus: From Bench to Bedside. Gastroenterology 2021, 160, 1502–1520.e1. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Pellicoro, A.; Fallowfield, J.A. Liver Fibrosis: Understanding the Dynamics of Bidirectional Wound Repair to Inform the Design of Markers and Therapies. Dig. Dis. 2017, 35, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Ellis, E.L.; Mann, D.A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 2012, 56, 1171–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufour, J.F.; DeLellis, R.; Kaplan, M.M. Reversibility of hepatic fibrosis in autoimmune hepatitis. Ann. Intern. Med. 1997, 127, 981–985. [Google Scholar] [CrossRef]
- Rockey, D.C. Translating an understanding of the pathogenesis of hepatic fibrosis to novel therapies. Clin. Gastroenterol. Hepatol. 2013, 11, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC. Global Viral Hepatitis: Millions of People are Affected. Available online: https://www.cdc.gov/hepatitis/global/index.htm (accessed on 8 November 2021).
- Campana, L.; Iredale, J. Regression of Liver Fibrosis. Semin. Liver Dis. 2017, 37, 001–010. [Google Scholar] [CrossRef]
- Kisseleva, T.; Brenner, D.A. Mechanisms of Fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Zimmermann, H.W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 2014, 60, 1090–1096. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.; Liu, C.; Zhou, D.; Zhang, L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J. Histochem. Cytochem. 2016, 64, 157–167. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- D’Amico, G.; Morabito, A.; D’Amico, M.; Pasta, L.; Malizia, G.; Rebora, P.; Valsecchi, M.G. New concepts on the clinical course and stratification of compensated and decompensated cirrhosis. Hepatol. Int. 2018, 12, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Habib, A.; Chokr, D.; Wan, J.; Hegde, P.; Mabire, M.; Siebert, M.; Ribeiro-Parenti, L.; Le Gall, M.; Lettéron, P.; Pilard, N.; et al. Inhibition of monoacylglycerol lipase, an anti-inflammatory and antifibrogenic strategy in the liver. Gut 2019, 68, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.-P.; Schwabe, R.F. Fate tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its aetiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.-H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [Green Version]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef] [PubMed]
- Karsdal, M.A.; Detlefsen, S.; Daniels, S.J.; Nielsen, M.J.; Krag, A.; Schuppan, D. Is the Total Amount as Important as Localization and Type of Collagen in Liver Fibrosis Attributable to Steatohepatitis? Hepatology 2020, 71, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Massard, J.; Ratziu, V.; Thabut, D.; Moussalli, J.; Lebray, P.; Benhamou, Y.; Poynard, T. Natural history and predictors of disease severity in chronic hepatitis C. J. Hepatol. 2006, 44, S19–S24. [Google Scholar] [CrossRef]
- Villesen, I.F.; Daniels, S.J.; Leeming, D.J.; Karsdal, M.A.; Nielsen, M.J. Review article: The signalling and functional role of the extracellular matrix in the development of liver fibrosis. Aliment. Pharmacol. Ther. 2020, 52, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P. Tissue inhibitors of metalloproteinases in liver fibrosis. Int. J. Biochem. Cell Biol. 1997, 29, 43–54. [Google Scholar] [CrossRef]
- Smigiel, K.S.; Parks, W.C. Matrix Metalloproteinases and Leukocyte Activation. Prog. Mol. Biol. Transl. Sci. 2017, 147, 167–195. [Google Scholar] [CrossRef]
- Poynard, T.; Bedossa, P.; Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. Lancet 1997, 349, 825–832. [Google Scholar] [CrossRef]
- Cheng, T.; Liu, Q.; Zhang, R.; Zhang, Y.; Chen, J.; Yu, R.; Ge, G. Lysyl oxidase promotes bleomycin-induced lung fibrosis through modulating inflammation. J. Mol. Cell Biol. 2014, 6, 506–515. [Google Scholar] [CrossRef]
