
Prolactin Rescues Immature B Cells from Apoptosis-Induced BCR-Aggregation through STAT3, Bcl2a1a, Bcl2l2, and Birc5 in Lupus-Prone MRL/lpr Mice

 , and
, and 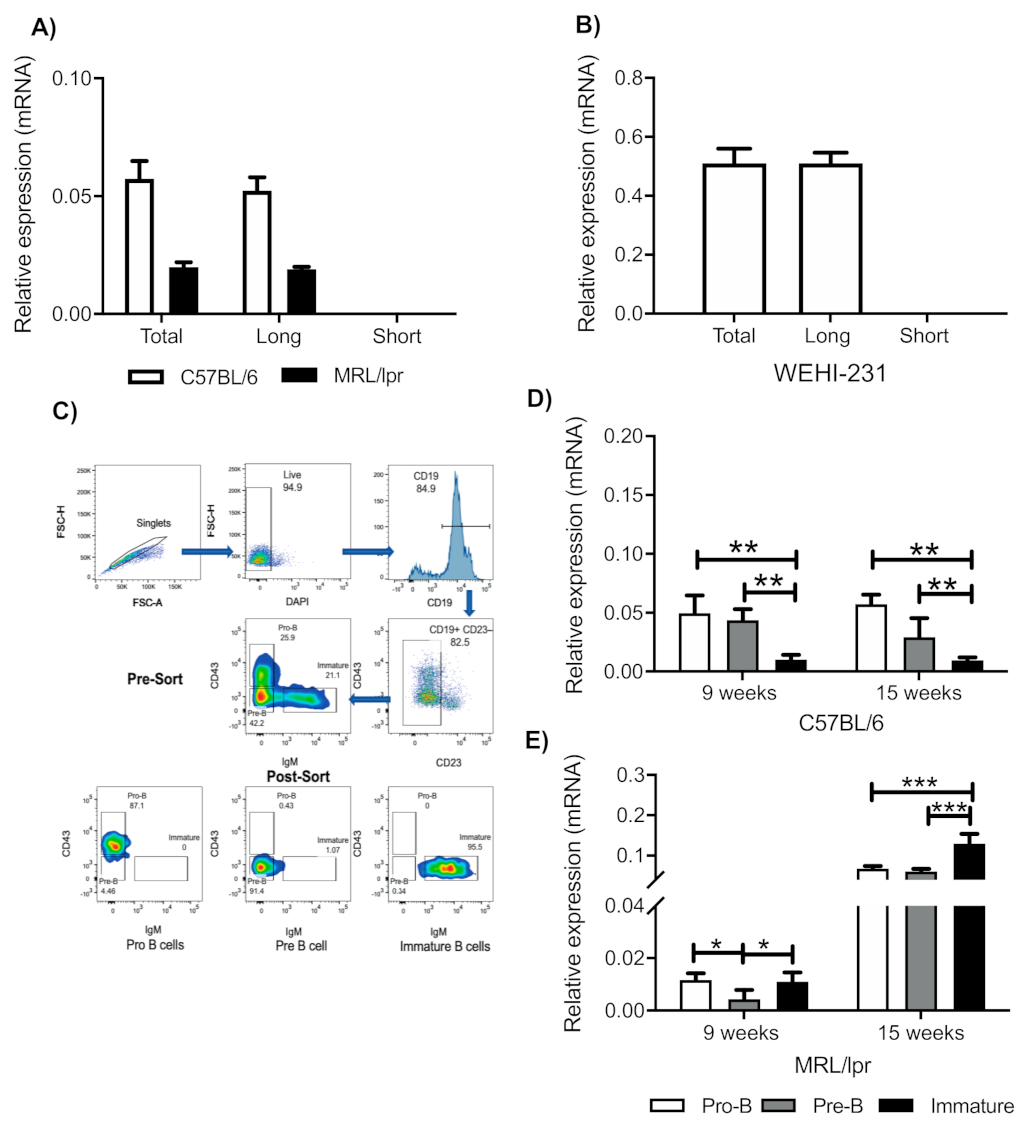{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Line
2.2. Mice
2.3. Prolactin Hormone and Inhibitors
2.4. Antibodies
2.5. Purification of B Cells (B220+CD23−) from the Bone Marrow
2.6. Analysis of the Signaling Pathways
2.7. Sorting B Cells from BM
2.8. RT-PCR for PRL-Receptor Isoforms
2.9. PCR Array and RT-PCR
2.10. Analysis of Gene Expression
2.11. Enrichment of Transcription Factors and Biological Processes
2.12. Chromatin Immunoprecipitation Assays
2.13. Apoptosis Assays
2.14. Statistical Analysis
3. Results
3.1. Immature B Cells from Mice and WEHI-231 Cells Express the Long Isoform of the PRL Receptor
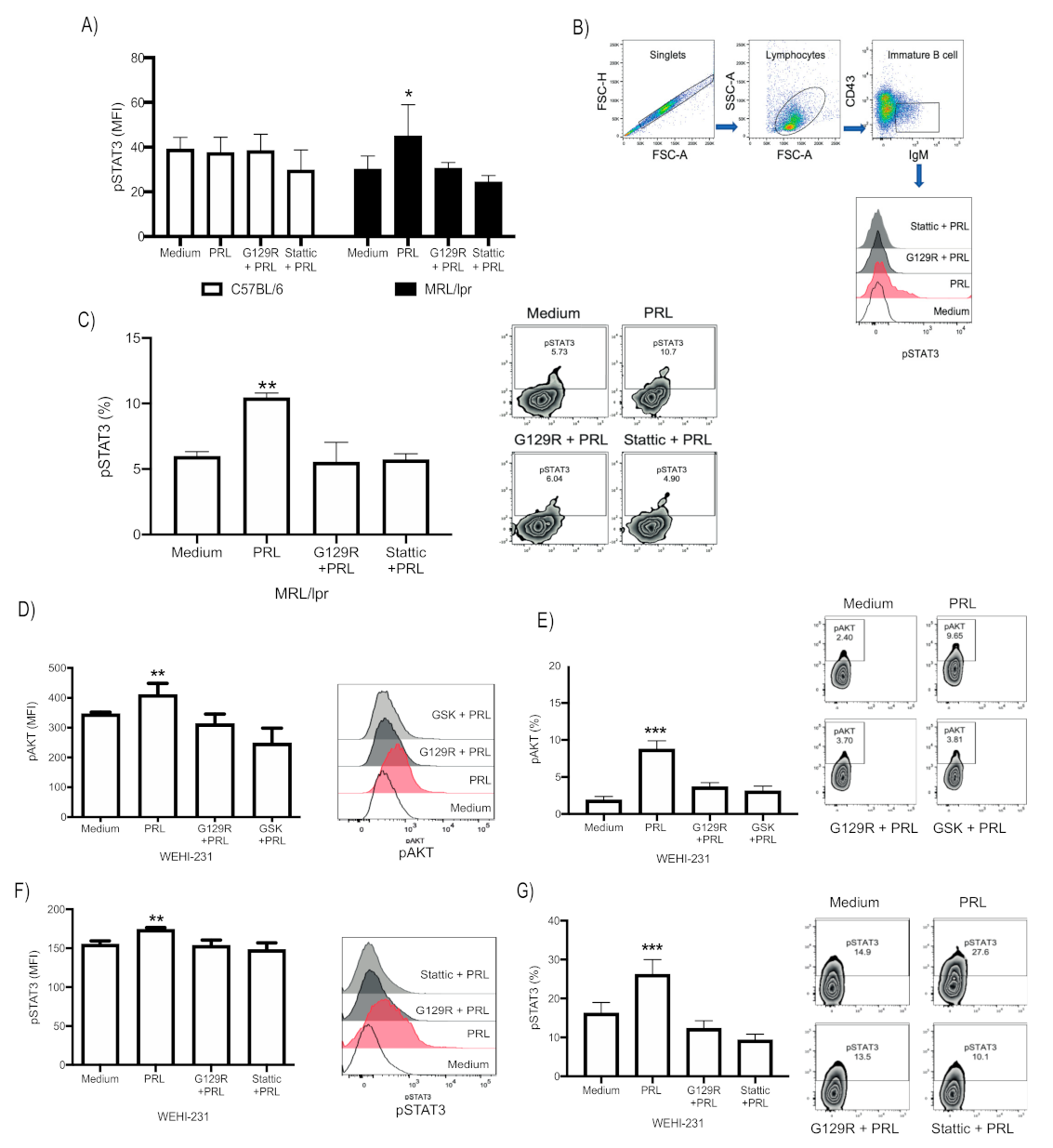
3.2. PRL Activates STAT3 in Immature B Cells from MRL/lpr Mice, Whereas in WEHI-231, It Activates the PI3K/AKT and STAT3 Signaling Pathways
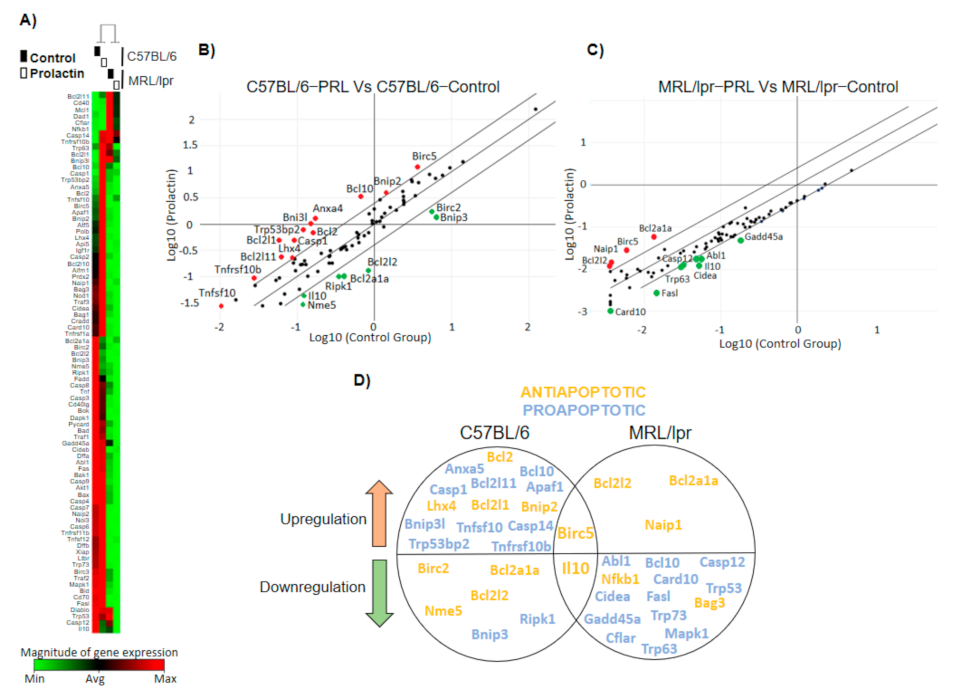
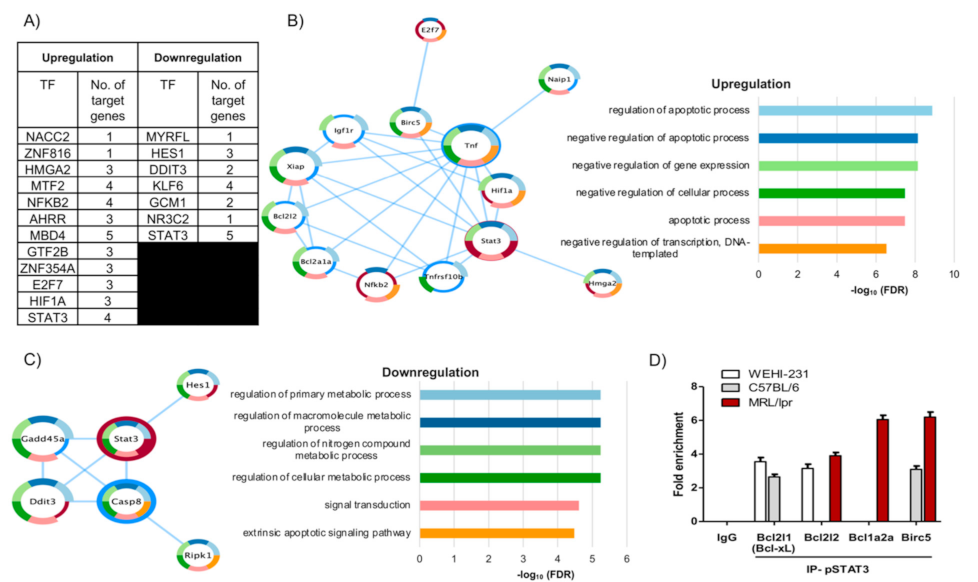
3.3. The Pattern of PRL-Regulated Genes Correlates with Antiapoptotic Activity and Places STAT3 as a Central Transcription Factor
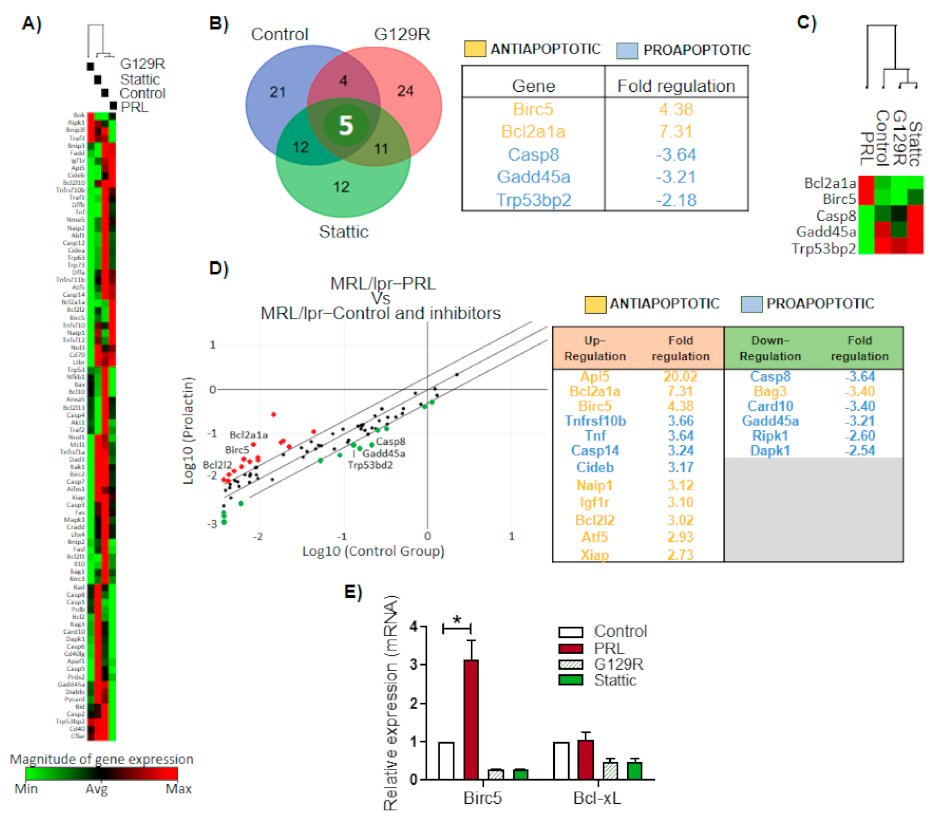
3.4. pSTAT3 Binds to Promoters of the Bcl2 Family and Birc5 of Antiapoptotic Genes
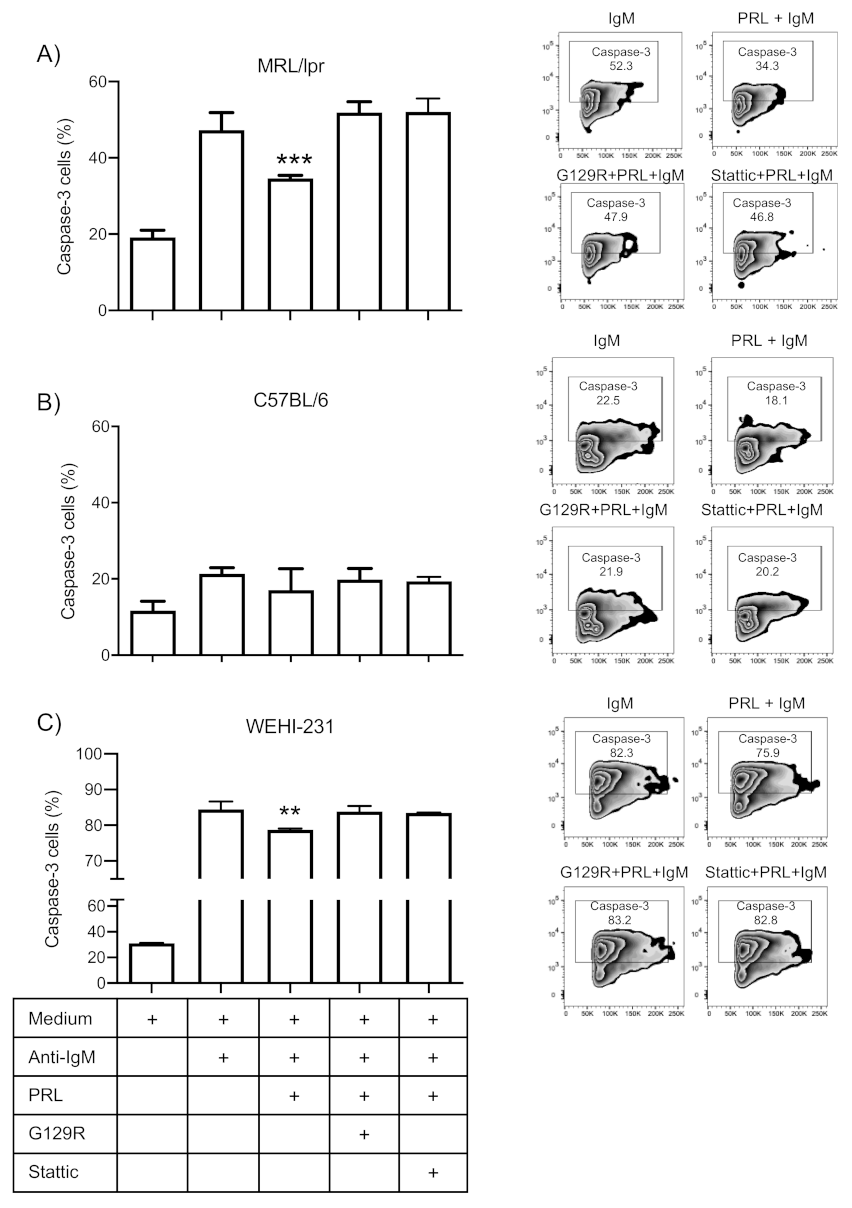
3.5. Prolactin Rescues Immature B Cells from Apoptosis via the STAT3 Pathway
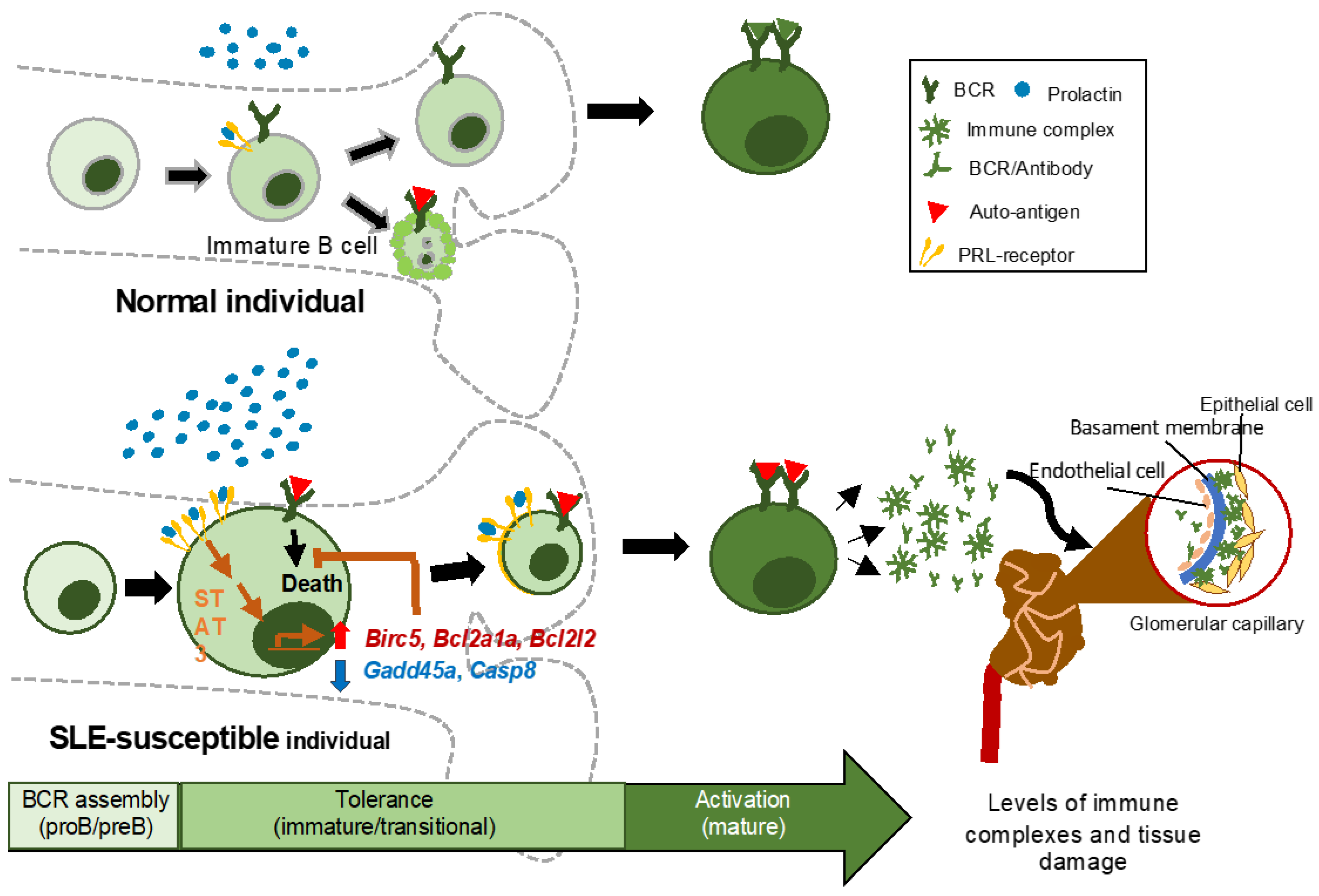
4. Discussion
5. Conclusions
6. Limitations of Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, R.R.; Hayakawa, K. B Cell development pathways. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef] [PubMed]
- Hobeika, E.; Dautzenberg, M.; Levit-Zerdoun, E.; Pelanda, R.; Reth, M. Conditional Selection of B Cells in Mice With an Inducible B Cell Development. Front. Immunol. 2018, 9, 1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemazee, D. Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 2017, 17, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Yurasov, S.; Wardemann, H.; Hammersen, J.; Tsuiji, M.; Meffre, E.; Pascual, V.; Nussenzweig, M.C. Defective B cell tolerance checkpoints in systemic lupus erythematosus. J. Exp. Med. 2005, 201, 703–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisnevskaia, L.; Murphy, G.; Isenberg, D. Systemic lupus erythematosus. Lancet 2014, 384, 1878–1888. [Google Scholar] [CrossRef]
