
Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe?



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Neuroinflammation in the Pathogenesis of ALS
3. Tumor Necrosis Factor Alpha
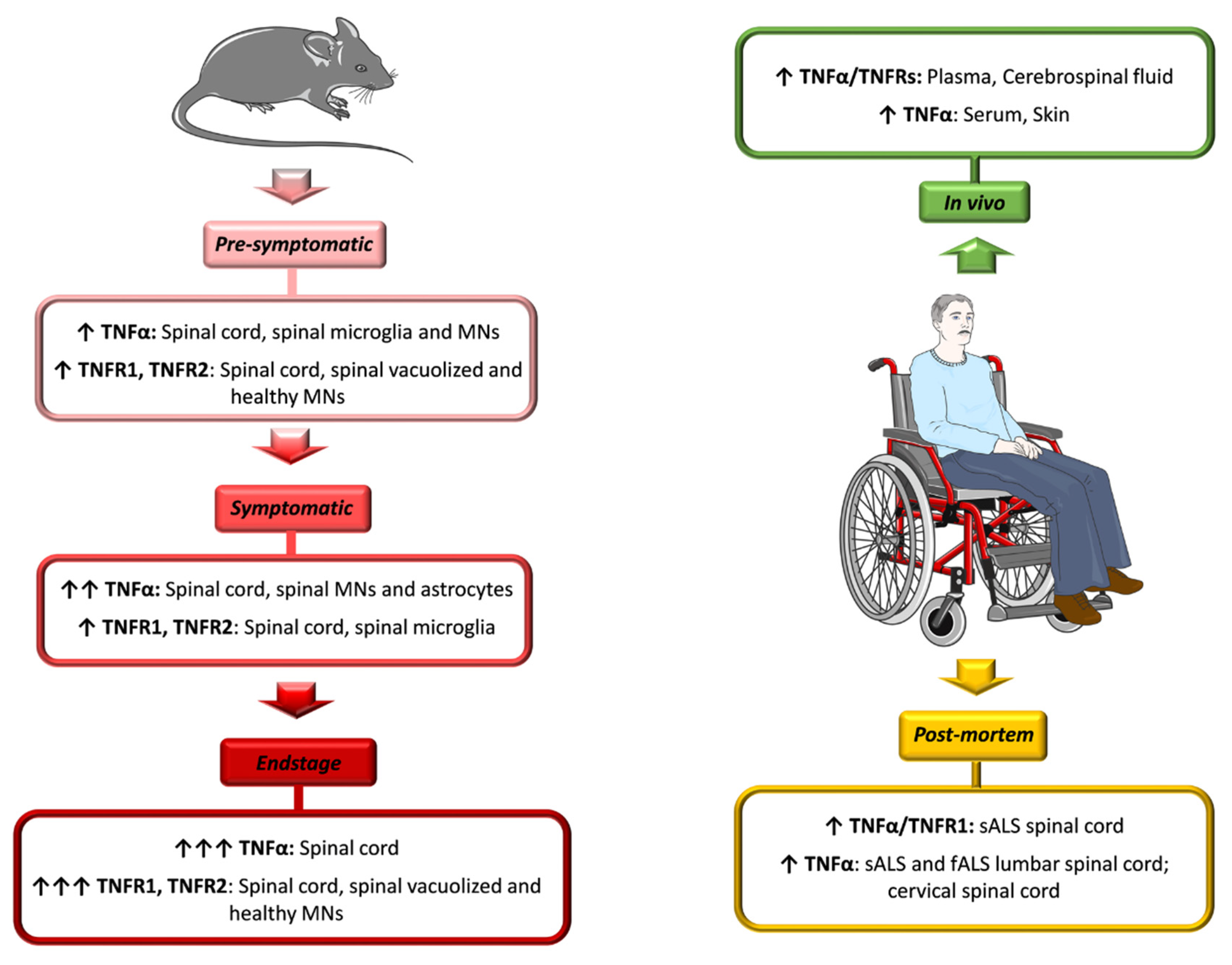
4. TNFα Role in ALS: Evidence from Patients and Animal Models
4.1. ALS Patients
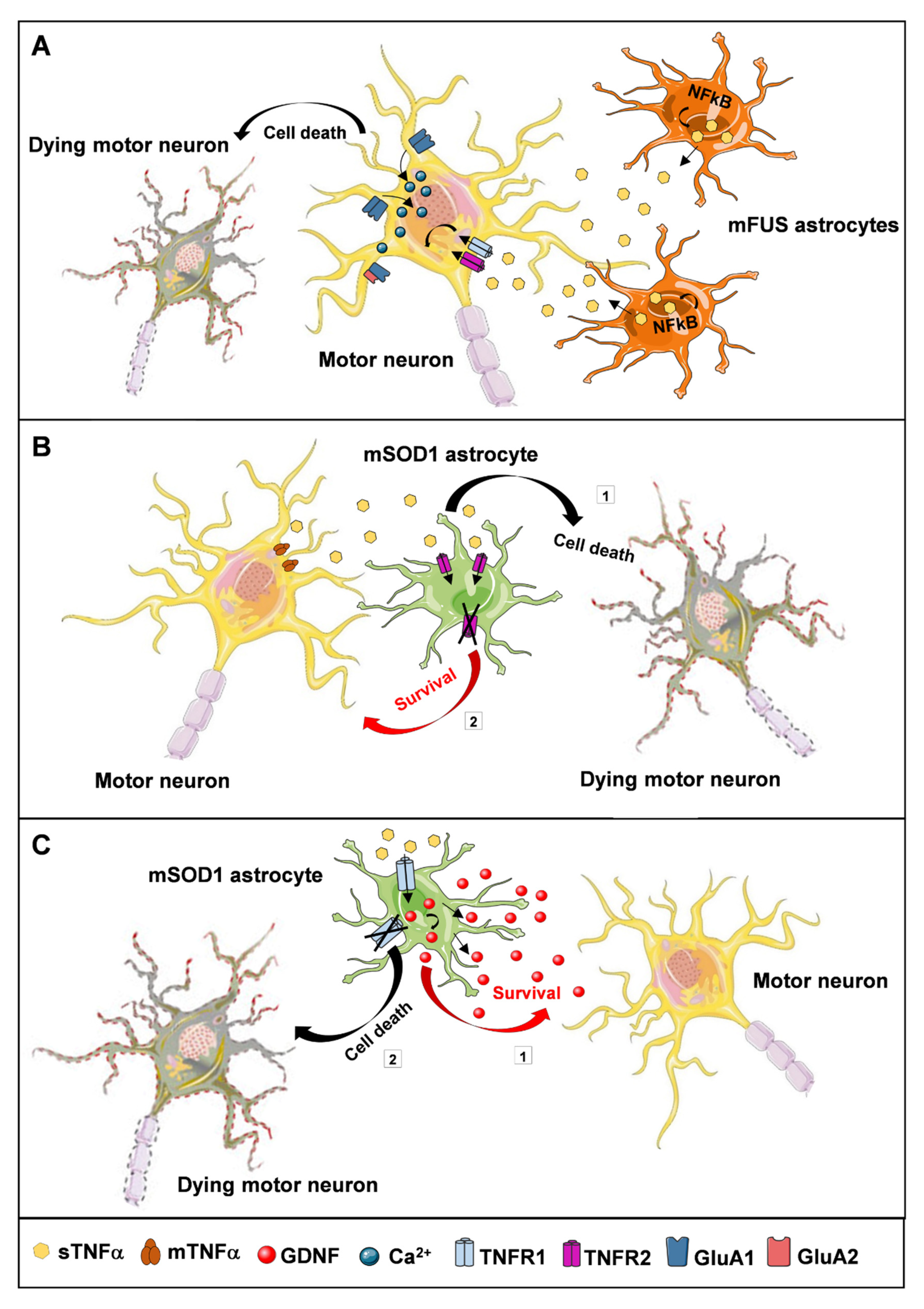
4.2. ALS Animal Models
5. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D. Astrocyte physiopathology: At the crossroads of intercellular networking, inflammation and cell death. Prog. Neurobiol. 2015, 130, 86–120. [Google Scholar] [CrossRef] [PubMed]
- Valori, C.F.; Guidotti, G.; Brambilla, L.; Rossi, D. Astrocytes in Motor Neuron Diseases. Adv. Exp. Med. Biol. 2019, 1175, 227–272. [Google Scholar] [CrossRef]
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Berg, L.H.V.D. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, nrdp201771-17071. [Google Scholar] [CrossRef]
- A van Es, M.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; Berg, L.H.V.D. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Murphy, J.; Factor-Litvak, P.; Goetz, R.R.; Lomen-Hoerth, C.; Nagy, P.L.; Hupf, J.; Singleton, J.; Woolley, S.; Andrews, J.A.; Heitzman, D.; et al. Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 2016, 86, 813–820. [Google Scholar] [CrossRef] [Green Version]
- Trojsi, F.; Siciliano, M.; Femiano, C.; Santangelo, G.; Lunetta, C.; Calvo, A.; Moglia, C.; Marinou, K.; Ticozzi, N.; Ferrante, G.D.; et al. Comorbidity of dementia with amyotrophic lateral sclerosis (ALS): Insights from a large multicenter Italian cohort. J. Neurol. 2017, 264, 2224–2231. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Della Gatta, P.; Foletta, V.; Russell, A. The Role of Exercise as a Non-pharmacological Therapeutic Approach for Amyotrophic Lateral Sclerosis: Beneficial or Detrimental? Front. Neurol. 2019, 10, 783. [Google Scholar] [CrossRef] [Green Version]
- Lunetta, C.; Lizio, A.; Sansone, V.A.; Cellotto, N.M.; Maestri, E.; Bettinelli, M.; Gatti, V.; Melazzini, M.G.; Meola, G.; Corbo, M. Strictly monitored exercise programs reduce motor deterioration in ALS: Preliminary results of a randomized controlled trial. J. Neurol. 2016, 263, 52–60. [Google Scholar] [CrossRef]
- Merico, A.; Cavinato, M.; Gregorio, C.; Lacatena, A.; Gioia, E.; Piccione, F.; Angelini, C. Effects of combined endurance and resistance training in Amyotrophic Lateral Sclerosis: A pilot, randomized, controlled study. Eur. J. Transl. Myol. 2018, 28, 7278. [Google Scholar] [CrossRef]
