
Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. BV-2 Microglia Culture
2.2. Primary Microglia Culture
2.3. Neutral Lipid Staining in Primary Microglia and BV-2 Cells
2.4. Confocal Microscopy
2.5. Lipid Droplet Quantification
2.6. Lipidomic Analysis
2.7. Cholesterol Efflux Assay
2.8. Isolation of Microglia from Adult Mouse Brain
2.9. RNA Isolation and RNAseq Library Preparation
2.10. Real-Time Quantitative Polymerase Chain Reaction (RT qPCR)
2.11. Western Blot Analysis of Protein Expression
2.12. PPAR Agonist Supplementation Experiments
2.13. Statistical Analysis
3. Results
3.1. LPL Depletion Leads to Increased Lipid Droplet Accumulation
3.2. LPL Deficient Microglia Show Marked Changes in Cholesterol and Phospholipid Metabolism
3.3. Transcriptomic Analysis of LPL KD Microglia Highlights Impaired FFA Uptake, Reduced Cholesterol Efflux, and Inflammation
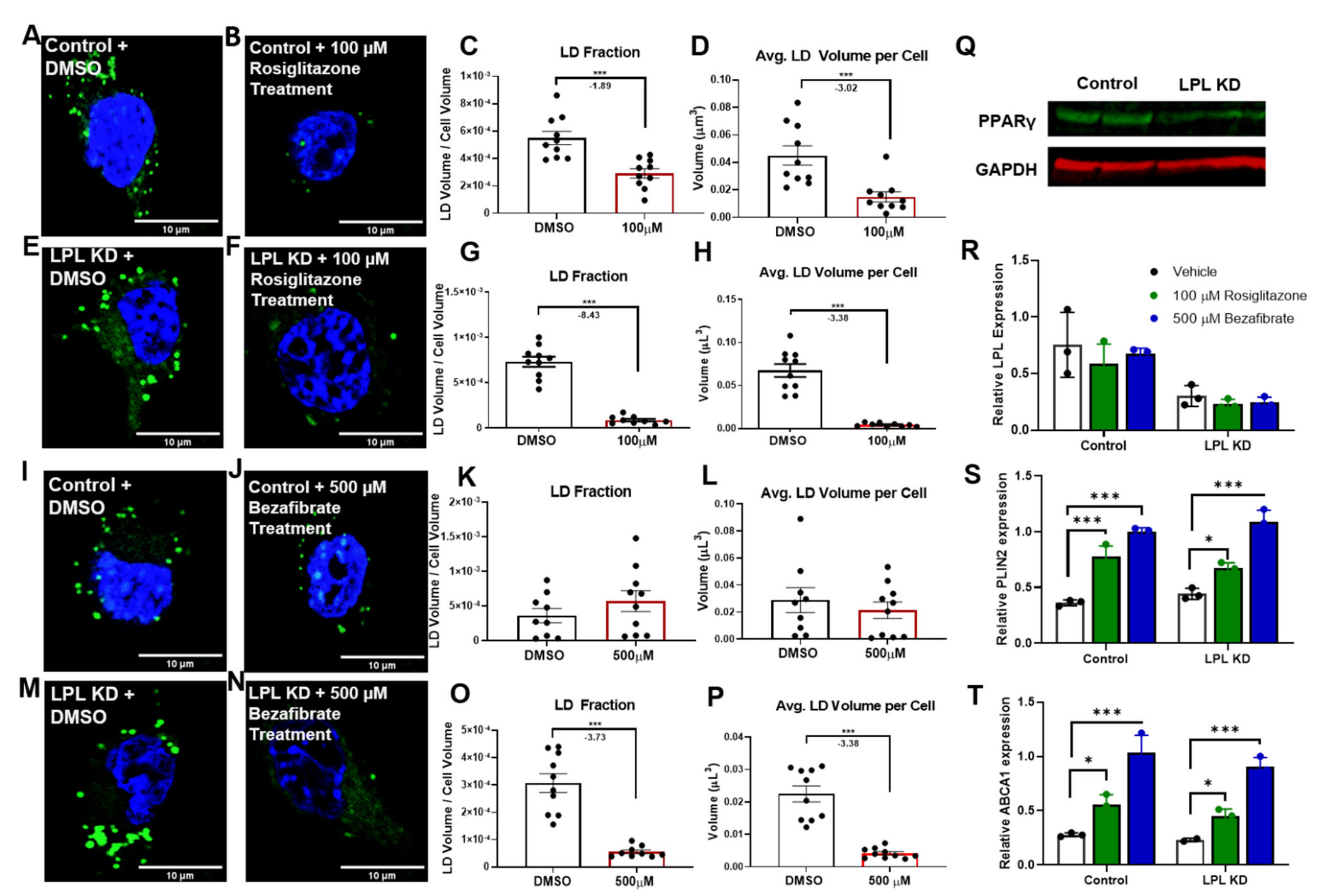
3.4. PPAR Activators Reduce LD Accumulation in Microglia
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shemer, A.; Erny, D.; Jung, S.; Prinz, M. Microglia Plasticity during Health and Disease: An Immunological Perspective. Trends Immunol. 2015, 36, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zhang, Y.-Q.; Qadri, Y.J.; Serhan, C.N.; Ji, R.-R. Microglia in Pain: Detrimental and Protective Roles in Pathogenesis and Resolution of Pain. Neuron 2018, 100, 1292–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Baillie, J.K.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain region-dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Loving, B.A.; Bruce, K.D. Lipid and Lipoprotein Metabolism in Microglia. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Kopper, T.J.; Gensel, J.C. Myelin as an inflammatory mediator: Myelin interactions with complement, macrophages, and microglia in spinal cord injury. J. Neurosci. Res. 2018, 96, 969–977. [Google Scholar] [CrossRef] [Green Version]
- Clarner, T.; Diederichs, F.; Berger, K.; Denecke, B.; Gan, L.; van der Valk, P.; Beyer, C.; Amor, S.; Kipp, M. Myelin debris regulates inflammatory responses in an experimental demyelination animal model and multiple sclerosis lesions. Glia 2012, 60, 1468–1480. [Google Scholar] [CrossRef]
- Bruce, K.D.; Gorkhali, S.; Given, K.; Coates, A.M.; Boyle, K.E.; Macklin, W.B.; Eckel, R.H. Lipoprotein Lipase Is a Feature of Alternatively-Activated Microglia and May Facilitate Lipid Uptake in the CNS During Demyelination. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakovcevski, I.; Filipovic, R.; Mo, Z.; Rakic, S.; Zecevic, N. Oligodendrocyte development and the onset of myelination in the human fetal brain. Front. Neuroanat. 2009, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barres, B.A.; Hart, I.K.; Coles, H.S.R.; Burne, J.F.; Voyvodic, J.T.; Richardson, W.D.; Raff, M.C. Cell death and control of cell survival in the oligodendrocyte lineage. Cell 1992, 70, 31–46. [Google Scholar] [CrossRef]
- Lloyd, A.F.; Miron, V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019, 15, 447–458. [Google Scholar] [CrossRef]
- Cantuti-Castelvetri, L.; Fitzner, D.; Bosch-Queralt, M.; Weil, M.-T.; Su, M.; Sen, P.; Ruhwedel, T.; Mitkovski, M.; Trendelenburg, G.; Lütjohann, D.; et al. Defective cholesterol clearance limits remyelination in the aged central nervous system. Science 2018, 359, 684. [Google Scholar] [CrossRef] [Green Version]
