
Phosphorylation of Microglial IRF5 and IRF4 by IRAK4 Regulates Inflammatory Responses to Ischemia


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Spontaneously Immortalized Murine Microglia Culture
2.2. Primary Microglia Culture
2.3. Cortical Neurons
2.4. ND2158 Treatment, Oxygen-Glucose Deprivation (OGD), and LPS or IL-4 Stimulation
2.5. Immunocytochemistry (ICC)
2.6. Co-Immunoprecipitation (Co-IP)
2.7. Cell Fractionation
2.8. Western Blotting
2.9. Enzyme-Linked Immunosorbent Assay (ELISA)
2.10. mRNA Extraction and Real-Time Polymerase Chain Reaction (RT-PCR)
2.11. Calcein Cell Viability Assay
2.12. Statistical Analysis
3. Results
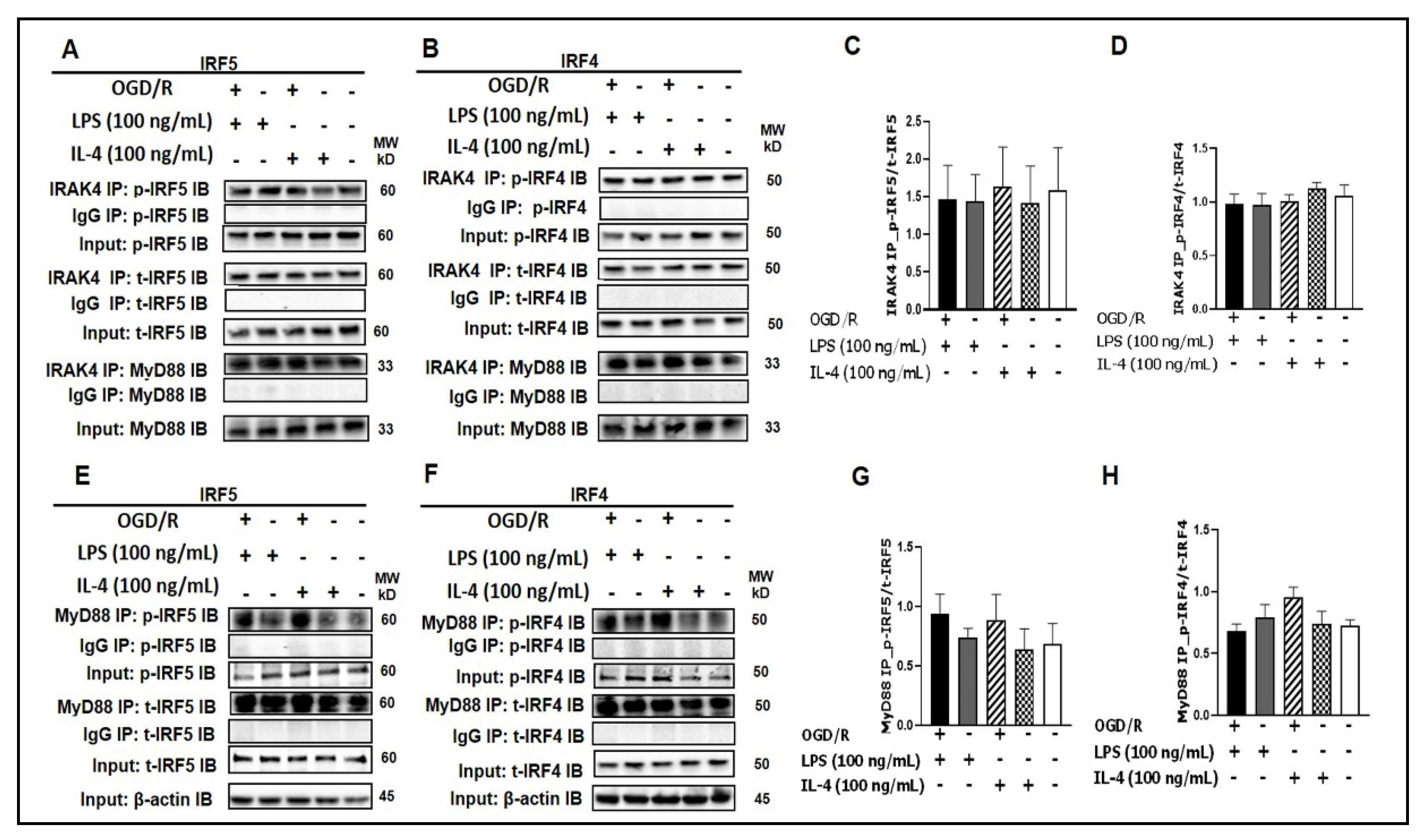
3.1. Interaction of IRF5/IRF4 with MyD88/IRAK4
3.2. IRAK4 Phosphorylation Triggers Phosphorylation of IRF5 and IRF4
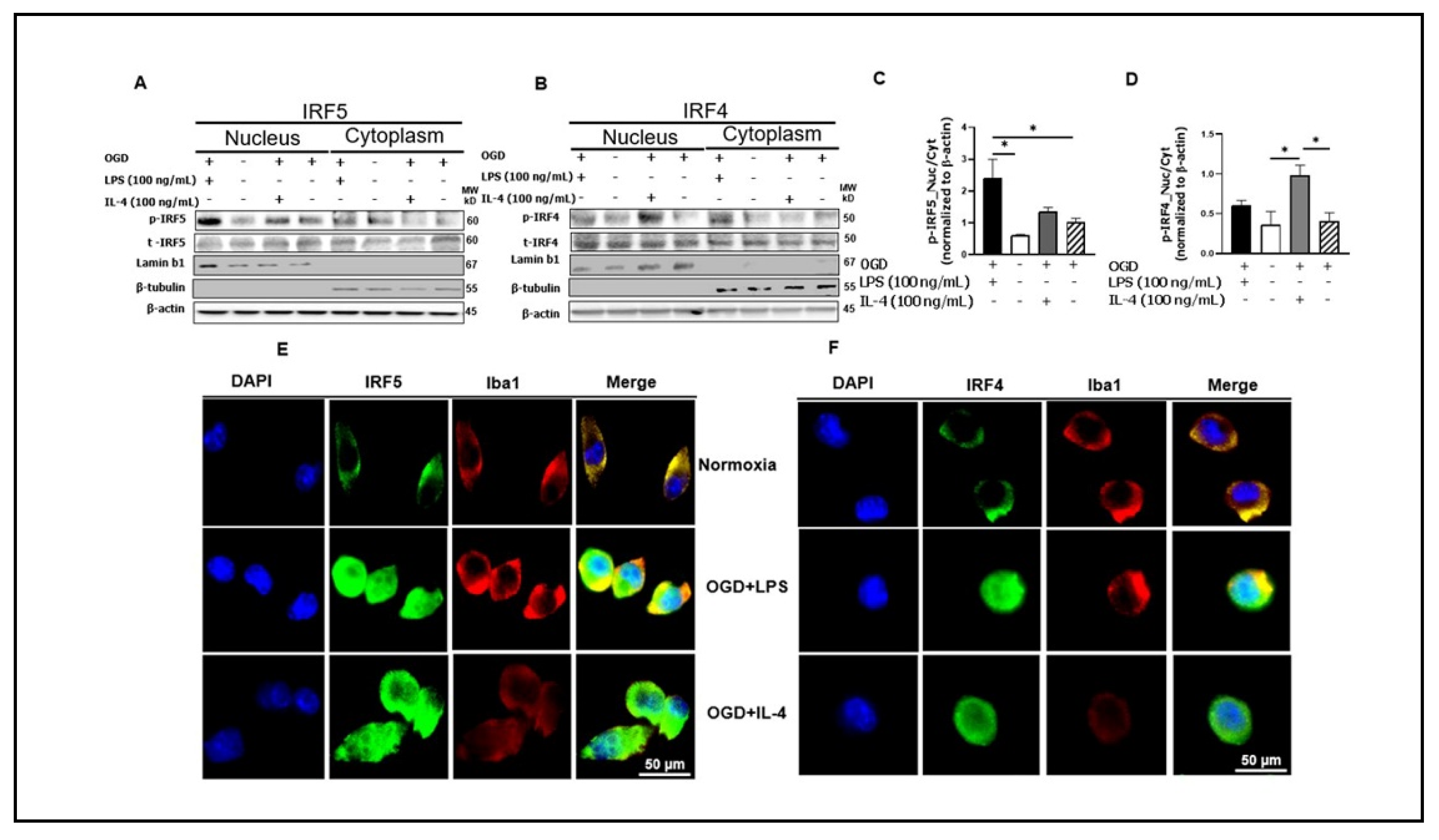
3.3. Phosphorylated IRF5 and IRF4 Translocate from the Cytoplasm to the Nucleus
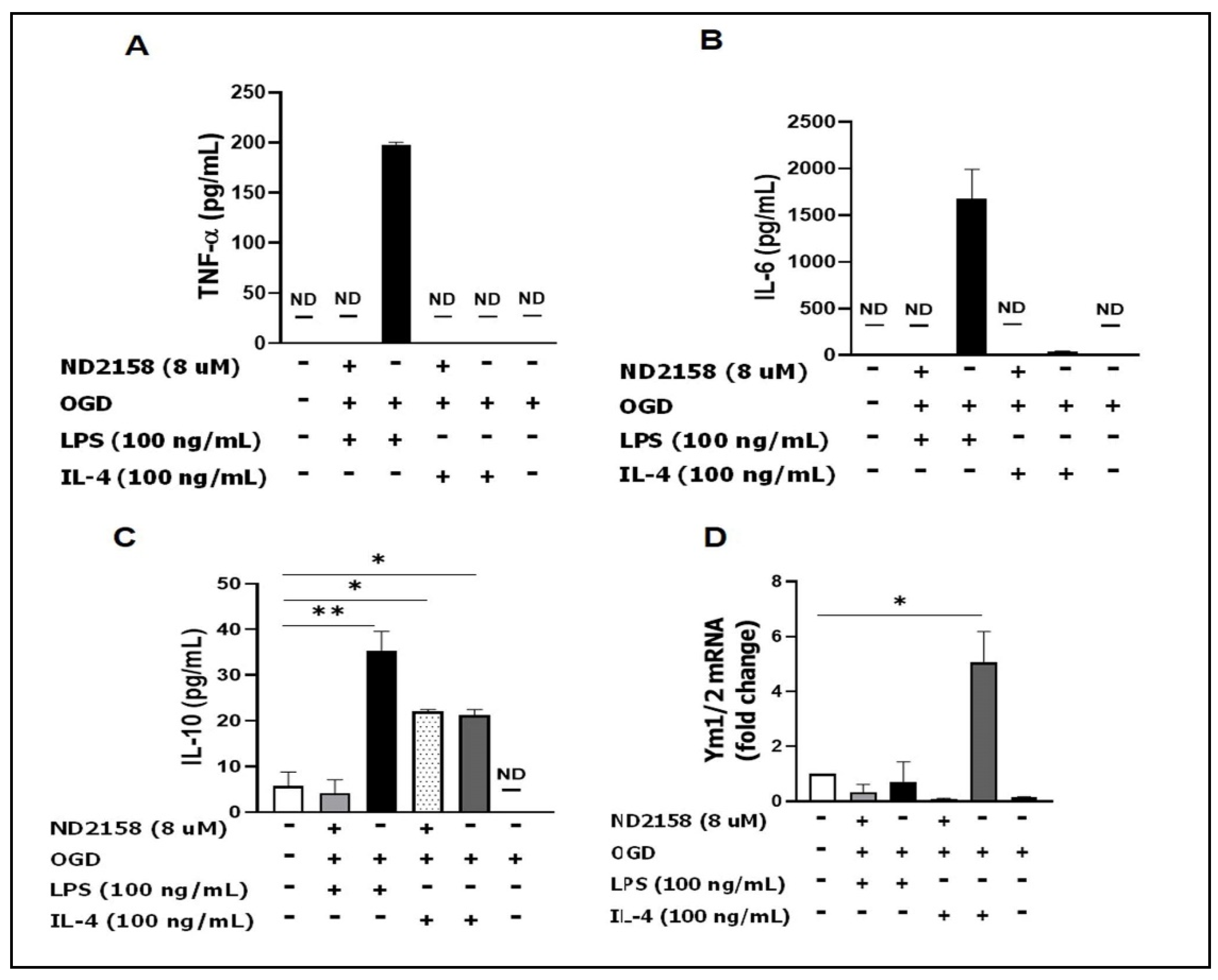
3.4. IRAK4 Modulates Microglial Activation
3.5. Microglial Culture Medium Conditioned by ND2158 Increases Neuronal Viability
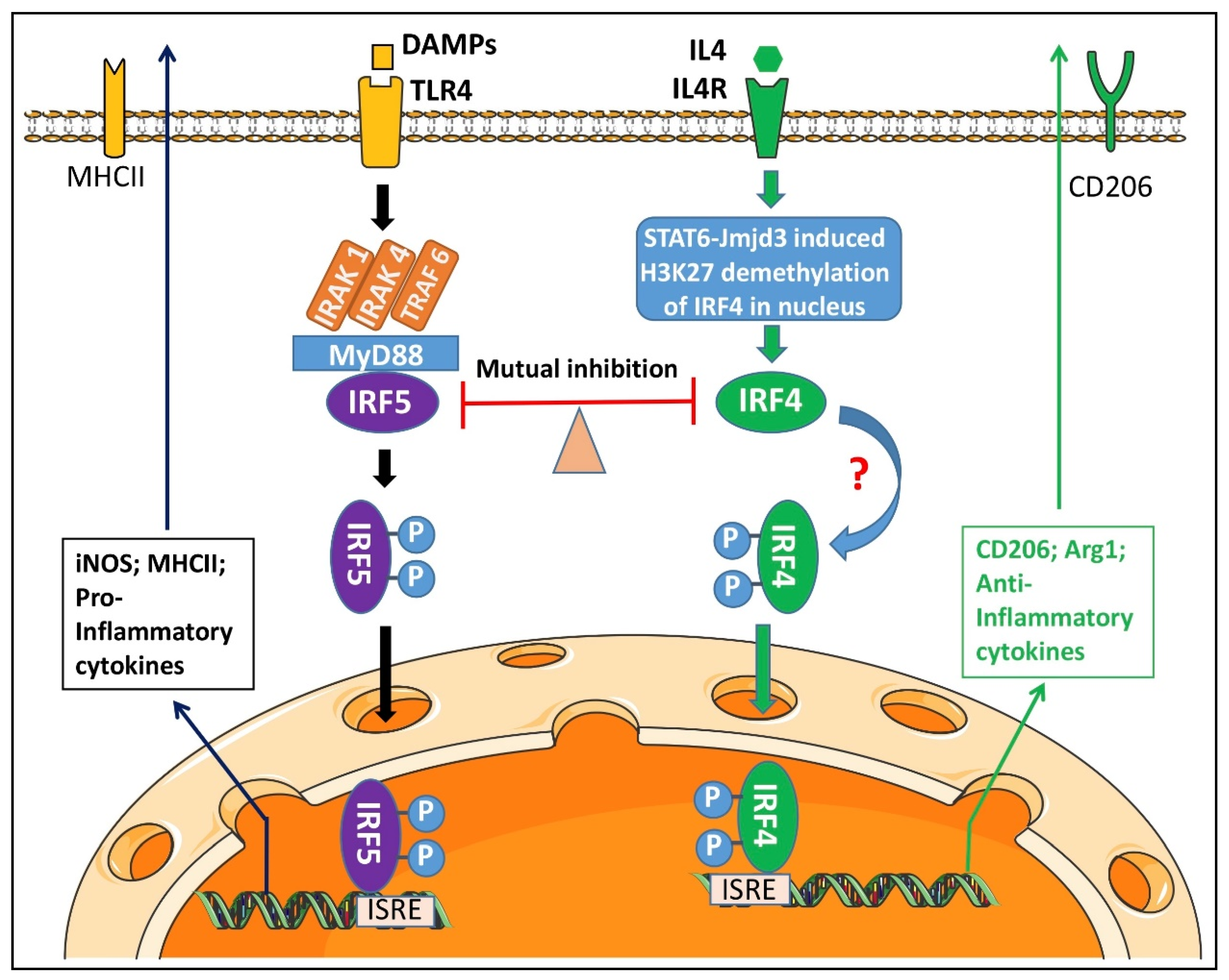
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ATCC | American Type Culture Collection |
| Co-IP | Co-Immunoprecipitation |
| DAMP | Damage associated molecular patterns |
| FBS | Fetal bovine serum |
| HS | Horse serum |
| ICC | Immunocytochemistry |
| IFN | Interferon |
| IL-4 | Interleukin-4 |
| IRF4 | Interferon regulatory factor 4 |
| IRF5 | Interferon regulatory factor 5 |
| IRAK-4 | Interleukin-1 receptor-associated kinase 4 |
| ISRE | IFN-stimulated response element |
| LPS | Lipopolysaccharide |
| JMJD3 | Jumonji domain-containing protein 3 |
| MAP2 | Microtubule-associated Protein 2 |
| OGD | Oxygen glucose deprivation |
| PBS | Phosphate buffered saline |
| PBST | Phosphate buffer saline tween-20 (0.05%) |
| P/S | Penicillin and streptomycin |
| ROCK2 | Rho-associated coiled-coil-containing protein kinase 2 |
| RT | Room temperature |
| STAT6 | Signal transducer and activator of transcription 6 |
| TLR | Toll-like receptor |
References
- Famakin, B.M. The Immune Response to Acute Focal Cerebral Ischemia and Associated Post-stroke Immunodepression: A Focused Review. Aging Dis. 2014, 5, 307–326. [Google Scholar] [PubMed]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef] [Green Version]