- Zhao, X.; Kwan, J.Y.Y.; Yip, K.; Liu, P.P.; Liu, F.F. Targeting metabolic dysregulation for fibrosis therapy. Nat. Rev. Drug Discov. 2020, 19, 57–75. [Google Scholar] [CrossRef] [PubMed]
- Mak, K.M.; Mei, R. Basement Membrane Type IV Collagen and Laminin: An Overview of Their Biology and Value as Fibrosis Biomarkers of Liver Disease. Anat. Rec. 2017, 300, 1371–1390. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.L.; Bloomer, S.A.; Chan, E.P.; Gaça, M.D.A.A.; Georges, P.C.; Sackey, B.; Uemura, M.; Janmey, P.A.; Wells, R.G. Hepatic stellate cells require a stiff environment for myofibroblastic differentiation. Am. J. Physiol. Liver Physiol. 2011, 301, G110–G118. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Dranoff, J.A.; Chan, E.P.; Uemura, M.; Sévigny, J.; Wells, R.G. Transforming growth factor-β and substrate stiffness regulate portal fibroblast activation in culture. Hepatology 2007, 46, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Herrera, J.; Henke, C.A.; Bitterman, P.B. Extracellular matrix as a driver of progressive fibrosis. J. Clin. Investig. 2018, 128, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Kupffer, C. Ueber Sternzellen der Leber-Briefliche Mittheilung an Prof. Waldeyer. Arch. Mikroskopische Anat. 1876, 12, 353–358. [Google Scholar] [CrossRef]
- Friedman, S.L. Hepatic Stellate Cells: Protean, Multifunctional, and Enigmatic Cells of the Liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Wake, K. “Sternzellen” in the liver: Perisinusoidal cells with special reference to storage of vitamin A. Am. J. Anat. 1971, 132, 429–461. [Google Scholar] [CrossRef] [PubMed]
- Zisser, A.; Ipsen, D.H.; Tveden-Nyborg, P. Hepatic Stellate Cell Activation and Inactivation in NASH-Fibrosis—Roles as Putative Treatment Targets? Biomedicines 2021, 9, 365. [Google Scholar] [CrossRef]
- Rohn, F.; Kordes, C.; Buschmann, T.; Reichert, D.; Wammers, M.; Poschmann, G.; Stühler, K.; Benk, A.S.; Geiger, F.; Spatz, J.P.; et al. Impaired integrin α 5 /β 1 -mediated hepatocyte growth factor release by stellate cells of the aged liver. Aging Cell 2020, 19, e13131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 10, 29. [Google Scholar] [CrossRef] [Green Version]
- Blomhoff, R.; Wake, K. Perisinusoidal stellate cells of the liver: Important roles in retinol metabolism and fibrosis. FASEB J. 1991, 5, 271–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirakami, Y.; Lee, S.-A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 2012, 1821, 124–136. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, D.N.; Clugston, R.D.; Blaner, W.S. Vitamin A metabolism: An update. Nutrients 2011, 3, 63–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomhoff, R.; Holte, K.; Næss, L.; Berg, T. Newly administered [3H]retinol is transferred from hepatocytes to stellate cells in liver for storage. Exp. Cell Res. 1984, 150, 186–193. [Google Scholar] [CrossRef]
- Blomhoff, R.; Helgerud, P.; Rasmussen, M.; Berg, T.; Norum, K.R. In vivo uptake of chylomicron [3H]retinyl ester by rat liver: Evidence for retinol transfer from parenchymal to nonparenchymal cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7326–7330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajendiran, P.; Vega, L.I.; Itoh, K.; Sesaki, H.; Vakili, M.R.; Lavasanifar, A.; Hong, K.; Mezey, E.; Ganapathy-Kanniappan, S. Elevated mitochondrial activity distinguishes fibrogenic hepatic stellate cells and sensitizes for selective inhibition by mitotropic doxorubicin. J. Cell. Mol. Med. 2018, 22, 2210–2219. [Google Scholar] [CrossRef] [Green Version]
- Molenaar, M.R.; Vaandrager, A.B.; Helms, J.B. Some Lipid Droplets Are More Equal Than Others: Different Metabolic Lipid Droplet Pools in Hepatic Stellate Cells. Lipid Insights 2017, 10, 117863531774728. [Google Scholar] [CrossRef] [Green Version]