- Eglys Gómez-Hernández, A.; José García-Mac Gregor, E.; Alfonso García-Montiel, D. Assessment of serum prolactin levels in patients with systemic lupus erythematosus. Investig. Clin. 2016, 57, 237–245. [Google Scholar]
- Song, G.G.; Lee, Y.H. Circulating prolactin level in systemic lupus erythematosus and its correlation with disease activity: A meta-analysis. Lupus 2017, 26, 1260–1268. [Google Scholar] [CrossRef]
- Yang, J.; Li, Q.; Yang, X.; Li, M. Increased serum level of prolactin is related to autoantibody production in systemic lupus erythematosus. Lupus 2016, 25, 513–519. [Google Scholar] [CrossRef]
- Blanco-Favela, F.; Quintal-Alvarez, G.; Leanos-Miranda, A. Association between prolactin and disease activity in systemic lupus erythematosus. Influence of statistical power. J. Rheumatol. 1999, 26, 55–59. [Google Scholar]
- Ledesma-Soto, Y.; Blanco-Favela, F.; Fuentes-Panana, E.M.; Tesoro-Cruz, E.; Hernandez-Gonzalez, R.; Arriaga-Pizano, L.; Legorreta-Haquet, M.V.; Montoya-Diaz, E.; Chavez-Sanchez, L.; Chavez-Rueda, A.K.; et al. Increased levels of prolactin receptor expression correlate with the early onset of lupus symptoms and increased numbers of transitional-1 B cells after prolactin treatment. BMC Immunol. 2012, 13, 11. [Google Scholar] [CrossRef] [Green Version]
- Legorreta-Haquet, M.V.; Flores-Fernandez, R.; Blanco-Favela, F.; Fuentes-Panana, E.M.; Chavez-Sanchez, L.; Hernandez-Gonzalez, R.; Tesoro-Cruz, E.; Arriaga-Pizano, L.; Chavez-Rueda, A.K. Prolactin levels correlate with abnormal B cell maturation in MRL and MRL/lpr mouse models of systemic lupus erythematosus-like disease. Clin. Dev. Immunol. 2013, 2013, 287469. [Google Scholar] [CrossRef] [PubMed]