- Nadjar, A. Role of metabolic programming in the modulation of microglia phagocytosis by lipids. Prostaglandins Leukot. Essent. Fat. Acids 2018, 135, 63–73. [Google Scholar] [CrossRef]
- Marschallinger, J.; Iram, T.; Zardeneta, M.; Lee, S.E.; Lehallier, B.; Haney, M.S.; Pluvinage, J.V.; Mathur, V.; Hahn, O.; Morgens, D.W.; et al. Lipid droplet accumulating microglia represent a dysfunctional and pro-inflammatory state in the aging brain. bioRxiv 2019. [Google Scholar] [CrossRef]
- Khatchadourian, A.; Bourque, S.D.; Richard, V.R.; Titorenko, V.I.; Maysinger, D. Dynamics and regulation of lipid droplet formation in lipopolysaccharide (LPS)-stimulated microglia. Biochim. Biophys. Acta 2012, 1821, 607–617. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Astarita, G.; Taussig, M.D.; Bharadwaj, K.G.; DiPatrizio, N.V.; Nave, K.A.; Piomelli, D.; Goldberg, I.J.; Eckel, R.H. Deficiency of lipoprotein lipase in neurons modifies the regulation of energy balance and leads to obesity. Cell Metab. 2011, 13, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J.; Soprano, D.R.; Wyatt, M.L.; Vanni, T.M.; Kirchgessner, T.G.; Schotz, M.C. Localization of lipoprotein lipase mRNA in selected rat tissues. J. Lipid Res. 1989, 30, 1569–1577. [Google Scholar] [CrossRef]
- Bessesen, D.H.; Richards, C.L.; Etienne, J.; Goers, J.W.; Eckel, R.H. Spinal cord of the rat contains more lipoprotein lipase than other brain regions. J. Lipid Res. 1993, 34, 229–238. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.B.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Clavel, S.; Julien, P.; Murthy, M.R.; de Bilbao, F.; Arsenijevic, D.; Giannakopoulos, P.; Vallet, P.; Richard, D. Lipoprotein lipase and endothelial lipase expression in mouse brain: Regional distribution and selective induction following kainic acid-induced lesion and focal cerebral ischemia. Neurobiol. Dis. 2004, 15, 312–325. [Google Scholar] [CrossRef]
- Ma, Y.; Bao, J.; Zhao, X.; Shen, H.; Lv, J.; Ma, S.; Zhang, X.; Li, Z.; Wang, S.; Wang, Q.; et al. Activated cyclin-dependent kinase 5 promotes microglial phagocytosis of fibrillar beta-amyloid by up-regulating lipoprotein lipase expression. Mol. Cell Proteom. 2013, 12, 2833–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, H.; Dong, W.; Rostad, S.W.; Marcovina, S.M.; Albers, J.J.; Brunzell, J.D.; Vuletic, S. Lipoprotein lipase (LPL) is associated with neurite pathology and its levels are markedly reduced in the dentate gyrus of Alzheimer’s disease brains. J. Histochem. Cytochem. 2013, 61, 857–868. [Google Scholar] [CrossRef] [Green Version]
- D Bruce, K.; Tang, M.; Reigan, P.; H Eckel, R. Genetic Variants of Lipoprotein Lipase and Regulatory Factors Associated with Alzheimer’s Disease Risk. Int. J. Mol. Sci. 2020, 21, 8338. [Google Scholar] [CrossRef]
- Hayne, C.K.; Lafferty, M.J.; Eglinger, B.J.; Kane, J.P.; Neher, S.B. Biochemical Analysis of the Lipoprotein Lipase Truncation Variant, LPL(S447X), Reveals Increased Lipoprotein Uptake. Biochemistry 2017, 56, 525–533. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Vidal-Itriago, A.; Kalsbeek, M.J.; Layritz, C.; Garcia-Caceres, C.; Tom, R.Z.; Eichmann, T.O.; Vaz, F.M.; Houtkooper, R.H.; van der Wel, N.; et al. Lipoprotein Lipase Maintains Microglial Innate Immunity in Obesity. Cell Rep. 2017, 20, 3034–3042. [Google Scholar] [CrossRef] [Green Version]
