
Role of Telomeres Shortening in Atherogenesis: An Overview

 , and
, and 
Abstract
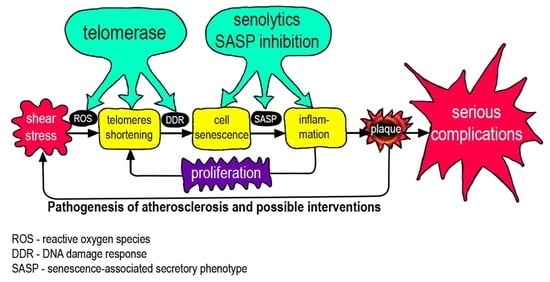
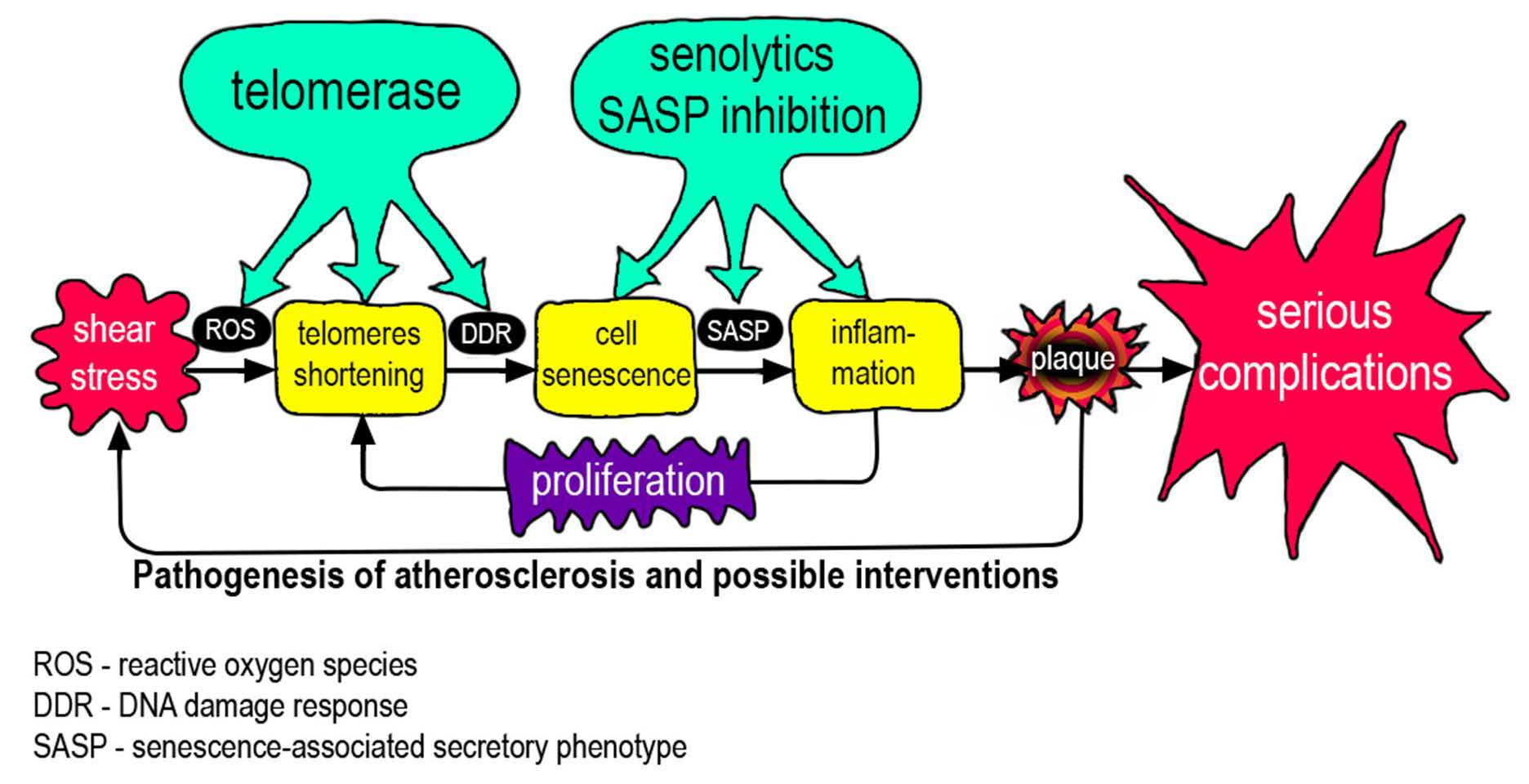
:
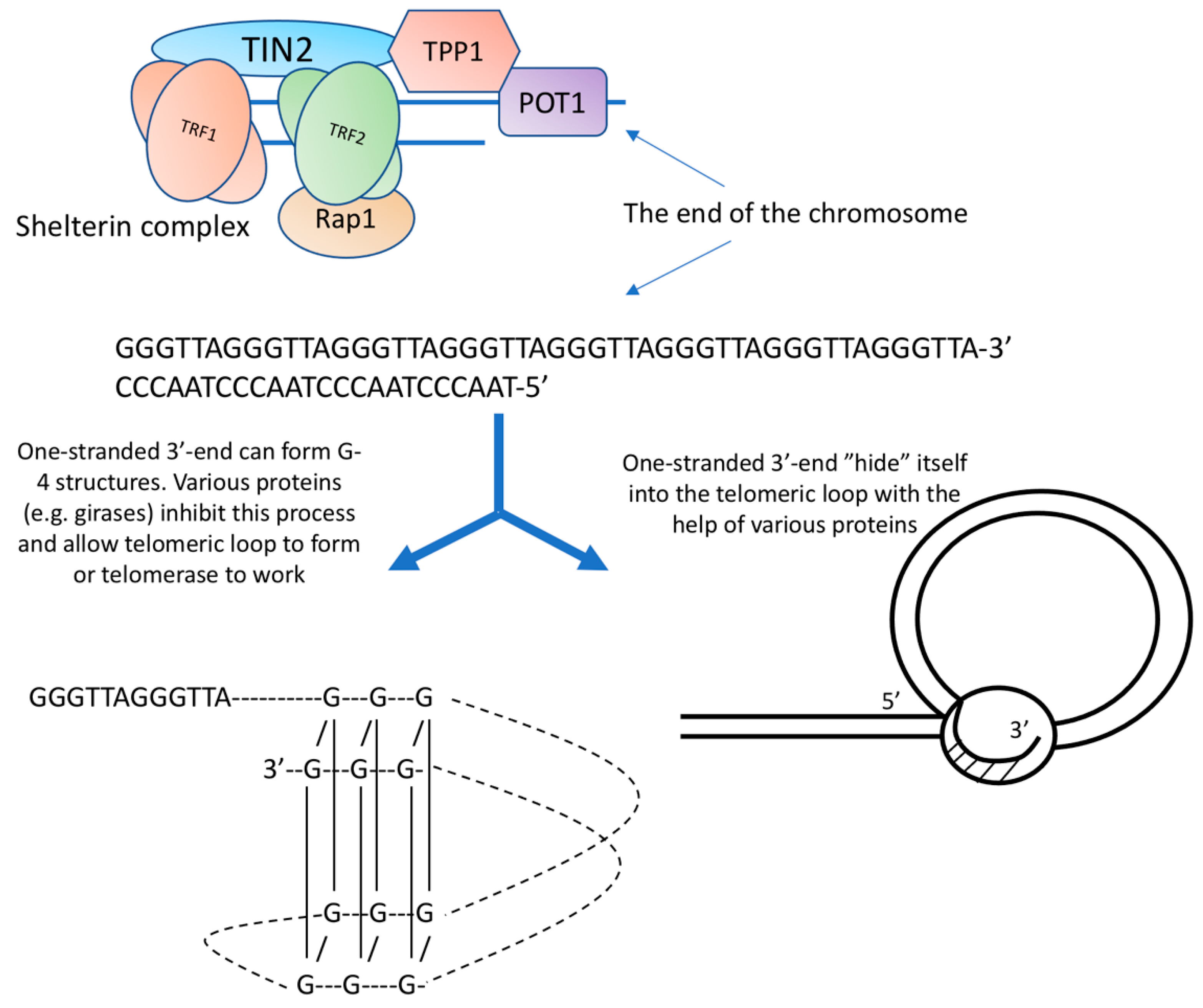
1. Telomeres
2. Telomerase
Non-Canonical Telomerase Activities
3. Cell Senescence
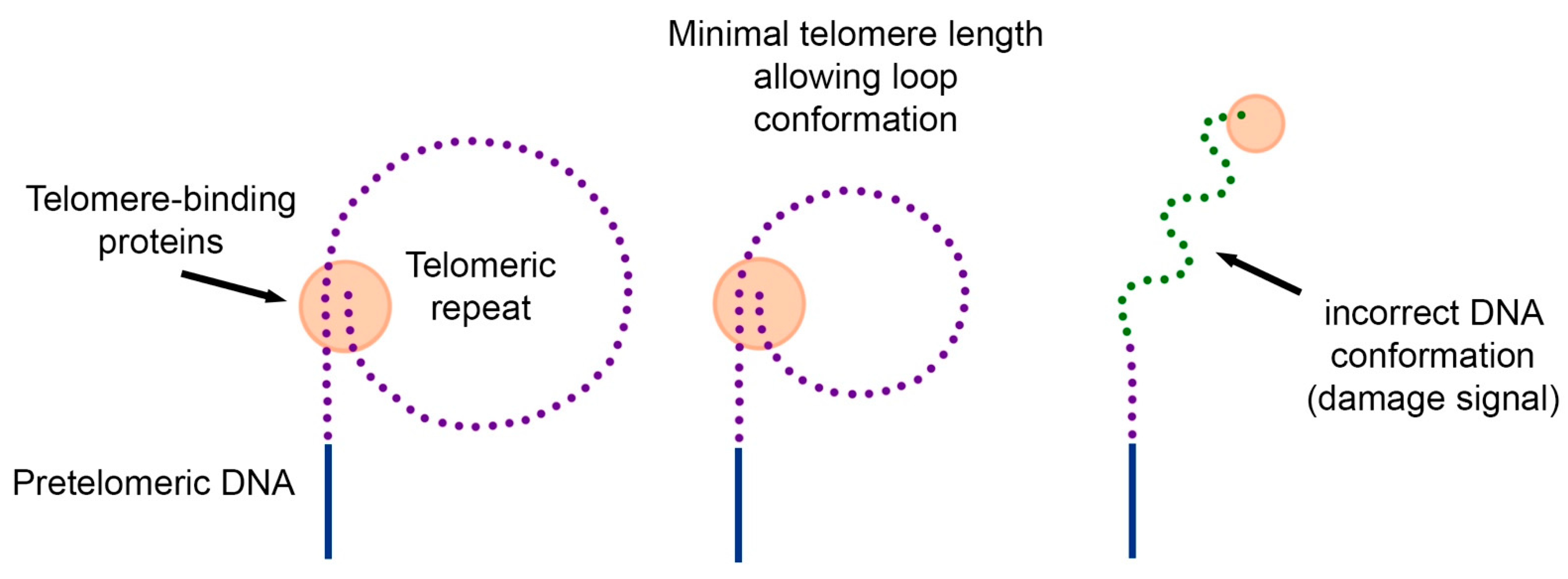
3.1. How Does a Cell Measure the Length of Its Telomeres?
3.2. DDR and SASP
4. Telomeres and Atherosclerosis
4.1. Atherosclerosis Is Associated with the Cell Senescence
4.2. Various Factors Affect the Length of Telomeres
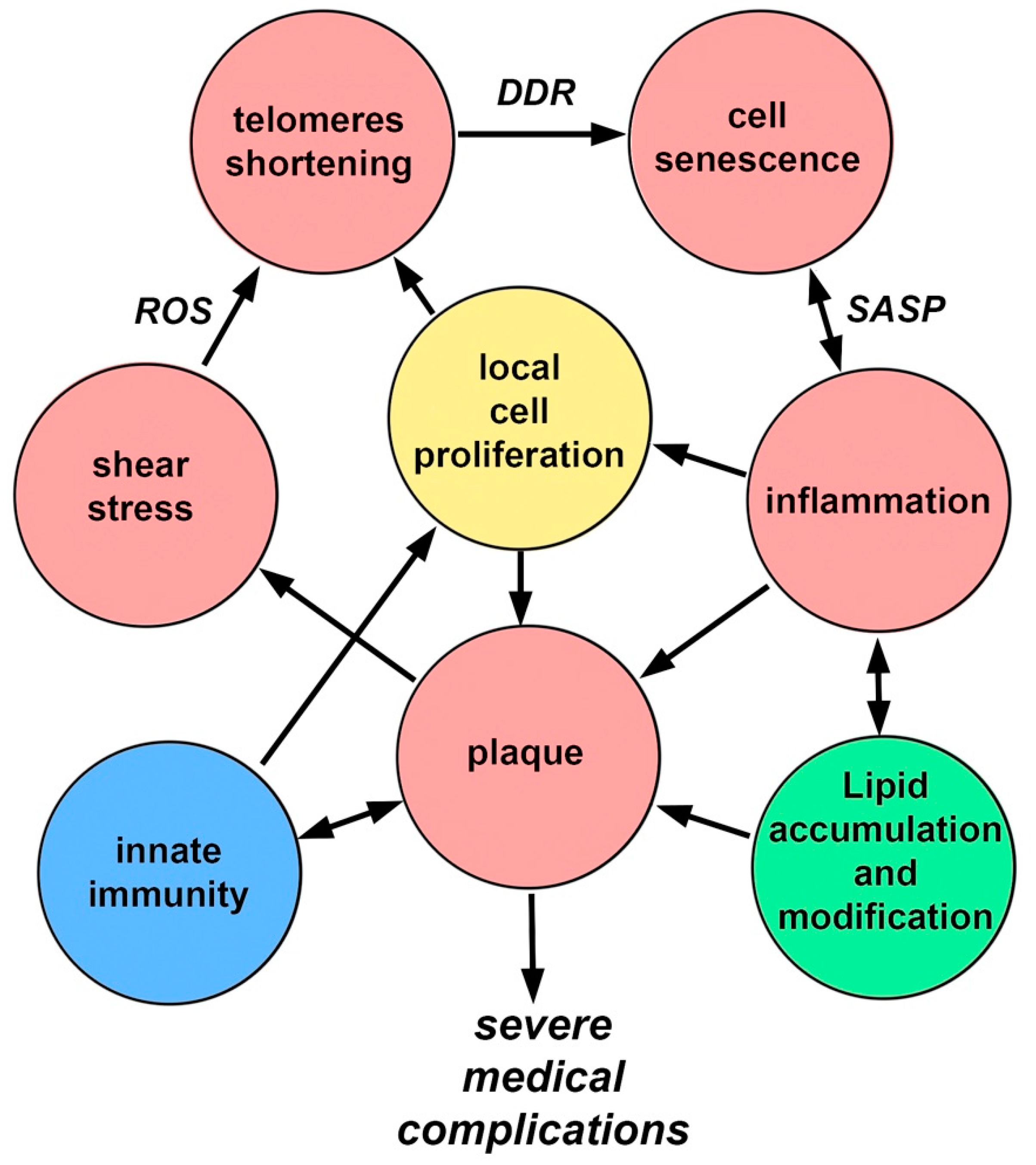
5. Pathogenetic Features of the Mechanisms Involved in the Development of Local Inflammation in Atherosclerosis
6. Consider Targeting Telomeres and Telomerase
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Doksani, Y. The Response to DNA Damage at Telomeric Repeats and Its Consequences for Telomere Function. Genes 2019, 10, 318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runnberg, R.; Narayanan, S.; Itriago, H.; Cohn, M. Either Rap1 or Cdc13 can protect telomeric single-stranded 3′ overhangs from degradation in vitro. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffith, J.D.; Comeau, L.; Rosenfield, S.; Stansel, R.M.; Bianchi, A.; Moss, H.; De Lange, T. Mammalian Telomeres End in a Large Duplex Loop. Cell 1999, 97, 503–514. [Google Scholar] [CrossRef] [Green Version]
- De Lange, T. Shelterin-Mediated Telomere Protection. Annu. Rev. Genet. 2018, 52, 223–247. [Google Scholar] [CrossRef]
- De Lange, T. What I got wrong about shelterin. J. Biol. Chem. 2018, 293, 10453–10456. [Google Scholar] [CrossRef] [Green Version]
- Smith, E.M.; Pendlebury, D.F.; Nandakumar, J. Structural biology of telomeres and telomerase. Cell. Mol. Life Sci. 2020, 77, 61–79. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Regulation of human telomerase in homeostasis and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Chang, G.; Wang, F.; Wang, F.; Geng, X. Alternative Splicing of hTERT Pre-mRNA: A Potential Strategy for the Regulation of Telomerase Activity. Int. J. Mol. Sci. 2017, 18, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeung, H.C.; Rha, S.Y.; Shin, S.J.; Ahn, J.B.; Park, K.H.; Kim, T.S.; Kim, J.J.; Roh, J.K.; Chung, H.C. Changes in telomerase activity due to alternative splicing of human telomerase reverse transcriptase in colorectal cancer. Oncol. Lett. 2017, 14, 2385–2392. [Google Scholar] [CrossRef] [Green Version]
- Ludlow, A.T.; Slusher, A.L.; Sayed, M.E. Insights into Telomerase/hTERT Alternative Splicing Regulation Using Bioinformatics and Network Analysis in Cancer. Cancers 2019, 11, 666. [Google Scholar] [CrossRef] [Green Version]
- Ramlee, M.K.; Wang, J.; Toh, W.X.; Li, S. Transcription Regulation of the Human Telomerase Reverse Transcriptase (hTERT) Gene. Genes 2016, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Abreu, E.; Terns, R.M.; Terns, M.P. Visualization of Human Telomerase Localization by Fluorescence Microscopy Techniques. Adv. Struct. Saf. Stud. 2017, 1587, 113–125. [Google Scholar] [CrossRef]
- Venteicher, A.S.; Artandi, S.E. TCAB1: Driving telomerase to Cajal bodies. Cell Cycle 2009, 8, 1329–1331. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, K.T.T.T.; Wong, J.M.Y. Telomerase Biogenesis and Activities from the Perspective of its Direct Interacting Partners. Cancers 2020, 12, 1679. [Google Scholar] [CrossRef]
- González-Suárez, E.; Samper, E.; Ramírez, A.; Flores, J.M.; Martín-Caballero, J.; Jorcano, J.L.; Blasco, M.A. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. EMBO J. 2001, 20, 2619–2630. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.-H.; Guo, Y.; Li, L.; Qu, S.; Zhao, W.; Lu, Q.-T.; Mo, Q.-Y.; Yu, B.-B.; Zhou, L.; Lin, G.-X.; et al. Cancer stem cell-like characteristics and telomerase activity of the nasopharyngeal carcinoma radioresistant cell line CNE -2R. Cancer Med. 2018, 7, 4755–4764. [Google Scholar] [CrossRef]
- Bagheri, S.; Nosrati, M.; Li, S.; Fong, S.; Torabian, S.; Rangel, J.; Moore, D.H.; Federman, S.; Laposa, R.R.; Baehner, F.L.; et al. Genes and pathways downstream of telomerase in melanoma metastasis. Proc. Natl. Acad. Sci. USA 2006, 103, 11306–11311. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Björkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial–mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2012, 32, 4203–4213. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.; Saginc, G.; Leow, S.C.; Khattar, E.; Shin, E.M.; Yan, T.D.; Wong, M.; Zhang, Z.; Li, G.; Sung, W.-K.; et al. Telomerase directly regulates NF-κB-dependent transcription. Nat. Cell Biol. 2012, 14, 1270–1281. [Google Scholar] [CrossRef]