- Flores-Fernandez, R.; Blanco-Favela, F.; Fuentes-Panana, E.M.; Chavez-Sanchez, L.; Gorocica-Rosete, P.; Pizana-Venegas, A.; Chavez-Rueda, A.K. Prolactin Rescues Immature B-Cells from Apoptosis Induced by B-Cell Receptor Cross-Linking. J. Immunol. Res. 2016, 2016, 3219017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binart, N.; Bachelot, A.; Bouilly, J. Impact of prolactin receptor isoforms on reproduction. Trends Endocrinol. Metab. 2010, 21, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Sangeeta Devi, Y.; Halperin, J. Reproductive actions of prolactin mediated through short and long receptor isoforms. Mol. Cell. Endocrinol. 2014, 382, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Abramicheva, P.A.; Smirnova, O.V. Prolactin Receptor Isoforms as the Basis of Tissue-Specific Action of Prolactin in the Norm and Pathology. Biochemistry 2019, 84, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Devi, Y.S.; Shehu, A.; Stocco, C.; Halperin, J.; Le, J.; Seibold, A.M.; Lahav, M.; Binart, N.; Gibori, G. Regulation of transcription factors and repression of Sp1 by prolactin signaling through the short isoform of its cognate receptor. Endocrinology 2009, 150, 3327–3335. [Google Scholar] [CrossRef] [Green Version]
- Pascual-Mathey, L.I.; Rojas-Duran, F.; Aranda-Abreu, G.E.; Manzo, J.; Herrera-Covarrubias, D.; Munoz-Zavaleta, D.A.; Garcia, L.I.; Hernandez, M.E. Effect of hyperprolactinemia on PRL-receptor expression and activation of Stat and Mapk cell signaling in the prostate of long-term sexually-active rats. Physiol. Behav. 2016, 157, 170–177. [Google Scholar] [CrossRef]
- Belugin, S.; Diogenes, A.R.; Patil, M.J.; Ginsburg, E.; Henry, M.A.; Akopian, A.N. Mechanisms of transient signaling via short and long prolactin receptor isoforms in female and male sensory neurons. J. Biol. Chem. 2013, 288, 34943–34955. [Google Scholar] [CrossRef] [Green Version]
- Creamer, B.A.; Sakamoto, K.; Schmidt, J.W.; Triplett, A.A.; Moriggl, R.; Wagner, K.U. Stat5 promotes survival of mammary epithelial cells through transcriptional activation of a distinct promoter in Akt1. Mol. Cell. Biol. 2010, 30, 2957–2970. [Google Scholar]
- Jabbour, H.N.; Critchley, H.O.; Boddy, S.C. Expression of functional prolactin receptors in nonpregnant human endometrium: Janus kinase-2, signal transducer and activator of transcription-1 (STAT1), and STAT5 proteins are phosphorylated after stimulation with prolactin. J. Clin. Endocrinol. Metab. 1998, 83, 2545–2553. [Google Scholar] [CrossRef]