- Tuohetahuntila, M.; Molenaar, M.R.; Spee, B.; Brouwers, J.F.; Wubbolts, R.; Houweling, M.; Yan, C.; Du, H.; VanderVen, B.C.; Vaandrager, A.B.; et al. Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation. J. Biol. Chem. 2017, 292, 12436–12448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, W.; Syn, W.-K. Role of Metabolism in Hepatic Stellate Cell Activation and Fibrogenesis. Front. Cell Dev. Biol. 2018, 6, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.F.; Mak, K.M.; Rackovsky, O.; Lin, Y.L.; Kwong, A.J.; Loke, J.C.; Friedman, S.L. Downregulation of hepatic stellate cell activation by retinol and palmitate mediated by Adipose Differentiation-Related Protein (ADRP). J. Cell. Physiol. 2010, 223, 648–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoneda, A.; Sakai-Sawada, K.; Niitsu, Y.; Tamura, Y. Vitamin A and insulin are required for the maintenance of hepatic stellate cell quiescence. Exp. Cell Res. 2016, 341, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Rennie Tankersley, L.; Tang, M.; Potter, J.J.; Mezey, E. Regulation of the murine α2(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys. 2002, 401, 262–270. [Google Scholar] [CrossRef]
- Shaw, I.; Rider, S.; Mullins, J.; Hughes, J.; Péault, B. Pericytes in the renal vasculature: Roles in health and disease. Nat. Rev. Nephrol. 2018, 14, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Dobie, R.; Wilson-Kanamori, J.R.; Dora, E.F.; Henderson, B.E.P.; Luu, N.T.; Portman, J.R.; Matchett, K.P.; Brice, M.; Marwick, J.A.; et al. Resolving the fibrotic niche of human liver cirrhosis at single-cell level. Nature 2019, 575, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Dobie, R.; Wilson-Kanamori, J.R.; Henderson, B.E.P.; Smith, J.R.; Matchett, K.P.; Portman, J.R.; Wallenborg, K.; Picelli, S.; Zagorska, A.; Pendem, S.V.; et al. Single-Cell Transcriptomics Uncovers Zonation of Function in the Mesenchyme during Liver Fibrosis. Cell Rep. 2019, 29, 1832–1847.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, S.B.; Liu, X.; Ganguly, S.; Dhar, D.; Pasillas, M.P.; Ricciardelli, E.; Li, R.Z.; Troutman, T.D.; Kisseleva, T.; Glass, C.K.; et al. Heterogeneity of HSCs in a Mouse Model of NASH. Hepatology 2021, 74, 667–685. [Google Scholar] [CrossRef]
- Zavadil, J.; Bitzer, M.; Liang, D.; Yang, Y.C.; Massimi, A.; Kneitz, S.; Piek, E.; Böttinger, E.P. Genetic programs of epithelial cell plasticity directed by transforming growth factor-β. Proc. Natl. Acad. Sci. USA 2001, 98, 6686–6691. [Google Scholar] [CrossRef] [Green Version]
- DeLeve, L.D.; Wang, X.; Guo, Y. Sinusoidal endothelial cells prevent rat stellate cell activation and promote reversion to quiescence. Hepatology 2008, 48, 920–930. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Wang, X.; Wang, L.; Wang, L.; Atkinson, R.D.; Kanel, G.C.; Gaarde, W.A.; DeLeve, L.D. Role of Differentiation of Liver Sinusoidal Endothelial Cells in Progression and Regression of Hepatic Fibrosis in Rats. Gastroenterology 2012, 142, 918–927.e6. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [Green Version]
- Wree, A.; Marra, F. The inflammasome in liver disease. J. Hepatol. 2016, 65, 1055–1056. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Kanzler, S.; Lohse, A.W.; Keil, A.; Henninger, J.; Dienes, H.P.; Schirmacher, P.; Rose-John, S.; Meyer Zum Büschenfelde, K.H.; Blessing, M. TGF-β1 in liver fibrosis: An inducible transgenic mouse model to study liver fibrogenesis. Am. J. Physiol. Liver Physiol. 1999, 276, G1059–G1068. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Seki, E. TNFα in Liver Fibrosis. Curr. Pathobiol. Rep. 2015, 3, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, N.K.; Anania, F.A. Adipocytokines and hepatic fibrosis. Trends Endocrinol. Metab. 2015, 26, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, F.; Wang, K.; Aoyama, T.; Grivennikov, S.I.; Paik, Y.; Scholten, D.; Cong, M.; Iwaisako, K.; Liu, X.; Zhang, M.; et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology 2012, 143, 765–776.e3. [Google Scholar] [CrossRef] [Green Version]