- Chen, K.; Chen, L.; Li, L.; Qu, S.; Yu, B.; Sun, Y.; Wan, F.; Chen, X.; Liang, R.; Zhu, X. A positive feedback loop between Wnt/β-catenin signaling and hTERT regulates the cancer stem cell-like traits in radioresistant nasopharyngeal carcinoma cells. J. Cell. Biochem. 2020, 121, 4612–4622. [Google Scholar] [CrossRef] [PubMed]
- Rosen, J.; Jakobs, P.; Ale-Agha, N.; Altschmied, J.; Haendeler, J. Non-canonical functions of Telomerase Reverse Transcriptase–Impact on redox homeostasis. Redox Biol. 2020, 34, 101543. [Google Scholar] [CrossRef]
- Zhang, Q.; Wang, H.Y.; Woetmann, A.; Raghunath, P.N.; Odum, N.; Wasik, M.A. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in malignant T lymphocytes. Blood 2006, 108, 1058–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-dependent RNA polymerase formed by TERT and the RMRP RNA. Nat. Cell Biol. 2009, 461, 230–235. [Google Scholar] [CrossRef]
- Ridanpää, M.; Van Eenennaam, H.; Pelin, K.; Chadwick, R.; Johnson, C.; Yuan, B.; Vanvenrooij, W.; Pruijn, G.; Salmela, R.; Rockas, S.; et al. Mutations in the RNA Component of RNase MRP Cause a Pleiotropic Human Disease, Cartilage-Hair Hypoplasia. Cell 2001, 104, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Chen, Z.; Wong, K.-K.; Masutomi, K.; Hahn, W.C. RMRP Is a Non-Coding RNA Essential for Early Murine Development. PLOS ONE 2011, 6, e26270. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.K.; Reyes, A.; Green, P.; Caron, M.J.; Bonini, M.G.; Gordon, D.M.; Holt, I.J.; Santos, J.H. Human telomerase acts as a hTR-independent reverse transcriptase in mitochondria. Nucleic Acids Res. 2011, 40, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Walter, M.; Davies, J.P.; Ioannou, Y.A. Telomerase immortalization upregulates Rab9 expression and restores LDL cholesterol egress from Niemann-Pick C1 late endosomes. J. Lipid Res. 2003, 44, 243–253. [Google Scholar] [CrossRef] [Green Version]
- E Yegorov, Y.; Chernov, D.N.; Akimov, S.S.; Akhmalisheva, A.K.; Smirnova, Y.B.; Shinkarev, D.B.; Semenova, I.V.; Yegorova, I.N.; Zelenin, A.V. Blockade of telomerase function by nucleoside analogs. Biochemistry (Mosc.) 1997, 62, 1296–1305. [Google Scholar]
- Sławińska, N.; Krupa, R. Molecular Aspects of Senescence and Organismal Ageing—DNA Damage Response, Telomeres, Inflammation and Chromatin. Int. J. Mol. Sci. 2021, 22, 590. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.-L.; Ding, J.; Meng, L.-H. Oncogene-induced senescence: A double edged sword in cancer. Acta Pharmacol. Sin. 2018, 39, 1553–1558. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The senescence-associated secretory phenotype and its regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; Van Deursen, J.M. Cellular senescence in aging and age-related disease: From mechanisms to therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Childs, B.G.; Baker, D.J.; Wijshake, T.; Conover, C.A.; Campisi, J.; Van Deursen, J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 2016, 354, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Ikram, S.; Bibi, N.; Mir, A. Hutchinson–Gilford Progeria Syndrome: A Premature Aging Disease. Mol. Neurobiol. 2017, 55, 1–11. [Google Scholar] [CrossRef]
- Bonello-Palot, N.; Simoncini, S.; Robert, S.; Bourgeois, P.; Sabatier, F.; Levy, N.; Dignat-George, F.; Badens, C. Prelamin A accumulation in endothelial cells induces premature senescence and functional impairment. Atherosclerosis 2014, 237, 45–52. [Google Scholar] [CrossRef]
- Gonzalo, S.; Kreienkamp, R. DNA repair defects and genome instability in Hutchinson–Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 2015, 34, 75–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudlow, B.A.; Stanfel, M.N.; Burtner, C.R.; Johnston, E.D.; Kennedy, B.K. Suppression of Proliferative Defects Associated with Processing-defective Lamin A Mutants by hTERT or Inactivation of p53. Mol. Biol. Cell 2008, 19, 5238–5248. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.; Lim, J.S.Y.; A Mutalif, R.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin reduces LAP2α-telomere association in Hutchinson-Gilford progeria. eLife 2015, 4, e07759. [Google Scholar] [CrossRef]