- Timmerman, R.; Burm, S.M.; Bajramovic, J.J. An Overview of in vitro Methods to Study Microglia. Front. Cell Neurosci. 2018, 12, 242. [Google Scholar] [CrossRef] [PubMed]
- Rueden, C.T.; Schindelin, J.; Hiner, M.C.; DeZonia, B.E.; Walter, A.E.; Arena, E.T.; Eliceiri, K.W. ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinform. 2017, 18, 529. [Google Scholar] [CrossRef] [PubMed]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Gross, R.W. Shotgun lipidomics: Electrospray ionization mass spectrometric analysis and quantitation of cellular lipidomes directly from crude extracts of biological samples. Mass Spectrom. Rev. 2005, 24, 367–412. [Google Scholar] [CrossRef] [PubMed]
- Low, H.; Hoang, A.; Sviridov, D. Cholesterol efflux assay. J. Vis. Exp. 2012, e3810. [Google Scholar] [CrossRef]
- Park, E.J.; Park, S.Y.; Joe, E.H.; Jou, I. 15d-PGJ2 and rosiglitazone suppress Janus kinase-STAT inflammatory signaling through induction of suppressor of cytokine signaling 1 (SOCS1) and SOCS3 in glia. J. Biol. Chem. 2003, 278, 14747–14752. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, K.; Yagi, T.; Guo, T.; Takeda, K.; Ohguchi, H.; Koyama, H.; Aotani, D.; Imaeda, K.; Kataoka, H.; Tanaka, T. Pemafibrate, a selective PPARalpha modulator, and fenofibrate suppress microglial activation through distinct PPARalpha and SIRT1-dependent pathways. Biochem. Biophys. Res. Commun. 2020, 524, 385–391. [Google Scholar] [CrossRef]
- Haj Ahmed, S.; Kharroubi, W.; Zarrouk, A.; Brahmi, F.; Nury, T.; Lizard, G.; Hammami, M. Protective effects of bezafibrate against elaidic acid-induced accumulation of lipid droplets in monocytic cells. Curr. Res. Transl. Med. 2017, 65, 20–30. [Google Scholar] [CrossRef]
- Xicoy, H.; Wieringa, B.; Martens, G.J.M. The Role of Lipids in Parkinson’s Disease. Cells 2019, 8, 27. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, J. Lipid metabolism in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 331–345. [Google Scholar] [CrossRef] [Green Version]
- Chausse, B.; Kakimoto, P.A.; Caldeira-da-Silva, C.C.; Chaves-Filho, A.B.; Yoshinaga, M.Y.; da Silva, R.P.; Miyamoto, S.; Kowaltowski, A.J. Distinct metabolic patterns during microglial remodeling by oleate and palmitate. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Geltinger, F.; Tevini, J.; Briza, P.; Geiser, A.; Bischof, J.; Richter, K.; Felder, T.; Rinnerthaler, M. The transfer of specific mitochondrial lipids and proteins to lipid droplets contributes to proteostasis upon stress and aging in the eukaryotic model system Saccharomyces cerevisiae. Geroscience 2020, 42, 19–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Veen, J.N.; Kennelly, J.P.; Wan, S.; Vance, J.E.; Vance, D.E.; Jacobs, R.L. The critical role of phosphatidylcholine and phosphatidylethanolamine metabolism in health and disease. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1558–1572. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Omura, T.; Masaki, N.; Arima, H.; Banno, T.; Okamoto, A.; Hanada, M.; Takei, S.; Matsushita, S.; Sugiyama, E.; et al. Increased arachidonic acid-containing phosphatidylcholine is associated with reactive microglia and astrocytes in the spinal cord after peripheral nerve injury. Sci. Rep. 2016, 6, 26427. [Google Scholar] [CrossRef] [Green Version]