- She, H.; Xiong, S.; Hazra, S.; Tsukamoto, H. Adipogenic transcriptional regulation of hepatic stellate cells. J. Biol. Chem. 2005, 280, 4959–4967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, F.; Meso, M.; Iredale, J.P.; Green, K.; Marek, C.J.; Zhou, X.; May, M.J.; Millward-Sadler, H.; Wright, M.C.; Mann, D.A. Inhibition of inhibitor of κb kinases stimulates hepatic stellate cell apoptosis and accelerated recovery from rat liver fibrosis. Gastroenterology 2005, 128, 108–120. [Google Scholar] [CrossRef]
- Jeong, W.-I.; Park, O.; Radaeva, S.; Gao, B. STAT1 inhibits liver fibrosis in mice by inhibiting stellate cell proliferation and stimulating NK cell cytotoxicity. Hepatology 2006, 44, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Correia, A.L.; Guimaraes, J.C.; Auf der Maur, P.; De Silva, D.; Trefny, M.P.; Okamoto, R.; Bruno, S.; Schmidt, A.; Mertz, K.; Volkmann, K.; et al. Hepatic stellate cells suppress NK cell-sustained breast cancer dormancy. Nature 2021, 594, 566–571. [Google Scholar] [CrossRef]
- Schnabl, B.; Purbeck, C.A.; Choi, Y.H.; Hagedorn, C.H.; Brenner, D.A. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003, 37, 653–664. [Google Scholar] [CrossRef]
- Lane, A.N.; Higashi, R.M.; Fan, T.W.M. Metabolic reprogramming in tumors: Contributions of the tumor microenvironment. Genes Dis. 2020, 7, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Choi, S.S.; Michelotti, G.A.; Chan, I.S.; Swiderska-Syn, M.; Karaca, G.F.; Xie, G.; Moylan, C.A.; Garibaldi, F.; Premont, R.; et al. Hedgehog Controls Hepatic Stellate Cell Fate by Regulating Metabolism. Gastroenterology 2012, 143, 1319–1329.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellerbrand, C.; Stefanovic, B.; Giordano, F.; Burchardt, E.R.; Brenner, D.A. The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J. Hepatol. 1999, 30, 77–87. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.; Rivera, S.; Monclus, E.A.; Synenki, L.; Zirk, A.; Eisenbart, J.; Feghali-Bostwick, C.; Mutlu, G.M.; Budinger, G.R.R.S.S.; Chandel, N.S. Mitochondrial Reactive Oxygen Species Regulate Transforming Growth Factor-β Signaling. J. Biol. Chem. 2013, 288, 770–777. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Chen, A. Curcumin diminishes the impacts of hyperglycemia on the activation of hepatic stellate cells by suppressing membrane translocation and gene expression of glucose transporter-2. Mol. Cell. Endocrinol. 2011, 333, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekaran, V.; Das, S.; Seth, R.K.; Dattaroy, D.; Alhasson, F.; Michelotti, G.; Nagarkatti, M.; Nagarkatti, P.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. Biochim. Biophys. Acta Mol. Basis Dis. 2016, 1862, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Mejias, M.; Gallego, J.; Naranjo-Suarez, S.; Ramirez, M.; Pell, N.; Manzano, A.; Suñer, C.; Bartrons, R.; Mendez, R.; Fernandez, M. CPEB4 Increases Expression of PFKFB3 to Induce Glycolysis and Activate Mouse and Human Hepatic Stellate Cells, Promoting Liver Fibrosis. Gastroenterology 2020, 159, 273–288. [Google Scholar] [CrossRef]