- Yrjanheikki, J.; Keinanen, R.; Pellikka, M.; Hokfelt, T.; Koistinaho, J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc. Natl. Acad. Sci. USA 1998, 95, 15769–15774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.J.; Wu, G.; Akhavan Sharif, M.R.; Baker, A.; Jia, Y.; Fahey, F.H.; Luo, H.R.; Feener, E.P.; Clapham, D.E. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 2012, 15, 565–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faustino, J.V.; Wang, X.; Johnson, C.E.; Klibanov, A.; Derugin, N.; Wendland, M.F.; Vexler, Z.S. Microglial cells contribute to endogenous brain defenses after acute neonatal focal stroke. J. Neurosci. 2011, 31, 12992–13001. [Google Scholar] [CrossRef] [Green Version]
- Lalancette-Hebert, M.; Gowing, G.; Simard, A.; Weng, Y.C.; Kriz, J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J. Neurosci. 2007, 27, 2596–2605. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Lombardi, M.; Gressens, P.; Verderio, C. How to reprogram microglia toward beneficial functions. Glia 2018, 66, 2531–2549. [Google Scholar] [CrossRef]
- Ouyang, X.; Negishi, H.; Takeda, R.; Fujita, Y.; Taniguchi, T.; Honda, K. Cooperation between MyD88 and TRIF pathways in TLR synergy via IRF5 activation. Biochem. Biophys. Res. Commun. 2007, 354, 1045–1051. [Google Scholar] [CrossRef]
- Al Mamun, A.; Chauhan, A.; Qi, S.; Ngwa, C.; Xu, Y.; Sharmeen, R.; Hazen, A.L.; Li, J.; Aronowski, J.A.; McCullough, L.D.; et al. Microglial IRF5-IRF4 regulatory axis regulates neuroinflammation after cerebral ischemia and impacts stroke outcomes. Proc. Natl. Acad. Sci. USA 2020, 117, 1742–1752. [Google Scholar] [CrossRef]
- Satoh, T.; Takeuchi, O.; Vandenbon, A.; Yasuda, K.; Tanaka, Y.; Kumagai, Y.; Miyake, T.; Matsushita, K.; Okazaki, T.; Saitoh, T.; et al. The Jmjd3-Irf4 axis regulates M2 macrophage polarization and host responses against helminth infection. Nat. Immunol. 2010, 11, 936–944. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, J.; Wang, Y.; Yang, G.Y. The biphasic function of microglia in ischemic stroke. Prog. Neurobiol. 2017, 157, 247–272. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Miura, M.; Toriyabe, M.; Koyama, M.; Hatakeyama, M.; Ishikawa, M.; Nakajima, T.; Onodera, O.; Takahashi, T.; Nishizawa, M.; et al. Microglia preconditioned by oxygen-glucose deprivation promote functional recovery in ischemic rats. Sci. Rep. 2017, 7, 42582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nardo, D.; Balka, K.R.; Cardona Gloria, Y.; Rao, V.R.; Latz, E.; Masters, S.L. Interleukin-1 receptor-associated kinase 4 (IRAK4) plays a dual role in myddosome formation and Toll-like receptor signaling. J. Biol. Chem. 2018, 293, 15195–15207. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Taniguchi, T. IRFs: Master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 2006, 6, 644–658. [Google Scholar] [CrossRef]
- Lawrence, T.; Natoli, G. Transcriptional regulation of macrophage polarization: Enabling diversity with identity. Nat. Rev. Immunol. 2011, 11, 750–761. [Google Scholar] [CrossRef]
- Wesche, H.; Henzel, W.J.; Shillinglaw, W.; Li, S.; Cao, Z. MyD88: An adapter that recruits IRAK to the IL-1 receptor complex. Immunity 1997, 7, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Motshwene, P.G.; Moncrieffe, M.C.; Grossmann, J.G.; Kao, C.; Ayaluru, M.; Sandercock, A.M.; Robinson, C.V.; Latz, E.; Gay, N.J. An oligomeric signaling platform formed by the Toll-like receptor signal transducers MyD88 and IRAK-4. J. Biol. Chem. 2009, 284, 25404–25411. [Google Scholar] [CrossRef] [Green Version]
- Lin, S.C.; Lo, Y.C.; Wu, H. Helical assembly in the MyD88-IRAK4-IRAK2 complex in TLR/IL-1R signalling. Nature 2010, 465, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Gay, N.J.; Gangloff, M.; O’Neill, L.A. What the Myddosome structure tells us about the initiation of innate immunity. Trends Immunol. 2011, 32, 104–109. [Google Scholar] [CrossRef]
- Nagamoto-Combs, K.; Kulas, J.; Combs, C.K. A novel cell line from spontaneously immortalized murine microglia. J. Neurosci. Methods 2014, 233, 187–198. [Google Scholar] [CrossRef] [Green Version]
- Draheim, H.J.; Prinz, M.; Weber, J.R.; Weiser, T.; Kettenmann, H.; Hanisch, U.K. Induction of potassium channels in mouse brain microglia: Cells acquire responsiveness to pneumococcal cell wall components during late development. Neuroscience 1999, 89, 1379–1390. [Google Scholar] [CrossRef]
- Ji, K.; Akgul, G.; Wollmuth, L.P.; Tsirka, S.E. Microglia actively regulate the number of functional synapses. PLoS ONE 2013, 8, e56293. [Google Scholar] [CrossRef] [Green Version]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar]