- Roy, G.; Lombardia, M.; Palacios, C.; Serrano, A.; Cespon, C.; Ortega, E.; Eiras, P.; Lujan, S.; Revilla, Y.; Gonzalez-Porque, P. Mechanistic aspects of the induction of apoptosis by lauryl gallate in the murine B-cell lymphoma line Wehi 231. Arch. Biochem. Biophys. 2000, 383, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Zand, B.; Ozpolat, B.; Szczepanski, M.J.; Lu, C.; Yuca, E.; Carroll, A.R.; Lutgendorf, S.K.; Liu, J.; Sood, A.K.; et al. Antagonism of tumoral prolactin receptor promotes autophagy-related cell death. Cell. Rep. 2014, 7, 488–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schust, J.; Sperl, B.; Hollis, A.; Mayer, T.U.; Berg, T. Stattic: A small-molecule inhibitor of STAT3 activation and dimerization. Chem. Biol. 2006, 13, 1235–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [PubMed] [Green Version]
- Krishnan, N.; Thellin, O.; Buckley, D.J.; Horseman, N.D.; Buckley, A.R. Prolactin suppresses glucocorticoid-induced thymocyte apoptosis in vivo. Endocrinology 2003, 144, 2102–2110. [Google Scholar] [CrossRef] [PubMed]
- Bauernhofer, T.; Kuss, I.; Friebe-Hoffmann, U.; Baum, A.S.; Dworacki, G.; Vonderhaar, B.K.; Whiteside, T.L. Role of prolactin receptor and CD25 in protection of circulating T lymphocytes from apoptosis in patients with breast cancer. Br. J. Cancer 2003, 88, 1301–1309. [Google Scholar] [CrossRef] [Green Version]
- Chou, W.C.; Levy, D.E.; Lee, C.K. STAT3 positively regulates an early step in B-cell development. Blood 2006, 108, 3005–3011. [Google Scholar] [CrossRef] [Green Version]
- Asai-Sato, M.; Nagashima, Y.; Miyagi, E.; Sato, K.; Ohta, I.; Vonderhaar, B.K.; Hirahara, F. Prolactin inhibits apoptosis of ovarian carcinoma cells induced by serum starvation or cisplatin treatment. Int. J. Cancer 2005, 115, 539–544. [Google Scholar] [CrossRef]
- Zhu, F.; Wang, K.B.; Rui, L. STAT3 Activation and Oncogenesis in Lymphoma. Cancers 2019, 12, 19. [Google Scholar] [CrossRef] [Green Version]
- Al-Sakkaf, K.A.; Mooney, L.M.; Dobson, P.R.; Brown, B.L. Possible role for protein kinase B in the anti-apoptotic effect of prolactin in rat Nb2 lymphoma cells. J. Endocrinol. 2000, 167, 85–92. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.F.; Yu-Lee, L.Y. Multiple stat complexes interact at the interferon regulatory factor-1 interferon-gamma activation sequence in prolactin-stimulated Nb2 T cells. Mol. Cell. Endocrinol. 1996, 121, 19–28. [Google Scholar] [CrossRef]