- Zheng, D.; Jiang, Y.; Qu, C.; Yuan, H.; Hu, K.; He, L.; Chen, P.; Li, J.; Tu, M.; Lin, L.; et al. Pyruvate Kinase M2 Tetramerization Protects against Hepatic Stellate Cell Activation and Liver Fibrosis. Am. J. Pathol. 2020, 190, 2267–2281. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T. Liver zonation in health and disease: Hypoxia and hypoxia-inducible transcription factors as concert masters. Int. J. Mol. Sci. 2019, 20, 2347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Hyun, J.; Premont, R.T.; Choi, S.S.; Michelotti, G.A.; Swiderska-Syn, M.; Dalton, G.D.; Thelen, E.; Rizi, B.S.; Jung, Y.; et al. Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 2018, 154, 1465–1479.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, K.; Chitneni, S.K.; Suzuki, A.; Wang, Y.; Henao, R.; Hyun, J.; Premont, R.T.; Naggie, S.; Moylan, C.A.; Bashir, M.R.; et al. Increased Glutaminolysis Marks Active Scarring in Nonalcoholic Steatohepatitis Progression. Cell. Mol. Gastroenterol. Hepatol. 2020, 10, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harvey, L.D.; Chan, S.Y. YAPping About Glutaminolysis in Hepatic Fibrosis. Gastroenterology 2018, 154, 1231–1233. [Google Scholar] [CrossRef] [Green Version]
- Bertero, T.; Cottrill, K.A.; Lu, Y.; Haeger, C.M.; Dieffenbach, P.; Annis, S.; Hale, A.; Bhat, B.; Kaimal, V.; Zhang, Y.Y.; et al. Matrix Remodeling Promotes Pulmonary Hypertension through Feedback Mechanoactivation of the YAP/TAZ-miR-130/301 Circuit. Cell Rep. 2015, 13, 1016–1032. [Google Scholar] [CrossRef] [Green Version]
- Esguerra, V.; Zhao, Y. Preventing Glutaminolysis: A Potential Therapy for Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 61, 408–409. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.-Y.; Dapito, D.H.; Pradere, J.-P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef]
- Kida, Y.; Xia, Z.; Zheng, S.; Mordwinkin, N.M.; Louie, S.G.; Zheng, S.G.; Feng, M.; Shi, H.; Duan, Z.; Han, Y.-P. Interleukin-1 as an Injury Signal Mobilizes Retinyl Esters in Hepatic Stellate Cells through Down Regulation of Lecithin Retinol Acyltransferase. PLoS ONE 2011, 6, e26644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández–Gea, V.; Ghiassi–Nejad, Z.; Rozenfeld, R.; Gordon, R.; Fiel, M.I.; Yue, Z.; Czaja, M.J.; Friedman, S.L. Autophagy Releases Lipid That Promotes Fibrogenesis by Activated Hepatic Stellate Cells in Mice and in Human Tissues. Gastroenterology 2012, 142, 938–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, X.; Wang, M.; Wei, X.; Deng, S.; Fu, N.; Peng, Q.; Jiang, Y.; Ye, L.; Xie, J.; Lin, Y. Peroxisome Proliferator-Activated Receptor-γ: Master Regulator of Adipogenesis and Obesity. Curr. Stem Cell Res. Ther. 2016, 11, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP transcription factors: Master regulators of lipid homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef]
- Tsukamoto, H. Adipogenic phenotype of hepatic stellate cells. Alcohol. Clin. Exp. Res. 2005, 29, 132S–133S. [Google Scholar] [CrossRef] [PubMed]
- Beaven, S.W.; Tontonoz, P. Nuclear Receptors in Lipid Metabolism: Targeting the Heart of Dyslipidemia. Annu. Rev. Med. 2006, 57, 313–329. [Google Scholar] [CrossRef] [PubMed]
- Beaven, S.W.; Wroblewski, K.; Wang, J.; Hong, C.; Bensinger, S.; Tsukamoto, H.; Tontonoz, P. Liver X Receptor Signaling Is a Determinant of Stellate Cell Activation and Susceptibility to Fibrotic Liver Disease. Gastroenterology 2011, 140, 1052–1062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, J.; Vijayakumar, A.; Ghoshal, S.; Marchand, B.; Yi, S.; Kornyeyev, D.; Zagorska, A.; Hollenback, D.; Walker, K.; Liu, K.; et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J. Hepatol. 2020, 73, 896–905. [Google Scholar] [CrossRef]
- Lally, J.S.V.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef] [Green Version]
- Marcher, A.B.; Bendixen, S.M.; Terkelsen, M.K.; Hohmann, S.S.; Hansen, M.H.; Larsen, B.D.; Mandrup, S.; Dimke, H.; Detlefsen, S.; Ravnskjaer, K. Transcriptional regulation of Hepatic Stellate Cell activation in NASH. Sci. Rep. 2019, 9, 2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilgenkrantz, H.; Mallat, A.; Moreau, R.; Lotersztajn, S. Targeting cell-intrinsic metabolism for antifibrotic therapy. J. Hepatol. 2021, 74, 1442–1454. [Google Scholar] [CrossRef]