- Okuda, K.; Khan, M.; Skurnick, J.; Kimura, M.; Aviv, H.; Aviv, A. Telomere attrition of the human abdominal aorta: Relationships with age and atherosclerosis. Atherosclerosis 2000, 152, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Ogami, M.; Ikura, Y.; Ohsawa, M.; Matsuo, T.; Kayo, S.; Yoshimi, N.; Hai, E.; Shirai, N.; Ehara, S.; Komatsu, R.; et al. Telomere Shortening in Human Coronary Artery Diseases. Arter. Thromb. Vasc. Biol. 2004, 24, 546–550. [Google Scholar] [CrossRef] [Green Version]
- Chang, E.; Harley, C.B. Telomere length and replicative aging in human vascular tissues. Proc. Natl. Acad. Sci. USA 1995, 92, 11190–11194. [Google Scholar] [CrossRef] [Green Version]
- Minamino, T.; Miyauchi, H.; Yoshida, T.; Ishida, Y.; Yoshida, H.; Komuro, I. Endothelial Cell Senescence in Human Atherosclerosis. Circulation 2002, 105, 1541–1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogendoorn, A.; Kok, A.M.; Hartman, E.M.J.; De Nisco, G.; Casadonte, L.; Chiastra, C.; Coenen, A.; Korteland, S.-A.; Van Der Heiden, K.; Gijsen, F.J.H.; et al. Multidirectional wall shear stress promotes advanced coronary plaque development: Comparing five shear stress metrics. Cardiovasc. Res. 2019, 116, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Baeyens, N.; Bandyopadhyay, C.; Coon, B.G.; Yun, S.; Schwartz, M.A. Endothelial fluid shear stress sensing in vascular health and disease. J. Clin. Investig. 2016, 126, 821–828. [Google Scholar] [CrossRef] [PubMed]
- McNally, J.S.; Davis, M.E.; Giddens, D.P.; Saha, A.; Hwang, J.; Dikalov, S.; Jo, H.; Harrison, D.G. Role of xanthine oxidoreductase and NAD(P)H oxidase in endothelial superoxide production in response to oscillatory shear stress. Am. J. Physiol. Circ. Physiol. 2003, 285, H2290–H2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouilette, S.; Singh, R.K.; Thompson, J.R.; Goodall, A.H.; Samani, N.J. White Cell Telomere Length and Risk of Premature Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2003, 23, 842–846. [Google Scholar] [CrossRef] [Green Version]
- Madrid, A.S.; Rode, L.; Nordestgaard, B.G.; Bojesen, S.E. Short Telomere Length and Ischemic Heart Disease: Observational and Genetic Studies in 290 022 Individuals. Clin. Chem. 2016, 62, 1140–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weischer, M.; Bojesen, S.E.; Cawthon, R.M.; Freiberg, J.J.; Tybjærg-Hansen, A.; Nordestgaard, B.G. Short Telomere Length, Myocardial Infarction, Ischemic Heart Disease, and Early Death. Arter. Thromb. Vasc. Biol. 2012, 32, 822–829. [Google Scholar] [CrossRef] [Green Version]
- Haycock, P.C.; E Heydon, E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Hu, H.; Lin, Y.; Huang, F.; Ji, H.; Li, Y.; Lin, S.; Chen, X.; Duan, S. Differences in Leukocyte Telomere Length between Coronary Heart Disease and Normal Population: A Multipopulation Meta-Analysis. BioMed Res. Int. 2019, 2019, 5046867. [Google Scholar] [CrossRef]
- Wang, X.-B.; Cui, N.-H.; Zhang, S.; Liu, Z.-J.; Ma, J.-F.; Ming, L. Leukocyte telomere length, mitochondrial DNA copy number, and coronary artery disease risk and severity: A two-stage case-control study of 3064 Chinese subjects. Atherosclerosis 2019, 284, 165–172. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, S.; Jiao, F.; Kong, Q.; Liu, C.; Wu, Y. Telomere Length: A Potential Biomarker for the Risk and Prognosis of Stroke. Front. Neurol. 2019, 10, 624. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Palumbo, L.; Fornengo, C.; Di Gaetano, C.; Ricceri, F.; Guarrera, S.; Critelli, R.; Anselmino, M.; Piazza, A.; Gaita, F.; et al. Telomere Length Variation in Juvenile Acute Myocardial Infarction. PLOS ONE 2012, 7, e49206. [Google Scholar] [CrossRef]
- Vasan, R.S.; Demissie, S.; Kimura, M.; Cupples, L.A.; White, C.; Gardner, J.P.; Cao, X.; Levy, D.; Benjamin, E.J.; Aviv, A. Association of Leukocyte Telomere Length With Echocardiographic Left Ventricular Mass. Circulation 2009, 120, 1195–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuznetsova, T.; Codd, V.; Brouilette, S.; Thijs, L.; González, A.; Jin, Y.; Richart, T.; Van Der Harst, P.; Díez, J.; Staessen, J.A.; et al. Association Between Left Ventricular Mass and Telomere Length in a Population Study. Am. J. Epidemiol. 2010, 172, 440–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, G.; Sun, J.; Zhang, L.; Li, R.; Wang, Y.; Cianflone, K.; Ding, H.; Wang, D.W. Lack of causal relationship between leukocyte telomere length and coronary heart disease. Atherosclerosis 2014, 233, 375–380. [Google Scholar] [CrossRef]