- Jensen, L.; Djurtoft, J.B.; Bech, R.D.; Nielsen, J.L.; Jørgensen, L.H.; Schrøder, H.D.; Frandsen, U.; Aagaard, P.; Hvid, L.G. Influence of Resistance Training on Neuromuscular Function and Physical Capacity in ALS Patients. J. Neurodegener. Dis. 2017, 2017, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, T.; Al Khleifat, A.; Meurgey, J.-H.; Jones, A.; Leigh, P.N.; Bensimon, G.; Al-Chalabi, A. Stage at which riluzole treatment prolongs survival in patients with amyotrophic lateral sclerosis: A retrospective analysis of data from a dose-ranging study. Lancet Neurol. 2018, 17, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: A randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P., Jr.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nat. Cell Biol. 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, M.R.; Hardiman, O.; Benatar, M.; Brooks, B.R.; Chio, A.; De Carvalho, M.; Ince, P.G.; Lin, C.; Miller, R.G.; Mitsumoto, H.; et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013, 12, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Pegoraro, V.; Merico, A.; Angelini, C. Micro-RNAs in ALS muscle: Differences in gender, age at onset and disease duration. J. Neurol. Sci. 2017, 380, 58–63. [Google Scholar] [CrossRef] [Green Version]
- Tasca, E.; Pegoraro, V.; Merico, A.; Angelini, C. Circulating microRNAs as biomarkers of muscle differentiation and atrophy in ALS. Clin. Neuropathol. 2016, 35, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Pegoraro, V.; Marozzo, R.; Angelini, C. MicroRNAs and HDAC4 protein expression in the skeletal muscle of ALS patients. Clin. Neuropathol. 2020, 39, 105–114. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammatory processes in amyotrophic lateral sclerosis. Muscle Nerve 2002, 26, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Clement, A.M. Wild-Type Nonneuronal Cells Extend Survival of SOD1 Mutant Motor Neurons in ALS Mice. Science 2003, 302, 113–117. [Google Scholar] [CrossRef] [Green Version]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valori, C.F.; Brambilla, L.; Martorana, F.; Rossi, D. The multifaceted role of glial cells in amyotrophic lateral sclerosis. Cell. Mol. Life Sci. 2013, 71, 287–297. [Google Scholar] [CrossRef]
- Ghezzi, P.; Bernardini, R.; Giuffrida, R.; Bellomo, M.; Manzoni, C.; Comoletti, D.; Di Santo, E.; Benigni, F.; Mennini, T. Tumor necrosis factor is increased in the spinal cord of an animal model of motor neuron degeneration. Eur. Cytokine Netw. 1998, 9, 139–144. [Google Scholar]
- Poloni, M.; Facchetti, D.; Mai, R.; Micheli, A.; Agnoletti, L.; Francolini, G.; Mora, G.; Camana, C.; Mazzini, L.; Bachetti, T. Circulating levels of tumour necrosis factor-α and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci. Lett. 2000, 287, 211–214. [Google Scholar] [CrossRef]
- Cereda, C.; Baiocchi, C.; Bongioanni, P.; Cova, E.; Guareschi, S.; Metelli, M.R.; Rossi, B.; Sbalsi, I.; Cuccia, M.C.; Ceroni, M. TNF and sTNFR1/2 plasma levels in ALS patients. J. Neuroimmunol. 2008, 194, 123–131. [Google Scholar] [CrossRef]