- Milner, R.; Hung, S.; Wang, X.; Berg, G.I.; Spatz, M.; del Zoppo, G.J. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke 2008, 39, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Del Zoppo, G.J.; Frankowski, H.; Gu, Y.H.; Osada, T.; Kanazawa, M.; Milner, R.; Wang, X.; Hosomi, N.; Mabuchi, T.; Koziol, J.A. Microglial cell activation is a source of metalloproteinase generation during hemorrhagic transformation. J. Cereb. Blood Flow Metab. 2012, 32, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Bohgaki, M.; Bohgaki, T.; El Ghamrasni, S.; Srikumar, T.; Maire, G.; Panier, S.; Fradet-Turcotte, A.; Stewart, G.S.; Raught, B.; Hakem, A.; et al. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA 2013, 110, 20982–20987. [Google Scholar] [CrossRef] [Green Version]
- Al Mamun, A.; Chauhan, A.; Yu, H.; Xu, Y.; Sharmeen, R.; Liu, F. Interferon regulatory factor 4/5 signaling impacts on microglial activation after ischemic stroke in mice. Eur. J. Neurosci. 2018, 47, 140–149. [Google Scholar] [CrossRef] [Green Version]
- Almuttaqi, H.; Udalova, I.A. Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J. 2019, 286, 1624–1637. [Google Scholar] [CrossRef]
- Pelka, K.; Latz, E. IRF5, IRF8, and IRF7 in human pDCs—The good, the bad, and the insignificant? Eur. J. Immunol. 2013, 43, 1693–1697. [Google Scholar] [CrossRef] [Green Version]
- Cushing, L.; Stochaj, W.; Siegel, M.; Czerwinski, R.; Dower, K.; Wright, Q.; Hirschfield, M.; Casanova, J.L.; Picard, C.; Puel, A.; et al. Interleukin 1/Toll-like receptor-induced autophosphorylation activates interleukin 1 receptor-associated kinase 4 and controls cytokine induction in a cell type-specific manner. J. Biol. Chem. 2014, 289, 10865–10875. [Google Scholar] [CrossRef] [Green Version]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNγ+TNFα) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell. Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [Green Version]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Pannell, M.; Szulzewsky, F.; Matyash, V.; Wolf, S.A.; Kettenmann, H. The subpopulation of microglia sensitive to neurotransmitters/neurohormones is modulated by stimulation with LPS, interferon-γ, and IL-4. Glia 2014, 62, 667–679. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, K.; Imagama, S.; Ohgomori, T.; Hirano, K.; Uchimura, K.; Sakamoto, K.; Hirakawa, A.; Takeuchi, H.; Suzumura, A.; Ishiguro, N.; et al. Minocycline selectively inhibits M1 polarization of microglia. Cell Death Dis. 2013, 4, e525. [Google Scholar] [CrossRef] [Green Version]