- Toupance, S.; Labat, C.; Temmar, M.; Rossignol, P.; Kimura, M.; Aviv, A.; Benetos, A. Short Telomeres, but Not Telomere Attrition Rates, Are Associated With Carotid Atherosclerosis. Hypertension 2017, 70, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Bateson, M.; Aviv, A.; Bendix, L.; Benetos, A.; Ben-Shlomo, Y.; Bojesen, S.E.; Cooper, C.; Cooper, R.; Deary, I.J.; Hägg, S.; et al. Smoking does not accelerate leucocyte telomere attrition: A meta-analysis of 18 longitudinal cohorts. R. Soc. Open Sci. 2019, 6, 190420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharifi-Sanjani, M.; Oyster, N.M.; Tichy, E.D.; Bedi, K.C.; Harel, O.; Margulies, K.B.; Mourkioti, F. Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. J. Am. Hear. Assoc. 2017, 6, e005086. [Google Scholar] [CrossRef] [Green Version]
- Verhulst, S.; Dalgård, C.; Labat, C.; Kark, J.D.; Kimura, M.; Christensen, K.; Toupance, S.; Aviv, A.; Kyvik, K.O.; Benetos, A. A short leucocyte telomere length is associated with development of insulin resistance. Diabetologia 2016, 59, 1258–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Mello, M.J.; Ross, S.A.; Briel, M.; Anand, S.S.; Gerstein, H.; Paré, G. Association Between Shortened Leukocyte Telomere Length and Cardiometabolic Outcomes. Circ. Cardiovasc. Genet. 2015, 8, 82–90. [Google Scholar] [CrossRef]
- Rehkopf, D.H.; Needham, B.L.; Lin, J.; Blackburn, E.H.; Zota, A.R.; Wojcicki, J.M.; Epel, E.S. Leukocyte Telomere Length in Relation to 17 Biomarkers of Cardiovascular Disease Risk: A Cross-Sectional Study of US Adults. PLoS Med. 2016, 13, e1002188. [Google Scholar] [CrossRef]
- Aviv, A.; Levy, D. Hemothelium, Clonal Hematopoiesis of Indeterminate Potential, and Atherosclerosis. Circulation 2019, 139, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Hjelmborg, J.B.; Dalgård, C.; Möller, S.; Steenstrup, T.; Kimura, M.; Christensen, K.; O Kyvik, K.; Aviv, A. The heritability of leucocyte telomere length dynamics. J. Med. Genet. 2015, 52, 297–302. [Google Scholar] [CrossRef] [Green Version]
- Benetos, A.; Verhulst, S.; Labat, C.; Lai, T.-P.; Girerd, N.; Toupance, S.; Zannad, F.; Rossignol, P.; Aviv, A. Telomere length tracking in children and their parents: Implications for adult onset diseases. FASEB J. 2019, 33, 14248–14253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benetos, A.; Kark, J.D.; Susser, E.; Kimura, M.; Sinnreich, R.; Chen, W.; Steenstrup, T.; Christensen, K.; Herbig, U.; Hjelmborg, J.V.B.; et al. Tracking and fixed ranking of leukocyte telomere length across the adult life course. Aging Cell 2013, 12, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Broer, L.; Codd, V.; Nyholt, D.R.; Deelen, J.; Mangino, M.; Willemsen, G.; Albrecht, E.; Amin, N.; Beekman, M.; De Geus, E.J.; et al. Meta-analysis of telomere length in 19 713 subjects reveals high heritability, stronger maternal inheritance and a paternal age effect. Eur. J. Hum. Genet. 2013, 21, 1163–1168. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.; Cai, G.; Xia, Y.; Chen, J.; Wu, P.; Wang, Z.; Li, G.; Wei, D. Mitochondrial Dysfunction in Atherosclerosis. DNA Cell Biol. 2019, 38, 597–606. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Shkurat, T.P.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. The role of mitochondrial dysfunction in cardiovascular disease: A brief review. Ann. Med. 2018, 50, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Volobueva, A.; Grechko, A.; Yet, S.-F.; Sobenin, I.; Orekhov, A. Changes in Mitochondrial Genome Associated with Predisposition to Atherosclerosis and Related Disease. Biomolecules 2019, 9, 377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniali, L.; Benetos, A.; Susser, E.; Kark, J.D.; Labat, C.; Kimura, M.; Desai, K.K.; Granick, M.; Aviv, A. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 2013, 4, 1597. [Google Scholar] [CrossRef]
- Aubert, G.; Baerlocher, G.M.; Vulto, I.; Poon, S.S.; Lansdorp, P.M. Collapse of Telomere Homeostasis in Hematopoietic Cells Caused by Heterozygous Mutations in Telomerase Genes. PLoS Genet. 2012, 8, e1002696. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Akawi, O.; Fox, S.A.; Li, F.; O’Neil, C.; Balint, B.; Arpino, J.-M.; Watson, A.; Wong, J.; Guo, L.; et al. Cardiac-Referenced Leukocyte Telomere Length and Outcomes After Cardiovascular Surgery. JACC Basic Transl. Sci. 2018, 3, 591–600. [Google Scholar] [CrossRef]