- Moreau, C.; Devos, D.; Brunaud-Danel, V.; Defebvre, L.; Perez, T.; Destee, A.; Tonnel, A.B.; Lassalle, P.; Just, N. Elevated IL-6 and TNF- levels in patients with ALS: Inflammation or hypoxia? Neurology 2005, 65, 1958–1960. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased Glutamate Transport by the Brain and Spinal Cord in Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar] [CrossRef]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as determinants of disease progression in inherited amyotrophic lateral sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Gutmann, D.H.; Roos, R.P. Astrocyte loss of mutant SOD1 delays ALS disease onset and progression in G85R transgenic mice. Hum. Mol. Genet. 2010, 20, 286–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papadeas, S.T.; Kraig, S.E.; O’Banion, C.; Lepore, A.C.; Maragakis, N.J. Astrocytes carrying the superoxide dismutase 1 (SOD1G93A) mutation induce wild-type motor neuron degeneration in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 17803–17808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, T.; Funayama, M.; Tsukita, K.; Hotta, A.; Yasuda, A.; Nori, S.; Kaneko, S.; Nakamura, M.; Takahashi, R.; Okano, H.; et al. Focal Transplantation of Human iPSC-Derived Glial-Rich Neural Progenitors Improves Lifespan of ALS Mice. Stem Cell Rep. 2014, 3, 242–249. [Google Scholar] [CrossRef] [Green Version]
- Lepore, A.C.; Rauck, B.; Dejea, C.; Pardo, A.C.; Rao, M.S.; Rothstein, J.D.; Maragakis, N.J. Focal transplantation–based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat. Neurosci. 2008, 11, 1294–1301. [Google Scholar] [CrossRef]
- Qian, K.; Huang, H.; Peterson, A.; Hu, B.; Maragakis, N.J.; Ming, G.-L.; Chen, H.; Zhang, S.-C. Sporadic ALS Astrocytes Induce Neuronal Degeneration In Vivo. Stem Cell Rep. 2017, 8, 843–855. [Google Scholar] [CrossRef]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar] [PubMed]
- Engelhardt, J.I.; Tajti, J.; Appel, S.H. Lymphocytic Infiltrates in the Spinal Cord in Amyotrophic Lateral Sclerosis. Arch. Neurol. 1993, 50, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Henkel, J.S.; Engelhardt, J.I.; Siklós, L.; Simpson, E.P.; Kim, S.H.; Pan, T.; Goodman, J.C.; Siddique, T.; Beers, D.R.; Appel, S.H. Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann. Neurol. 2003, 55, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Nagy, D.; Kato, T.; Kushner, P.D. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. Neurosci. Res. 1994, 38, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Kushner, P.D.; Stephenson, D.T.; Wright, S. Reactive Astrogliosis is Widespread in the Subcortical White Matter of Amyotrophic Lateral Sclerosis Brain. J. Neuropathol. Exp. Neurol. 1991, 50, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive astrogliosis of the spinal cord in amyotrophic lateral sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar] [CrossRef]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.J.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. Neurobiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef]