- Girard, S.; Brough, D.; Lopez-Castejon, G.; Giles, J.; Rothwell, N.J.; Allan, S.M. Microglia and macrophages differentially modulate cell death after brain injury caused by oxygen-glucose deprivation in organotypic brain slices. Glia 2013, 61, 813–824. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Wang, J.; Fang, Y.; Liu, Y.; Chen, T.; Sun, H.; Zhou, X.F.; Liao, H. Nafamostat mesilate improves function recovery after stroke by inhibiting neuroinflammation in rats. Brain Behav. Immun. 2016, 56, 230–245. [Google Scholar] [CrossRef]
- Ji, J.; Xiang, P.; Li, T.; Lan, L.; Xu, X.; Lu, G.; Ji, H.; Zhang, Y.; Li, Y. NOSH-NBP, a Novel Nitric Oxide and Hydrogen Sulfide—Releasing Hybrid, Attenuates Ischemic Stroke-Induced Neuroinflammatory Injury by Modulating Microglia Polarization. Front. Cell. Neurosci. 2017, 11, 154. [Google Scholar] [CrossRef] [Green Version]
- Barakat, R.; Redzic, Z. Differential cytokine expression by brain microglia/macrophages in primary culture after oxygen glucose deprivation and their protective effects on astrocytes during anoxia. Fluids Barriers CNS 2015, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Churchward, M.A.; Tchir, D.R.; Todd, K.G. Microglial Function during Glucose Deprivation: Inflammatory and Neuropsychiatric Implications. Mol. Neurobiol. 2018, 55, 1477–1487. [Google Scholar] [CrossRef] [Green Version]
- Tanifum, E.A.; Devkota, L.; Ngwa, C.; Badachhape, A.A.; Ghaghada, K.B.; Romero, J.; Pautler, R.G.; Annapragada, A.V. A Hyperfluorinated Hydrophilic Molecule for Aqueous (19)F MRI Contrast Media. Contrast Media Mol. Imaging 2018, 2018, 1693513. [Google Scholar] [CrossRef]
- Trask, O.J., Jr. Nuclear Factor Kappa B (NF-kappaB) Translocation Assay Development and Validation for High Content Screening. In Assay Guidance Manual; Sittampalam, G.S., Grossman, A., Brimacombe, K., Arkin, M., Auld, D., Austin, C.P., Baell, J., Bejcek, B., Caaveiro, J.M.M., Chung, T.D.Y., et al., Eds.; Eli Lilly & Company and the National Center for Advancing Translational Sciences: Bethesda, MD, USA, 2004. [Google Scholar]
- Gimenez, N.; Schulz, R.; Higashi, M.; Aymerich, M.; Villamor, N.; Delgado, J.; Juan, M.; Lopez-Guerra, M.; Campo, E.; Rosich, L.; et al. Targeting IRAK4 disrupts inflammatory pathways and delays tumor development in chronic lymphocytic leukemia. Leukemia 2020, 34, 100–114. [Google Scholar] [CrossRef] [Green Version]
- Malich, G.; Markovic, B.; Winder, C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology 1997, 124, 179–192. [Google Scholar] [CrossRef]