- Hwang, S.M.; Kim, S.Y.; Kim, J.A.; Park, H.-S.; Park, S.N.; Im, K.; Kim, K.; Kim, S.-M.; Lee, D.S. Short telomere length and its correlation with gene mutations in myelodysplastic syndrome. J. Hematol. Oncol. 2016, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ebert, B.L. CHIP (Clonal Hematopoiesis of Indeterminate Potential). Circulation 2018, 138, 666–668. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and Cardiac Aging: Major Shareholders in Cardiovascular Disease Enterprises. Circulation 2003, 107, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Vangaveti, V.; Baune, B.T.; Kennedy, R.L. Review: Hydroxyoctadecadienoic acids: Novel regulators of macrophage differentiation and atherogenesis. Ther. Adv. Endocrinol. Metab. 2010, 1, 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochkov, V.; Gesslbauer, B.; Mauerhofer, C.; Philippova, M.; Erne, P.; Oskolkova, O.V. Pleiotropic effects of oxidized phospholipids. Free. Radic. Biol. Med. 2017, 111, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, J.; Jiang, L.-Q.; Spinetti, G.; Pintus, G.; Monticone, R.; Kolodgie, F.D.; Virmani, R.; Lakatta, E.G. Proinflammatory Profile Within the Grossly Normal Aged Human Aortic Wall. Hypertension 2007, 50, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Donato, A.J.; Morgan, R.G.; Walker, A.E.; Lesniewski, L.A. Cellular and molecular biology of aging endothelial cells. J. Mol. Cell. Cardiol. 2015, 89, 122–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Liu, H.; Lian, G.; Zhang, S.-Y.; Wang, X.; Jiang, C. HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediat. Inflamm. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Vecoli, C.; Borghini, A.; Andreassi, M.G. The molecular biomarkers of vascular aging and atherosclerosis: Telomere length and mitochondrial DNA4977 common deletion. Mutat. Res. Mutat. Res. 2020, 784, 108309. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Van Tuijl, J.; Joosten, L.A.B.; Netea, M.G.; Bekkering, S.; Riksen, N.P. Immunometabolism orchestrates training of innate immunity in atherosclerosis. Cardiovasc. Res. 2019, 115, 1416–1424. [Google Scholar] [CrossRef]
- Ungvari, Z.; Tarantini, S.; Donato, A.J.; Galvan, V.; Csiszar, A. Mechanisms of Vascular Aging. Circ. Res. 2018, 123, 849–867. [Google Scholar] [CrossRef]
- Davidson, S.M.; Yellon, D.M. Mitochondrial DNA damage, oxidative stress, and atherosclerosis: Where there is smoke there is not always fire. Circulation 2013, 128, 681–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, L.A. Milk Fat Intake and Telomere Length in U.S. Women and Men: The Role of the Milk Fat Fraction. Oxidative Med. Cell. Longev. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.; Poznyak, A.; Nikiforov, N.; Sobenin, I.; Orekhov, A. The Link between Chronic Stress and Accelerated Aging. Biomedicines 2020, 8, 198. [Google Scholar] [CrossRef]
- Nazari-Shafti, T.Z.; Cooke, J.P. Telomerase Therapy to Reverse Cardiovascular Senescence. Methodist DeBakey Cardiovasc. J. 2015, 11, 172–175. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Bloom, S.I.; Donato, A.J. The role of senescence, telomere dysfunction and shelterin in vascular aging. Microcirculation 2019, 26, e12487. [Google Scholar] [CrossRef] [PubMed]
- Matthews, C.; Gorenne, I.; Scott, S.; Figg, N.; Kirkpatrick, P.; Ritchie, A.; Goddard, M.; Bennett, M. Vascular Smooth Muscle Cells Undergo Telomere-Based Senescence in Human Atherosclerosis. Circ. Res. 2006, 99, 156–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bressler, J.; Franceschini, N.; Demerath, E.W.; Mosley, T.H.; Folsom, A.R.; Boerwinkle, E. Sequence variation in telomerase reverse transcriptase (TERT) as a determinant of risk of cardiovascular disease: The Atherosclerosis Risk in Communities (ARIC) study. BMC Med. Genet. 2015, 16, 52. [Google Scholar] [CrossRef] [Green Version]
- Morales, C.P.; Holt, S.E.; Ouellette, M.; Kaur, K.J.; Yan, Y.; Wilson, K.S.; White, M.A.; Wright, W.E.; Shay, J.W. Absence of cancer–associated changes in human fibroblasts immortalized with telomerase. Nat. Genet. 1999, 21, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Yegorov, Y.E.; Moldaver, M.V.; Vishnyakova, K.S.; Terekhov, S.M.; Dashinimaev, E.B.; Cheglakov, I.B.; Toropygin, I.Y.; Yarygin, K.N.; Chumakov, P.M.; Korochkin, L.I.; et al. Enhanced control of proliferation in telomerized cells. Russ. J. Dev. Biol. 2007, 38, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Sidorov, I.A.; Hirsch, K.S.; Harley, C.B.; Dimitrov, D.S. Cancer Cell Dynamics in Presence of Telomerase Inhibitors: Analysis of In Vitro Data. J. Theor. Biol. 2002, 219, 225–233. [Google Scholar] [CrossRef]