- Wang, S.; Jung, Y.; Hyun, J.; Friedersdorf, M.; Oh, S.-H.H.; Kim, J.; Premont, R.T.T.; Keene, J.D.D.; Diehl, A.M.M. RNA Binding Proteins Control Transdifferentiation of Hepatic Stellate Cells into Myofibroblasts. Cell. Physiol. Biochem. 2018, 48, 1215–1229. [Google Scholar] [CrossRef] [PubMed]
- Jonkhout, N.; Tran, J.; Smith, M.A.; Schonrock, N.; Mattick, J.S.; Novoa, E.M. The RNA modification landscape in human disease. RNA 2017, 23, 1754–1769. [Google Scholar] [CrossRef] [Green Version]
- Salem, E.S.B.; Vonberg, A.D.; Borra, V.J.; Gill, R.K.; Nakamura, T. RNAs and RNA-Binding Proteins in Immuno-Metabolic Homeostasis and Diseases. Front. Cardiovasc. Med. 2019, 6, 106. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef]
- Singh, G.; Pratt, G.; Yeo, G.W.; Moore, M.J. The Clothes Make the mRNA: Past and Present Trends in mRNP Fashion. Annu. Rev. Biochem. 2015, 84, 325–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Li, S.; Chen, D.; Chen, B.; Yu, T.; Zhao, F.; Wang, Q.; Yao, M.; Huang, S.; Chen, Z.; et al. Transcriptomic analyses of RNA-binding proteins reveal eIF3c promotes cell proliferation in hepatocellular carcinoma. Cancer Sci. 2017, 108, 877–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutschner, T.; Hämmerle, M.; Pazaitis, N.; Bley, N.; Fiskin, E.; Uckelmann, H.; Heim, A.; Groβ, M.; Hofmann, N.; Geffers, R.; et al. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) is an important protumorigenic factor in hepatocellular carcinoma. Hepatology 2014, 59, 1900–1911. [Google Scholar] [CrossRef]
- Mejias, M.; Coch, L.; Berzigotti, A.; Garcia-Pras, E.; Gallego, J.; Bosch, J.; Fernandez, M. Antiangiogenic and antifibrogenic activity of pigment epithelium-derived factor (PEDF) in bile duct-ligated portal hypertensive rats. Gut 2015, 64, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Dolicka, D.; Sobolewski, C.; Correia de Sousa, M.; Gjorgjieva, M.; Foti, M. mRNA Post-Transcriptional Regulation by AU-Rich Element-Binding Proteins in Liver Inflammation and Cancer. Int. J. Mol. Sci. 2020, 21, 6648. [Google Scholar] [CrossRef] [PubMed]
- Doller, A.; Pfeilschifter, J.; Eberhardt, W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell. Signal. 2008, 20, 2165–2173. [Google Scholar] [CrossRef] [PubMed]
- Woodhoo, A.; Iruarrizaga-Lejarreta, M.; Beraza, N.; García-Rodríguez, J.L.; Embade, N.; Fernández-Ramos, D.; Matinez-Lopez, N.; Gutiérrez, V.; Arteta, B.; Caballeria, J.; et al. HuR contributes to Hepatic Stellate Cell activation and liver fibrosis. Hepatology 2012, 56, 1870. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.; Ge, J.; Xiu, L.; Zhao, Z.; Duan, X.; Tian, L.; Xie, J.; Yang, L.; Li, L. HuR mediates motility of human bone marrow-derived mesenchymal stem cells triggered by sphingosine 1-phosphate in liver fibrosis. J. Mol. Med. 2017, 95, 69–82. [Google Scholar] [CrossRef]
- Cartier, A.; Hla, T. Sphingosine 1-phosphate: Lipid signaling in pathology and therapy. Science 2019, 366. [Google Scholar] [CrossRef] [PubMed]
- Xiu, L.; Chang, N.; Yang, L.; Liu, X.; Yang, L.; Ge, J.; Li, L. Intracellular Sphingosine 1-Phosphate Contributes to Collagen Expression of Hepatic Myofibroblasts in Human Liver Fibrosis Independent of Its Receptors. Am. J. Pathol. 2015, 185, 387–398. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yue, S.; Yang, L.; Liu, X.; Han, Z.; Zhang, Y.; Li, L. Sphingosine kinase/sphingosine 1-phosphate (S1P)/S1P receptor axis is involved in liver fibrosis-associated angiogenesis. J. Hepatol. 2013, 59, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Chang, N.; Zhao, Z.; Tian, L.; Duan, X.; Yang, L.; Li, L. Essential Roles of RNA-binding Protein HuR in Activation of Hepatic Stellate Cells Induced by Transforming Growth Factor-β. Sci. Rep. 2016, 6, 22141. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yao, Z.; Wang, L.; Ding, H.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy 2018, 14, 2083–2103. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Luo, Y.; Xia, Q.; He, K. Ferroptosis and Liver Fibrosis. Int. J. Med. Sci. 2021, 18, 3361–3366. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Shen, Y.; Chen, C.; Sui, X.; Yang, J.; Wang, L.; Zhou, J. The crosstalk between autophagy and ferroptosis: What can we learn to target drug resistance in cancer? Cancer Biol. Med. 2019, 16, 630–646. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, V.; Hawkins, W.D.; Klionsky, D.J. Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab. 2019, 29, 803–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiedje, C.; Diaz-Muñoz, M.D.; Trulley, P.; Ahlfors, H.; Laaß, K.; Blackshear, P.J.; Turner, M.; Gaestel, M. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucl. Acids Res. 2016, 44, 7418–7440. [Google Scholar] [CrossRef] [PubMed]
- Lykke-Andersen, J. Recruitment and activation of mRNA decay enzymes by two ARE-mediated decay activation domains in the proteins TTP and BRF-1. Genes Dev. 2005, 19, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, J.; Chen, Q.; Tang, D.; Ou, W.; Wang, J.; Mo, Z.; Tang, C.; Peng, L.; Wang, D. Activation of liver X receptors promotes inflammatory cytokine mRNA degradation by upregulation of tristetraprolin. Acta Biochim. Biophys. Sin. 2017, 49, 277–283. [Google Scholar] [CrossRef]
- Kröhler, T.; Kessler, S.M.; Hosseini, K.; List, M.; Barghash, A.; Patial, S.; Laggai, S.; Gemperlein, K.; Haybaeck, J.; Müller, R.; et al. The mRNA-binding Protein TTP/ZFP36 in Hepatocarcinogenesis and Hepatocellular Carcinoma. Cancers 2019, 11, 1754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Guo, M.; Li, Y.; Shen, M.; Kong, D.; Shao, J.; Ding, H.; Tan, S.; Chen, A.; Zhang, F.; et al. RNA-binding protein ZFP36/TTP protects against ferroptosis by regulating autophagy signaling pathway in hepatic stellate cells. Autophagy 2020, 16, 1482–1505. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-C.; Luo, S.-Z.; Liu, T.; Lu, L.-G.; Xu, M.-Y. linc-SCRG1 accelerates liver fibrosis by decreasing RNA-binding protein tristetraprolin. FASEB J. 2019, 33, 2105–2115. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Miranda, G.; Méndez, R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012, 11, 460–472. [Google Scholar] [CrossRef]
- Balvey, A.; Fernandez, M. Translational Control in Liver Disease. Front. Physiol. 2021, 12, 2144. [Google Scholar] [CrossRef] [PubMed]
- Pell, N.; Garcia-Pras, E.; Gallego, J.; Naranjo-Suarez, S.; Balvey, A.; Suñer, C.; Fernandez-Alfara, M.; Chanes, V.; Carbo, J.; Ramirez-Pedraza, M.; et al. Targeting the cytoplasmic polyadenylation element-binding protein CPEB4 protects against diet-induced obesity and microbiome dysbiosis. Mol. Metab. 2021, 54, 101388. [Google Scholar] [CrossRef]
- Schwaderer, J.; Phan, T.S.; Glöckner, A.; Delp, J.; Leist, M.; Brunner, T.; Delgado, M.E. Pharmacological LRH-1/Nr5a2 inhibition limits pro-inflammatory cytokine production in macrophages and associated experimental hepatitis. Cell Death Dis. 2020, 11, 154. [Google Scholar] [CrossRef]
- Leroux, A.; Ferrere, G.; Godie, V.; Cailleux, F.; Renoud, M.L.; Gaudin, F.; Naveau, S.; Prévot, S.; Makhzami, S.; Perlemuter, G.; et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J. Hepatol. 2012, 57, 141–149. [Google Scholar] [CrossRef]
- Bieghs, V.; Walenbergh, S.M.A.; Hendrikx, T.; van Gorp, P.J.; Verheyen, F.; Olde Damink, S.W.; Masclee, A.A.; Koek, G.H.; Hofker, M.H.; Binder, C.J.; et al. Trapping of oxidized LDL in lysosomes of Kupffer cells is a trigger for hepatic inflammation. Liver Int. 2013, 33, 1056–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Huangyang, P.; Burrows, M.; Guo, K.; Riscal, R.; Godfrey, J.; Lee, K.E.; Lin, N.; Lee, P.; Blair, I.A.; et al. FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat. Cell Biol. 2020, 22, 728–739. [Google Scholar] [CrossRef]