- Franklin, C.C.; Adler, V.; Kraft, A.S. Phosphorylation of transcription factors. Methods Enzymol. 1995, 254, 550–564. [Google Scholar]
- Shrum, C.K.; Defrancisco, D.; Meffert, M.K. Stimulated nuclear translocation of NF-kappaB and shuttling differentially depend on dynein and the dynactin complex. Proc. Natl. Acad. Sci. USA 2009, 106, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Stone, R.C.; Feng, D.; Deng, J.; Singh, S.; Yang, L.; Fitzgerald-Bocarsly, P.; Eloranta, M.L.; Ronnblom, L.; Barnes, B.J. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheum. 2012, 64, 788–798. [Google Scholar] [CrossRef]
- Rayasam, A.; Hsu, M.; Kijak, J.A.; Kissel, L.; Hernandez, G.; Sandor, M.; Fabry, Z. Immune responses in stroke: How the immune system contributes to damage and healing after stroke and how this knowledge could be translated to better cures? Immunology 2018, 154, 363–376. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Li, P.; Guo, Y.; Wang, H.; Leak, R.K.; Chen, S.; Gao, Y.; Chen, J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke 2012, 43, 3063–3070. [Google Scholar] [CrossRef] [Green Version]
- Takaoka, A.; Tamura, T.; Taniguchi, T. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 2008, 99, 467–478. [Google Scholar] [CrossRef]
- Gunthner, R.; Anders, H.J. Interferon-regulatory factors determine macrophage phenotype polarization. Mediators Inflamm 2013, 2013, 731023. [Google Scholar] [CrossRef] [Green Version]
- De Lima, T.M.; Sampaio, S.C.; Petroni, R.; Brigatte, P.; Velasco, I.T.; Soriano, F.G. Phagocytic activity of LPS tolerant macrophages. Mol. Immunol. 2014, 60, 8–13. [Google Scholar] [CrossRef]
- Van Hal, P.T.; Hopstaken-Broos, J.P.; Prins, A.; Favaloro, E.J.; Huijbens, R.J.; Hilvering, C.; Figdor, C.G.; Hoogsteden, H.C. Potential indirect anti-inflammatory effects of IL-4. Stimulation of human monocytes, macrophages, and endothelial cells by IL-4 increases aminopeptidase-N activity (CD13; EC 3.4.11.2). J. Immunol. 1994, 153, 2718–2728. [Google Scholar]
- Rey-Giraud, F.; Hafner, M.; Ries, C.H. In vitro generation of monocyte-derived macrophages under serum-free conditions improves their tumor promoting functions. PLoS ONE 2012, 7, e42656. [Google Scholar] [CrossRef] [Green Version]
- LoPresti, S.T.; Popovic, B.; Kulkarni, M.; Skillen, C.D.; Brown, B.N. Free radical-decellularized tissue promotes enhanced antioxidant and anti-inflammatory macrophage response. Biomaterials 2019, 222, 119376. [Google Scholar] [CrossRef]