- Hiyama, E.; Hiyama, K. Telomerase as tumor marker. Cancer Lett. 2003, 194, 221–233. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Hiyama, E.; Hiyama, K.; Yokoyama, T.; Matsuura, Y.; Piatyszek, M.A.; Shay, J.W. Correlating telomerase activity levels with human neuroblastoma outcomes. Nat. Med. 1995, 1, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Hiyama, E.; Kodama, T.; Shinbara, K.; Iwao, T.; Itoh, M.; Hiyama, K.; Shay, J.W.; Matsuura, Y.; Yokoyama, T. Telomerase activity is detected in pancreatic cancer but not in benign tumors. Cancer Res. 1997, 57, 9000577. [Google Scholar]
- Naito, Y.; Takagi, T.; Handa, O.; Ishikawa, T.; Matsumoto, N.; Yoshida, N.; Kato, H.; Ando, T.; Takemura, T.; Itani, K.; et al. Telomerase Activity and Expression of Telomerase RNA Component and Catalytic Subunits in Precancerous and Cancerous Colorectal Lesions. Tumor Biol. 2001, 22, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, M.M.; Liao, M.; Herbert, B.-S.; Johnson, M.; Holt, S.E.; Liss, H.S.; Shay, J.W.; Wright, W.E. Subsenescent Telomere Lengths in Fibroblasts Immortalized by Limiting Amounts of Telomerase. J. Biol. Chem. 2000, 275, 10072–10076. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Madu, C.O.; Lu, Y. Telomere and Its Role in Diseases. Oncomedicine 2019, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- De Jesus, B.B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef]
- De Jesus, B.B.; Schneeberger, K.; Vera, E.; Tejera, A.; Harley, C.B.; Blasco, M.A. The telomerase activator TA-65 elongates short telomeres and increases health span of adult/old mice without increasing cancer incidence. Aging Cell 2011, 10, 604–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez, M.L.; Thomas, M.S.; Lemos, B.S.; DiMarco, D.M.; Missimer, A.; Melough, M.; Chun, O.K.; Murillo, A.G.; Alyousef, H.M.; Medina-Vera, I. TA-65, A Telomerase Activator improves Cardiovascular Markers in Patients with Metabolic Syndrome. Curr. Pharm. Des. 2018, 24, 1905–1911. [Google Scholar] [CrossRef]
- Dookun, E.; Passos, J.F.; Arthur, H.M.; Richardson, G.D. Therapeutic Potential of Senolytics in Cardiovascular Disease. Cardiovasc. Drugs Ther. 2020, 1–10. [Google Scholar] [CrossRef]
- Makpol, S.; Durani, L.W.; Chua, K.H.; Yusof, Y.A.M.; Ngah, W.Z.W. Tocotrienol-Rich Fraction Prevents Cell Cycle Arrest and Elongates Telomere Length in Senescent Human Diploid Fibroblasts. J. Biomed. Biotechnol. 2011, 2011, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nzietchueng, R.; Elfarra, M.; Nloga, J.; Labat, C.; Carteaux, J.; Maureira, P.; Lacolley, P.; Villemot, J.; Benetos, A. Telomere length in vascular tissues from patients with atherosclerotic disease. J. Nutr. Health Aging 2011, 15, 153–156. [Google Scholar] [CrossRef]
- Panayiotou, A.; Nicolaides, A.; Griffin, M.; Tyllis, T.; Georgiou, N.; Bond, D.; Martin, R.; Hoppensteadt, D.; Fareed, J.; E Humphries, S. Leukocyte telomere length is associated with measures of subclinical atherosclerosis. Atherosclerosis 2010, 211, 176–181. [Google Scholar] [CrossRef]
- Shen, X.-H.; Xu, S.-J.; Jin, C.-Y.; Ding, F.; Zhou, Y.-C.; Fu, G.-S. Interleukin-8 prevents oxidative stress-induced human endothelial cell senescence via telomerase activation. Int. Immunopharmacol. 2013, 16, 261–267. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Ramunas, J.; Yakubov, E.; Brady, J.J.; Corbel, S.Y.; Holbrook, C.; Brandt, M.; Stein, J.; Santiago, J.G.; Cooke, J.P.; Blau, H.M. Transient delivery of modified mRNA encoding TERT rapidly extends telomeres in human cells. FASEB J. 2015, 29, 1930–1939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parodi, A.; Quattrocchi, N.; Van De Ven, A.L.; Chiappini, C.; Evangelopoulos, M.; Martinez, J.O.; Brown, B.S.; Khaled, S.Z.; Yazdi, I.K.; Enzo, M.V.; et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013, 8, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; E Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Martin-Ruiz, C.; Hoffmann, J.; Shmeleva, E.; Von Zglinicki, T.; Richardson, G.; Draganova, L.; Redgrave, R.; Collerton, J.; Arthur, H.; Keavney, B.; et al. CMV-independent increase in CD27−CD28+ CD8+ EMRA T cells is inversely related to mortality in octogenarians. npj Aging Mech. Dis. 2020, 6, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguado, J.; Sola-Carvajal, A.; Cancila, V.; Revêchon, G.; Ong, P.F.; Jones-Weinert, C.W.; Arzt, E.W.; Lattanzi, G.; Dreesen, O.; Tripodo, C.; et al. Inhibition of DNA damage response at telomeres improves the detrimental phenotypes of Hutchinson–Gilford Progeria Syndrome. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Yafit, H.; Amir, H.; Ramzia, A.H.; Malka, D.-K.; Merav, C.; Gregory, F.; Erez, L.; Nir, P.; Keren, D.; Mony, F.; et al. Hyperbaric oxygen therapy increases telomere length and decreases immunosenescence in isolated blood cells: A prospective trial. Aging 2020, 12, 22445–22456. [Google Scholar] [CrossRef]