- Corcia, P.; Tauber, C.; Vercoullie, J.; Arlicot, N.; Prunier, C.; Praline, J.; Nicolas, G.; Venel, Y.; Hommet, C.; Baulieu, J.-L.; et al. Molecular Imaging of Microglial Activation in Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e52941. [Google Scholar] [CrossRef] [PubMed]
- Alexianu, M.E.; Kozovska, M.; Appel, S.H. Immune reactivity in a mouse model of familial ALS correlates with disease progression. Neurology 2001, 57, 1282–1289. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Sanagi, T.; Yuasa, S.; Nakamura, Y.; Suzuki, E.; Aoki, M.; Warita, H.; Itoyama, Y.; Uchino, S.; Kohsaka, S.; Ohsawa, K. Appearance of phagocytic microglia adjacent to motoneurons in spinal cord tissue from a presymptomatic transgenic rat model of amyotrophic lateral sclerosis. J. Neurosci. Res. 2010, 88, 2736–2746. [Google Scholar] [CrossRef]
- Gerber, Y.N.; Sabourin, J.-C.; Rábano, M.; Vivanco, M.D.M.; Perrin, F.E. Early Functional Deficit and Microglial Disturbances in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2012, 7, e36000. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal degeneration of astrocytes in amyotrophic lateral sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef] [Green Version]
- Beers, D.R.; Zhao, W.; Liao, B.; Kano, O.; Wang, J.; Huang, A.; Appel, S.H.; Henkel, J.S. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 2011, 25, 1025–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated Mechanisms in the Pathoprogression of Amyotrophic Lateral Sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [Green Version]
- Chiu, I.M.; Chen, A.; Zheng, Y.; Kosaras, B.; Tsiftsoglou, S.A.; Vartanian, T.K.; Brown, R.H.; Carroll, M.C. T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 17913–17918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Beers, D.R.; Wen, S.; Rivera, A.L.; Toennis, K.M.; Appel, J.E.; Zhao, W.; Moore, D.H.; Powell, S.Z.; Appel, S.H. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol. Med. 2012, 5, 64–79. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Wang, J.; Zhang, X.; Wen, S.; Neal, D.; Thonhoff, J.R.; Alsuliman, A.S.; Shpall, E.J.; Rezvani, K.; et al. ALS patients’ regulatory T lymphocytes are dysfunctional, and correlate with disease progression rate and severity. JCI Insight 2017, 2, e89530. [Google Scholar] [CrossRef]
- Chen, X.; Oppenheim, J.J. Contrasting effects of TNF and anti-TNF on the activation of effector T cells and regulatory T cells in autoimmunity. FEBS Lett. 2011, 585, 3611–3618. [Google Scholar] [CrossRef] [Green Version]