- Cushing, L.; Winkler, A.; Jelinsky, S.A.; Lee, K.; Korver, W.; Hawtin, R.; Rao, V.R.; Fleming, M.; Lin, L.-L. IRAK4 kinase activity controls Toll-like receptor-induced inflammation through the transcription factor IRF5 in primary human monocytes. J. Biol. Chem. 2017, 292, 18689–18698. [Google Scholar] [CrossRef] [Green Version]
- Corzo, C.A.; Varfolomeev, E.; Setiadi, A.F.; Francis, R.; Klabunde, S.; Senger, K.; Sujatha-Bhaskar, S.; Drobnick, J.; Do, S.; Suto, E.; et al. The kinase IRAK4 promotes endosomal TLR and immune complex signaling in B cells and plasmacytoid dendritic cells. Sci. Signal. 2020, 13, eaaz1053. [Google Scholar] [CrossRef]
- Pennini, M.E.; Perkins, D.J.; Salazar, A.M.; Lipsky, M.; Vogel, S.N. Complete dependence on IRAK4 kinase activity in TLR2, but not TLR4, signaling pathways underlies decreased cytokine production and increased susceptibility to Streptococcus pneumoniae infection in IRAK4 kinase-inactive mice. J. Immunol. 2013, 190, 307–316. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Kopp, E.; Stadlen, A.; Chen, C.; Ghosh, S.; Janeway, C.A., Jr. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 1998, 2, 253–258. [Google Scholar] [CrossRef]
- Deguine, J.; Barton, G.M. MyD88: A central player in innate immune signaling. F1000Prime Rep. 2014, 6, 97. [Google Scholar] [CrossRef]
- Abd-El-Basset, E.; Fedoroff, S. Effect of bacterial wall lipopolysaccharide (LPS) on morphology, motility, and cytoskeletal organization of microglia in cultures. J. Neurosci. Res. 1995, 41, 222–237. [Google Scholar] [CrossRef]
- Ren, J.; Chen, X.; Chen, Z.J. IKKβ is an IRF5 kinase that instigates inflammation. Proc. Natl. Acad. Sci. USA 2014, 111, 17438–17443. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Sato, G.R.; Tamura, T. Regulation and role of the transcription factor IRF5 in innate immune responses and systemic lupus erythematosus. Int. Immunol. 2018, 30, 529–536. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Pelaez, M.; Lamont, D.J.; Peggie, M.; Shpiro, N.; Gray, N.S.; Cohen, P. Protein kinase IKKβ-catalyzed phosphorylation of IRF5 at Ser462 induces its dimerization and nuclear translocation in myeloid cells. Proc. Natl. Acad. Sci. USA 2014, 111, 17432–17437. [Google Scholar] [CrossRef] [Green Version]