- Huebener, P.; Pradere, J.-P.; Hernandez, C.; Gwak, G.-Y.; Caviglia, J.M.; Mu, X.; Loike, J.D.; Jenkins, R.E.; Antoine, D.J.; Schwabe, R.F. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Investig. 2019, 129, 1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with ‘Warburg- like’ cancer metabolism and L-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [Green Version]
- Phillips, K.; Kedersha, N.; Shen, L.; Blacksheart, P.J.; Anderson, P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2004, 101, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Gong, J.; Han, J.; He, J.; Liu, J.; Han, P.; Wang, Y.; Li, M.; Li, D.; Ding, X.; Du, Z.; et al. Paired related homeobox protein 1 regulates PDGF-induced chemotaxis of hepatic stellate cells in liver fibrosis. Lab. Investig. 2017, 97, 1020–1032. [Google Scholar] [CrossRef] [PubMed]
- Fritz, D.; Stefanovic, B. RNA binding protein RBMS3 is expressed in activated hepatic stellate cells and liver fibrosis and increases expression of transcription factor Prx. J. Mol. Biol. 2007, 371, 585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefanovic, B.; Hellerbrand, C.; Holcik, M.; Briendl, M.; Aliebhaber, S.; Brenner, D.A. Posttranscriptional regulation of collagen alpha1(I) mRNA in hepatic stellate cells. Mol. Cell. Biol. 1997, 17, 5201–5209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, H.; Berkova, Z.; Mathur, R.; Sehgal, L.; Khashab, T.; Tao, R.-H.; Ao, X.; Feng, L.; Sabichi, A.L.; Blechacz, B.; et al. HuR Suppresses Fas Expression and Correlates with Patient Outcome in Liver Cancer. Mol. Cancer Res. 2015, 13, 809–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullan, J.E.; Confeld, M.I.; Osborn, J.K.; Kim, J.; Sarkar, K.; Mallik, S. Exosomes as Drug Carriers for Cancer Therapy. Mol. Pharm. 2019, 16, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Maillo, C.; Martín, J.; Sebastián, D.; Hernández-Alvarez, M.; García-Rocha, M.; Reina, O.; Zorzano, A.; Fernandez, M.; Méndez, R. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cell Biol. 2017, 19, 94–105. [Google Scholar] [CrossRef]
- Calderone, V.; Gallego, J.; Fernandez-Miranda, G.; Garcia-Pras, E.; Maillo, C.; Berzigotti, A.; Mejias, M.; Bava, F.-A.A.; Angulo-Urarte, A.; Graupera, M.; et al. Sequential Functions of CPEB1 and CPEB4 Regulate Pathologic Expression of Vascular Endothelial Growth Factor and Angiogenesis in Chronic Liver Disease. Gastroenterology 2016, 150, 982–997.e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Pras, E.; Gallego, J.; Coch, L.; Mejias, M.; Fernandez-Miranda, G.; Pardal, R.; Bosch, J.; Mendez, R.; Fernandez, M. Role and therapeutic potential of vascular stem/progenitor cells in pathological neovascularisation during chronic portal hypertension. Gut 2017, 66, 1306–1320. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado, M.E.; Cárdenas, B.I.; Farran, N.; Fernandez, M. Metabolic Reprogramming of Liver Fibrosis. Cells 2021, 10, 3604. https://doi.org/10.3390/cells10123604
Delgado ME, Cárdenas BI, Farran N, Fernandez M. Metabolic Reprogramming of Liver Fibrosis. Cells. 2021; 10(12):3604. https://doi.org/10.3390/cells10123604
Chicago/Turabian StyleDelgado, M. Eugenia, Beatriz I. Cárdenas, Núria Farran, and Mercedes Fernandez. 2021. "Metabolic Reprogramming of Liver Fibrosis" Cells 10, no. 12: 3604. https://doi.org/10.3390/cells10123604
APA StyleDelgado, M. E., Cárdenas, B. I., Farran, N., & Fernandez, M. (2021). Metabolic Reprogramming of Liver Fibrosis. Cells, 10(12), 3604. https://doi.org/10.3390/cells10123604