- Tallima, H.; El Ridi, R. Arachidonic acid: Physiological roles and potential health benefits—A review. J. Adv. Res. 2018, 11, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Dyall, S.C. Long-chain omega-3 fatty acids and the brain: A review of the independent and shared effects of EPA, DPA and DHA. Front. Aging Neurosci. 2015, 7, 52. [Google Scholar] [CrossRef] [Green Version]
- Simopoulos, A.P. Evolutionary aspects of diet: The omega-6/omega-3 ratio and the brain. Mol. Neurobiol. 2011, 44, 203–215. [Google Scholar] [CrossRef]
- Bernardo, A.; Levi, G.; Minghetti, L. Role of the peroxisome proliferator-activated receptor-gamma (PPAR-gamma) and its natural ligand 15-deoxy-Delta12, 14-prostaglandin J2 in the regulation of microglial functions. Eur. J. Neurosci. 2000, 12, 2215–2223. [Google Scholar] [CrossRef]
- Bernardo, A.; Ajmone-Cat, M.A.; Levi, G.; Minghetti, L. 15-deoxy-delta12,14-prostaglandin J2 regulates the functional state and the survival of microglial cells through multiple molecular mechanisms. J. Neurochem. 2003, 87, 742–751. [Google Scholar] [CrossRef]
- Lemay, D.G.; Hwang, D.H. Genome-wide identification of peroxisome proliferator response elements using integrated computational genomics. J. Lipid Res. 2006, 47, 1583–1587. [Google Scholar] [CrossRef] [Green Version]
- Heinaniemi, M.; Uski, J.O.; Degenhardt, T.; Carlberg, C. Meta-analysis of primary target genes of peroxisome proliferator-activated receptors. Genome Biol. 2007, 8, R147. [Google Scholar] [CrossRef] [Green Version]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: The bezafibrate lessons. Cardiovasc. Diabetol. 2005, 4, 14. [Google Scholar] [CrossRef] [Green Version]
- Schott, M.B.; Weller, S.G.; Schulze, R.J.; Krueger, E.W.; Drizyte-Miller, K.; Casey, C.A.; McNiven, M.A. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J. Cell Biol. 2019, 218, 3320–3335. [Google Scholar] [CrossRef]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef]
- Pradines-Figueres, A.; Vannier, C.; Ailhaud, G. Lipoprotein lipase stored in adipocytes and muscle cells is a cryptic enzyme. J. Lipid Res. 1990, 31, 1467–1476. [Google Scholar] [CrossRef]
- Sundberg, E.L.; Deng, Y.; Burd, C.G. Syndecan-1 Mediates Sorting of Soluble Lipoprotein Lipase with Sphingomyelin-Rich Membrane in the Golgi Apparatus. Dev. Cell 2019, 51, 387–398.e384. [Google Scholar] [CrossRef]
- Gunn, K.H.; Roberts, B.S.; Wang, F.; Strauss, J.D.; Borgnia, M.J.; Egelman, E.H.; Neher, S.B. The structure of helical lipoprotein lipase reveals an unexpected twist in lipase storage. Proc. Natl. Acad. Sci. USA 2020, 117, 10254–10264. [Google Scholar] [CrossRef]
- Butovsky, O.; Weiner, H.L. Microglial signatures and their role in health and disease. Nat. Rev. Neurosci. 2018, 19, 622–635. [Google Scholar] [CrossRef]