- Amir, H.; Shai, E. The Hyperoxic-Hypoxic Paradox. Biomolecules 2020, 10, 958. [Google Scholar] [CrossRef]
- Gutsaeva, D.; Suliman, H.; Carraway, M.; Demchenko, I.; Piantadosi, C. Oxygen-induced mitochondrial biogenesis in the rat hippocampus. Neuroscience 2006, 137, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, A. Mild hyperbaric oxygen: Mechanisms and effects. J. Physiol. Sci. 2019, 69, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Gardin, C.; Bosco, G.; Ferroni, L.; Quartesan, S.; Rizzato, A.; Tatullo, M.; Zavan, B. Hyperbaric Oxygen Therapy Improves the Osteogenic and Vasculogenic Properties of Mesenchymal Stem Cells in the Presence of Inflammation In Vitro. Int. J. Mol. Sci. 2020, 21, 1452. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Sample Size | Reference |
|---|---|---|
| Terminal restriction fragment using Southern analysis | 51 subjects (23 males and 28 females, 37 White Americans, 14 African Americans) between ages of 1 month and 80 years. | [43] |
| Terminal restriction fragment using Southern analysis | 11 patients with CAD and 22 patients without CAD | [44] |
| Terminal restriction fragment using Southern analysis | 13 patients | [45] |
| Terminal restriction fragment using Southern analysis | 203 cases with a premature MI (50 years) and 180 controls | [51] |
| Modified monochrome multiplex quantitative PCR method | 105,055 individuals from Copenhagen (17,235 were diagnosed with ischemic heart disease between 1977 and 2013) Coronary ARtery DIsease Genome wide Replication and Meta-analysis (CARDIoGRAM) consortium dataset (184,967 participants, 60,837 cases of ischemic heart disease) | [52] |
| Modified monochrome multiplex quantitative polymerase chain reaction method | 19,838 Danish general population participants | [53] |
| qPCR | 1511 CAD patients; 1553 control | [56] |
| quantitative PCR-based method | 199 patients from 18 to 48 years old with first diagnosis of acute myocardial infarction; 190 control | [58] |
| Southern blot analysis | 850 Framingham Heart Study participants (mean age 58 years, 58% women) | [59] |
| quantitative polymerase chain reaction | 334 randomly selected Flemish participants (mean age 1⁄4 46.5 years; 52.5% women) | [60] |
| quantitative polymerase chain reaction (qPCR)-based assay | 2211 healthy individuals and 2140 CHD patients | [61] |
| Southern blots | 154 French men and women (aged 31–76 years at baseline) | [62] |
| Q-FISH | 63 samples | [64] |
| Southern blots | 756 intact twins pairs | [65] |
| quantitative PCR | 9191 participants aged 20–84 | [67] |
| Southern blots of the terminal restriction fragments (TRFs) | 1156 adult (44% women) | [71] |
| Terminal restriction fragment using Southern analysis | 87 adults (aged 19–77 years) | [76] |
| FISH | 835 healthy individuals and 60 individuals with reduced telomerase activity | [77] |
| qPCR | 163 patients who underwent cardiac surgery | [78] |
| quantitative fluorescence in situ hybridization | 58 patients with myelodysplastic syndrome | [79] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yegorov, Y.E.; Poznyak, A.V.; Nikiforov, N.G.; Starodubova, A.V.; Orekhov, A.N. Role of Telomeres Shortening in Atherogenesis: An Overview. Cells 2021, 10, 395. https://doi.org/10.3390/cells10020395
Yegorov YE, Poznyak AV, Nikiforov NG, Starodubova AV, Orekhov AN. Role of Telomeres Shortening in Atherogenesis: An Overview. Cells. 2021; 10(2):395. https://doi.org/10.3390/cells10020395
Chicago/Turabian StyleYegorov, Yegor E., Anastasia V. Poznyak, Nikita G. Nikiforov, Antonina V. Starodubova, and Alexander N. Orekhov. 2021. "Role of Telomeres Shortening in Atherogenesis: An Overview" Cells 10, no. 2: 395. https://doi.org/10.3390/cells10020395
APA StyleYegorov, Y. E., Poznyak, A. V., Nikiforov, N. G., Starodubova, A. V., & Orekhov, A. N. (2021). Role of Telomeres Shortening in Atherogenesis: An Overview. Cells, 10(2), 395. https://doi.org/10.3390/cells10020395