- Ban, T.; Sato, G.R.; Nishiyama, A.; Akiyama, A.; Takasuna, M.; Umehara, M.; Suzuki, S.; Ichino, M.; Matsunaga, S.; Kimura, A.; et al. Lyn Kinase Suppresses the Transcriptional Activity of IRF5 in the TLR-MyD88 Pathway to Restrain the Development of Autoimmunity. Immunity 2016, 45, 319–332. [Google Scholar] [CrossRef] [Green Version]
- Negishi, H.; Ohba, Y.; Yanai, H.; Takaoka, A.; Honma, K.; Yui, K.; Matsuyama, T.; Taniguchi, T.; Honda, K. Negative regulation of Toll-like-receptor signaling by IRF-4. Proc. Natl. Acad. Sci. USA 2005, 102, 15989–15994. [Google Scholar] [CrossRef] [Green Version]
- Negishi, H.; Fujita, Y.; Yanai, H.; Sakaguchi, S.; Ouyang, X.; Shinohara, M.; Takayanagi, H.; Ohba, Y.; Taniguchi, T.; Honda, K. Evidence for licensing of IFN-gamma-induced IFN regulatory factor 1 transcription factor by MyD88 in Toll-like receptor-dependent gene induction program. Proc. Natl. Acad. Sci. USA 2006, 103, 15136–15141. [Google Scholar] [CrossRef] [Green Version]
- Hsu, A.T.; Lupancu, T.J.; Lee, M.C.; Fleetwood, A.J.; Cook, A.D.; Hamilton, J.A.; Achuthan, A. Epigenetic and transcriptional regulation of IL4-induced CCL17 production in human monocytes and murine macrophages. J. Biol. Chem. 2018, 293, 11415–11423. [Google Scholar] [CrossRef] [Green Version]
- Biswas, P.S.; Gupta, S.; Chang, E.; Song, L.; Stirzaker, R.A.; Liao, J.K.; Bhagat, G.; Pernis, A.B. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Investig. 2010, 120, 3280–3295. [Google Scholar] [CrossRef] [Green Version]
- Balka, K.R.; De Nardo, D. Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 2019, 105, 339–351. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ngwa, C.; Mamun, A.A.; Xu, Y.; Sharmeen, R.; Liu, F. Phosphorylation of Microglial IRF5 and IRF4 by IRAK4 Regulates Inflammatory Responses to Ischemia. Cells 2021, 10, 276. https://doi.org/10.3390/cells10020276
Ngwa C, Mamun AA, Xu Y, Sharmeen R, Liu F. Phosphorylation of Microglial IRF5 and IRF4 by IRAK4 Regulates Inflammatory Responses to Ischemia. Cells. 2021; 10(2):276. https://doi.org/10.3390/cells10020276
Chicago/Turabian StyleNgwa, Conelius, Abdullah Al Mamun, Yan Xu, Romana Sharmeen, and Fudong Liu. 2021. "Phosphorylation of Microglial IRF5 and IRF4 by IRAK4 Regulates Inflammatory Responses to Ischemia" Cells 10, no. 2: 276. https://doi.org/10.3390/cells10020276
APA StyleNgwa, C., Mamun, A. A., Xu, Y., Sharmeen, R., & Liu, F. (2021). Phosphorylation of Microglial IRF5 and IRF4 by IRAK4 Regulates Inflammatory Responses to Ischemia. Cells, 10(2), 276. https://doi.org/10.3390/cells10020276