- Pedraz-Cuesta, E.; Fredsted, J.; Jensen, H.H.; Bornebusch, A.; Nejsum, L.N.; Kragelund, B.B.; Pedersen, S.F. Prolactin Signaling Stimulates Invasion via Na(+)/H(+) Exchanger NHE1 in T47D Human Breast Cancer Cells. Mol. Endocrinol. 2016, 30, 693–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haxholm, G.W.; Nikolajsen, L.F.; Olsen, J.G.; Fredsted, J.; Larsen, F.H.; Goffin, V.; Pedersen, S.F.; Brooks, A.J.; Waters, M.J.; Kragelund, B.B. Intrinsically disordered cytoplasmic domains of two cytokine receptors mediate conserved interactions with membranes. Biochem. J. 2015, 468, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Legorreta-Haquet, M.V.; Chávez-Rueda, K.; Chávez-Sánchez, L.; Cervera-Castillo, H.; Zenteno-Galindo, E.; Barile-Fabris, L.; Burgos-Vargas, R.; Álvarez-Hernández, E.; Blanco-Favela, F. Function of Treg Cells Decreased in Patients With Systemic Lupus Erythematosus Due To the Effect of Prolactin. Medicine 2016, 95, e2384. [Google Scholar] [CrossRef] [PubMed]
- Neradugomma, N.K.; Subramaniam, D.; Tawfik, O.W.; Goffin, V.; Kumar, T.R.; Jensen, R.A.; Anant, S. Prolactin signaling enhances colon cancer stemness by modulating Notch signaling in a Jak2-STAT3/ERK manner. Carcinogenesis 2014, 35, 795–806. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Briggs, J.; Park, S.; Niu, G.; Kortylewski, M.; Zhang, S.; Gritsko, T.; Turkson, J.; Kay, H.; Yu, H.; et al. Targeting Stat3 blocks both HIF-1 and VEGF expression induced by multiple oncogenic growth signaling pathways. Oncogene 2005, 24, 5552–5560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, R.; Chen, X.; Chen, L.; Wan, F.; Chen, K.; Sun, Y.; Zhu, X. STAT3 signaling in ovarian cancer: A potential therapeutic target. J. Cancer 2020, 11, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Hillmer, E.J.; Zhang, H.; Li, H.S.; Watowich, S.S. STAT3 signaling in immunity. Cytokine Growth Factor Rev 2016, 31, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Diehl, S.A.; Schmidlin, H.; Nagasawa, M.; Van Haren, S.D.; Kwakkenbos, M.J.; Yasuda, E.; Beaumont, T.; Scheeren, F.A.; Spits, H. STAT3-mediated up-regulation of BLIMP1 Is coordinated with BCL6 down-regulation to control human plasma cell differentiation. J. Immunol. 