- Valencia, X.; Stephens, G.; Goldbach-Mansky, R.; Wilson, M.; Shevach, E.M.; Lipsky, P.E. TNF downmodulates the function of human CD4+CD25hi T-regulatory cells. Blood 2006, 108, 253–261. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2018, 23, 5010–5021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brambilla, L.; Guidotti, G.; Martorana, F.; Iyer, A.M.; Aronica, E.; Valori, C.F.; Rossi, D. Disruption of the astrocytic TNFR1-GDNF axis accelerates motor neuron degeneration and disease progression in amyotrophic lateral sclerosis. Hum. Mol. Genet. 2016, 25, 3080–3095. [Google Scholar] [CrossRef] [Green Version]
- Endo, F.; Komine, O.; Fujimori-Tonou, N.; Katsuno, M.; Jin, S.; Watanabe, S.; Sobue, G.; Dezawa, M.; Wyss-Coray, T.; Yamanaka, K. Astrocyte-Derived TGF-β1 Accelerates Disease Progression in ALS Mice by Interfering with the Neuroprotective Functions of Microglia and T Cells. Cell Rep. 2015, 11, 592–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, F.; Yamanaka, K. Astrocytic TGF-β1: Detrimental factor in ALS. Oncotarget 2015, 6, 15728–15729. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K. Message and protein-level elevation of tumor necrosis factor α (TNFα) and TNFα-modulating cytokines in spinal cords of the G93A-SOD1 mouse model for amyotrophic lateral sclerosis. Neurobiol. Dis. 2003, 14, 74–80. [Google Scholar] [CrossRef]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Keiner, S.; Wurm, F.; Kunze, A.; Witte, O.W.; Redecker, C. Rehabilitative therapies differentially alter proliferation and survival of glial cell populations in the perilesional zone of cortical infarcts. Glia 2008, 56, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Latimer, C.S.; Searcy, J.L.; Bridges, M.T.; Brewer, L.D.; Popovic, J.; Blalock, E.M.; Landfield, P.W.; Thibault, O.; Porter, N.M. Reversal of Glial and Neurovascular Markers of Unhealthy Brain Aging by Exercise in Middle-Aged Female Mice. PLoS ONE 2011, 6, e26812. [Google Scholar] [CrossRef]
- Rodríguez, J.J.; Terzieva, S.; Olabarria, M.; Lanza, R.G.; Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 2013, 4, e678. [Google Scholar] [CrossRef] [PubMed]
- Saur, L.; Baptista, P.P.A.; De Senna, P.N.; Paim, M.F.; Nascimento, P.D.; Ilha, J.; Bagatini, P.B.; Achaval, M.; Xavier, L.L. Physical exercise increases GFAP expression and induces morphological changes in hippocampal astrocytes. Brain Struct. Funct. 2014, 219, 293–302. [Google Scholar] [CrossRef]
- Mee-Inta, O.; Zhao, Z.-W.; Kuo, Y.-M. Physical Exercise Inhibits Inflammation and Microglial Activation. Cells 2019, 8, 691. [Google Scholar] [CrossRef] [Green Version]