- Borst, K.; Schwabenland, M.; Prinz, M. Microglia metabolism in health and disease. Neurochem. Int. 2019, 130, 104331. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Huynh, K.; Pernes, G.; Mellett, N.A.; Meikle, P.J.; Murphy, A.J.; Lancaster, G.I. Lipidomic Profiling of Murine Macrophages Treated with Fatty Acids of Varying Chain Length and Saturation Status. Metabolites 2018, 8, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenegro-Burke, J.R.; Sutton, J.A.; Rogers, L.M.; Milne, G.L.; McLean, J.A.; Aronoff, D.M. Lipid profiling of polarized human monocyte-derived macrophages. Prostaglandins Lipid Mediat. 2016, 127, 1–8. [Google Scholar] [CrossRef]
- Tu, T.H.; Kim, H.; Yang, S.; Kim, J.K.; Kim, J.G. Linoleic acid rescues microglia inflammation triggered by saturated fatty acid. Biochem. Biophys. Res. Commun. 2019, 513, 201–206. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, D.; Wang, F.; Liu, S.; Zhao, S.; Ling, E.A.; Hao, A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br. J. Nutr. 2012, 107, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diestel, A.; Aktas, O.; Hackel, D.; Hake, I.; Meier, S.; Raine, C.S.; Nitsch, R.; Zipp, F.; Ullrich, O. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: A link between demyelination and neuronal damage. J. Exp. Med. 2003, 198, 1729–1740. [Google Scholar] [CrossRef] [Green Version]
- Lavrnja, I.; Smiljanic, K.; Savic, D.; Mladenovic-Djordjevic, A.; Tesovic, K.; Kanazir, S.; Pekovic, S. Expression profiles of cholesterol metabolism-related genes are altered during development of experimental autoimmune encephalomyelitis in the rat spinal cord. Sci. Rep. 2017, 7, 2702. [Google Scholar] [CrossRef] [Green Version]
- Di Paolo, G.; Kim, T.W. Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat. Rev. Neurosci. 2011, 12, 284–296. [Google Scholar] [CrossRef]
- Foley, P. Lipids in Alzheimer’s disease: A century-old story. Biochim. Biophys. Acta 2010, 1801, 750–753. [Google Scholar] [CrossRef]
- Lalancette-Hébert, M.; Moquin, A.; Choi, A.O.; Jasna, K.; Maysinger, D. Lipopolysaccharide-QD micelles induce marked induction of TLR2 and lipid droplet accumulation in olfactory bulb microglia. Mol. Pharm. 2010, 7. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Zhang, I.; Bisht, K.; Savage, J.C.; Lecours, C.; Parent, M.; Titorenko, V.; Maysinger, D. Remodeling of lipid bodies by docosahexaenoic acid in activated microglial cells. J. Neuroinflamm. 2016, 13, 116. [Google Scholar] [CrossRef] [Green Version]
- Pacheco, P.; Vieira-de-Abreu, A.; Gomes, R.N.; Barbosa-Lima, G.; Wermelinger, L.B.; Maya-Monteiro, C.M.; Silva, A.R.; Bozza, M.T.; Castro-Faria-Neto, H.C.; Bandeira-Melo, C.; et al. Monocyte chemoattractant protein-1/CC chemokine ligand 2 controls microtubule-driven biogenesis and leukotriene B4-synthesizing function of macrophage lipid bodies elicited by innate immune response. J. Immunol. 2007, 179, 8500–8508. [Google Scholar] [CrossRef] [Green Version]
- Gschwandtner, M.; Derler, R.; Midwood, K.S. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front. Immunol. 2019, 10, 2759. [Google Scholar] [CrossRef] [Green Version]
- den Brok, M.H.; Raaijmakers, T.K.; Collado-Camps, E.; Adema, G.J. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol. 2018, 39, 380–392. [Google Scholar] [CrossRef]