2008, 180, 4805–4815. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Nemeth, J.; O’Brien, C.; Susa, M.; Liu, X.; Zhang, Z.; Choy, E.; Mankin, H.; Hornicek, F.; Duan, Z. Effects of siltuximab on the IL-6-induced signaling pathway in ovarian cancer. Clin. Cancer Res. 2010, 16, 5759–5769. [Google Scholar] [CrossRef] [Green Version]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Gao, Q.; et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Diaz, N.; Minton, S.; Cox, C.; Bowman, T.; Gritsko, T.; Garcia, R.; Eweis, I.; Wloch, M.; Sullivan, D.; Muro-Cacho, C.A.; et al. Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin. Cancer Res. 2006, 12, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritsko, T.; Williams, A.; Turkson, J.; Kaneko, S.; Bowman, T.; Huang, M.; Nam, S.; Eweis, I.; Diaz, N.; Jove, R.; et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin. Cancer Res. 2006, 12, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Zhu, F.; Zhang, M.; Li, Y.; Drennan, A.C.; Kimpara, S.; Rumball, I.; Selzer, C.; Cameron, H.; Kellicut, A.; et al. Gene regulation and suppression of type I interferon signaling by STAT3 in diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2018, 115, E498–E505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathania, A.S.; Kumar, S.; Guru, S.K.; Bhushan, S.; Sharma, P.R.; Aithagani, S.K.; Singh, P.P.; Vishwakarma, R.A.; Kumar, A.; Malik, F. The synthetic tryptanthrin analogue suppresses STAT3 signaling and induces caspase dependent apoptosis via ERK up regulation in human leukemia HL-60 cells. PLoS ONE 2014, 9, e110411. [Google Scholar] [CrossRef]
- Zhou, J.; Bi, C.; Janakakumara, J.V.; Liu, S.C.; Chng, W.J.; Tay, K.G.; Poon, L.F.; Xie, Z.; Albert, D.H.; Chen, C.S.; et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood 2009, 113, 4052–4062. [Google Scholar] [CrossRef]
- Gharibi, T.; Babaloo, Z.; Hosseini, A.; Abdollahpour-Alitappeh, M.; Hashemi, V.; Marofi, F.; Nejati, K.; Baradaran, B. Targeting STAT3 in cancer and autoimmune diseases. Eur. J. Pharmacol. 2020, 878, 173107. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Morgan, N.G.; Hattersley, A.T.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef]
- Haapaniemi, E.M.; Kaustio, M.; Rajala, H.L.; Van Adrichem, A.J.; Kainulainen, L.; Glumoff, V.; Doffinger, R.; Kuusanmäki, H.; Mustjoki, S.; Kere, J.; et al. Autoimmunity, hypogammaglobulinemia, lymphoproliferation, and mycobacterial disease in patients with activating mutations in STAT3. Blood 2015, 125, 639–648. [Google Scholar] [CrossRef] [Green Version]
- De La Varga Martínez, R.; Rodríguez-Bayona, B.; Añez, G.A.; Medina Varo, F.; Pérez Venegas, J.J.; Brieva, J.A.; Rodríguez, C. Clinical relevance of circulating anti-ENA and anti-dsDNA secreting cells from SLE patients and their dependence on STAT-3 activation. Eur. J. Immunol. 2017, 47, 1211–1219. [Google Scholar] [CrossRef] [Green Version]
- Edwards, L.J.; Mizui, M.; Kyttaris, V. Signal transducer and activator of transcription (STAT) 3 inhibition delays the onset of lupus nephritis in MRL/lpr mice. Clin. Immunol. 2015, 158, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Zhang, W.; Liu, S.; Feng, X.; Gao, F.; Liu, Q. S3I-201 ameliorates tubulointerstitial lesion of the kidneys in MRL/lpr mice. Biochem. Biophys. Res. Commun. 2018, 503, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.J.; Furie, R.A.; Tanaka, Y.; Kalunian, K.C.; Mosca, M.; Petri, M.A.; Dörner, T.; DeLozier, A.M.; Janes, J.M.; Hoffman, R.W.; et al. Baricitinib for systemic lupus erythematosus: A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet 2018, 392, 222–231. [Google Scholar] [CrossRef]
- Miletic, A.V.; Jellusova, J.; Cato, M.H.; Lee, C.R.; Baracho, G.V.; Conway, E.M.; Rickert, R.C. Essential Role for Survivin in the Proliferative Expansion of Progenitor and Mature B Cells. J. Immunol. 2016, 196, 2195–2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Métais, J.Y.; Winkler, T.; Geyer, J.T.; Calado, R.T.; Aplan, P.D.; Eckhaus, M.A.; Dunbar, C.E. BCL2A1a over-expression in murine hematopoietic stem and progenitor cells decreases apoptosis and results in hematopoietic transformation. PLoS ONE 2012, 7, e48267. [Google Scholar] [CrossRef] [Green Version]
- Adams, C.M.; Kim, A.S.; Mitra, R.; Choi, J.K.; Gong, J.Z.; Eischen, C.M. BCL-W has a fundamental role in B cell survival and lymphomagenesis. J. Clin. Investig. 2017, 127, 635–650. [Google Scholar] [CrossRef] [Green Version]