- Churchward, M.A.; Tchir, D.R.; Todd, K.G. Microglial Function during Glucose Deprivation: Inflammatory and Neuropsychiatric Implications. Mol. Neurobiol. 2018, 55, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Nakachi, Y.; Yagi, K.; Nikaido, I.; Bono, H.; Tonouchi, M.; Schonbach, C.; Okazaki, Y. Identification of novel PPARgamma target genes by integrated analysis of ChIP-on-chip and microarray expression data during adipocyte differentiation. Biochem. Biophys. Res. Commun. 2008, 372, 362–366. [Google Scholar] [CrossRef]
- Rakhshandehroo, M.; Knoch, B.; Muller, M.; Kersten, S. Peroxisome proliferator-activated receptor alpha target genes. PPAR Res. 2010, 2010. [Google Scholar] [CrossRef] [Green Version]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Ueki, S.; Mahemuti, G.; Chiba, T.; Oyamada, H.; Saito, N.; Kanda, A.; Kayaba, H.; Chihara, J. Physiological levels of 15-deoxy-Delta12,14-prostaglandin J2 prime eotaxin-induced chemotaxis on human eosinophils through peroxisome proliferator-activated receptor-gamma ligation. J. Immunol. 2005, 175, 5744–5750. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Jackson, R.L.; Demel, R.A. Lipoprotein lipase-catalyzed hydrolysis of phospholipid monolayers: Effect of fatty acyl composition on enzyme activity. Biochem. Biophys. Res. Commun. 1985, 128, 670–675. [Google Scholar] [CrossRef]
- Mandrekar-Colucci, S.; Sauerbeck, A.; Popovich, P.G.; McTigue, D.M. PPAR agonists as therapeutics for CNS trauma and neurological diseases. ASN Neuro. 2013, 5, e00129. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, C.; Bright, J.J. Peroxisome proliferator-activated receptor-gamma agonists inhibit experimental allergic encephalomyelitis by blocking IL-12 production, IL-12 signaling and Th1 differentiation. Genes Immun. 2002, 3, 59–70. [Google Scholar] [CrossRef] [Green Version]
- Horvath, R.J.; Nutile-McMenemy, N.; Alkaitis, M.S.; Deleo, J.A. Differential migration, LPS-induced cytokine, chemokine, and NO expression in immortalized BV-2 and HAPI cell lines and primary microglial cultures. J. Neurochem. 2008, 107, 557–569. [Google Scholar] [CrossRef] [Green Version]
- Stansley, B.; Post, J.; Hensley, K. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 115. [Google Scholar] [CrossRef] [Green Version]
- Trent, C.M.; Yu, S.; Hu, Y.; Skoller, N.; Huggins, L.A.; Homma, S.; Goldberg, I.J. Lipoprotein lipase activity is required for cardiac lipid droplet production. J. Lipid Res. 2014, 55, 645–658. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loving, B.A.; Tang, M.; Neal, M.C.; Gorkhali, S.; Murphy, R.; Eckel, R.H.; Bruce, K.D. Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells 2021, 10, 198. https://doi.org/10.3390/cells10020198
Loving BA, Tang M, Neal MC, Gorkhali S, Murphy R, Eckel RH, Bruce KD. Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells. 2021; 10(2):198. https://doi.org/10.3390/cells10020198
Chicago/Turabian StyleLoving, Bailey A., Maoping Tang, Mikaela C. Neal, Sachi Gorkhali, Robert Murphy, Robert H. Eckel, and Kimberley D. Bruce. 2021. "Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation" Cells 10, no. 2: 198. https://doi.org/10.3390/cells10020198
APA StyleLoving, B. A., Tang, M., Neal, M. C., Gorkhali, S., Murphy, R., Eckel, R. H., & Bruce, K. D. (2021). Lipoprotein Lipase Regulates Microglial Lipid Droplet Accumulation. Cells, 10(2), 198. https://doi.org/10.3390/cells10020198