- Pahlavan, Y.; Kahroba, H.; Samadi, N.; Karimi, A.; Ansarin, K.; Khabbazi, A. Survivin modulatory role in autoimmune and autoinflammatory diseases. J. Cell. Physiol. 2019, 234, 19440–19450. [Google Scholar] [CrossRef]
- Gravina, G.; Wasén, C.; Garcia-Bonete, M.J.; Turkkila, M.; Erlandsson, M.C.; Töyrä Silfverswärd, S.; Brisslert, M.; Pullerits, R.; Andersson, K.M.; Bokarewa, M.I.; et al. Survivin in autoimmune diseases. Autoimmun. Rev. 2017, 16, 845–855. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flores-Fernández, R.; Aponte-López, A.; Suárez-Arriaga, M.C.; Gorocica-Rosete, P.; Pizaña-Venegas, A.; Chávez-Sanchéz, L.; Blanco-Favela, F.; Fuentes-Pananá, E.M.; Chávez-Rueda, A.K. Prolactin Rescues Immature B Cells from Apoptosis-Induced BCR-Aggregation through STAT3, Bcl2a1a, Bcl2l2, and Birc5 in Lupus-Prone MRL/lpr Mice. Cells 2021, 10, 316. https://doi.org/10.3390/cells10020316
Flores-Fernández R, Aponte-López A, Suárez-Arriaga MC, Gorocica-Rosete P, Pizaña-Venegas A, Chávez-Sanchéz L, Blanco-Favela F, Fuentes-Pananá EM, Chávez-Rueda AK. Prolactin Rescues Immature B Cells from Apoptosis-Induced BCR-Aggregation through STAT3, Bcl2a1a, Bcl2l2, and Birc5 in Lupus-Prone MRL/lpr Mice. Cells. 2021; 10(2):316. https://doi.org/10.3390/cells10020316
Chicago/Turabian StyleFlores-Fernández, Rocio, Angélica Aponte-López, Mayra C. Suárez-Arriaga, Patricia Gorocica-Rosete, Alberto Pizaña-Venegas, Luis Chávez-Sanchéz, Francico Blanco-Favela, Ezequiel M. Fuentes-Pananá, and Adriana K. Chávez-Rueda. 2021. "Prolactin Rescues Immature B Cells from Apoptosis-Induced BCR-Aggregation through STAT3, Bcl2a1a, Bcl2l2, and Birc5 in Lupus-Prone MRL/lpr Mice" Cells 10, no. 2: 316. https://doi.org/10.3390/cells10020316
APA StyleFlores-Fernández, R., Aponte-López, A., Suárez-Arriaga, M. C., Gorocica-Rosete, P., Pizaña-Venegas, A., Chávez-Sanchéz, L., Blanco-Favela, F., Fuentes-Pananá, E. M., & Chávez-Rueda, A. K. (2021). Prolactin Rescues Immature B Cells from Apoptosis-Induced BCR-Aggregation through STAT3, Bcl2a1a, Bcl2l2, and Birc5 in Lupus-Prone MRL/lpr Mice. Cells, 10(2), 316. https://doi.org/10.3390/cells10020316