- Bernardes, D.; Oliveira-Lima, O.C.; Da Silva, T.V.; Faraco, C.C.F.; Leite, H.R.; Juliano, M.A.; Dos Santos, D.M.; Bethea, J.R.; Brambilla, R.; Orian, J.M.; et al. Differential brain and spinal cord cytokine and BDNF levels in experimental autoimmune encephalomyelitis are modulated by prior and regular exercise. J. Neuroimmunol. 2013, 264, 24–34. [Google Scholar] [CrossRef] [Green Version]
- Leem, Y.-H.; Lee, Y.-I.; Son, H.-J.; Lee, S.-H. Chronic exercise ameliorates the neuroinflammation in mice carrying NSE/htau23. Biochem. Biophys. Res. Commun. 2011, 406, 359–365. [Google Scholar] [CrossRef]
- Piao, C.-S.; Stoica, B.A.; Wu, J.; Sabirzhanov, B.; Zhao, Z.; Cabatbat, R.; Loane, D.J.; Faden, A.I. Late exercise reduces neuroinflammation and cognitive dysfunction after traumatic brain injury. Neurobiol. Dis. 2013, 54, 252–263. [Google Scholar] [CrossRef] [Green Version]
- McCoy, M.K.; Tansey, M.G. TNF signaling inhibition in the CNS: Implications for normal brain function and neurodegenerative disease. J. Neuroinflam. 2008, 5, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabal-Hierro, L.; Lazo, P.S. Signal transduction by tumor necrosis factor receptors. Cell. Signal. 2012, 24, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Mohler, K.M.; Torrance, D.S.; A Smith, C.; Goodwin, R.G.; E Stremler, K.; Fung, V.P.; Madani, H.; Widmer, M.B. Soluble tumor necrosis factor (TNF) receptors are effective therapeutic agents in lethal endotoxemia and function simultaneously as both TNF carriers and TNF antagonists. J. Immunol. 1993, 151, 1548–1561. [Google Scholar] [PubMed]
- Aderka, D.; Engelmann, H.; Maor, Y.; Brakebusch, C.; Wallach, D. Stabilization of the bioactivity of tumor necrosis factor by its soluble receptors. J. Exp. Med. 1992, 175, 323–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baud, V.; Karin, M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001, 11, 372–377. [Google Scholar] [CrossRef]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants–past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [Green Version]
- Pekalski, J.; Zuk, P.J.; Kochańczyk, M.; Junkin, M.; Kellogg, R.; Tay, S.; Lipniacki, T. Spontaneous NF-κB Activation by Autocrine TNFα Signaling: A Computational Analysis. PLoS ONE 2013, 8, e78887. [Google Scholar] [CrossRef] [Green Version]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.J.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Swarup, V.; Phaneuf, D.; Dupré, N.; Petri, S.; Strong, M.; Kriz, J.; Julien, J.-P. Deregulation of TDP-43 in amyotrophic lateral sclerosis triggers nuclear factor κB–mediated pathogenic pathways. J. Exp. Med. 2011, 208, 2429–2447. [Google Scholar] [CrossRef]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia Induce Motor Neuron Death via the Classical NF-κB Pathway in Amyotrophic Lateral Sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Crosio, C.; Valle, C.; Casciati, A.; Iaccarino, C.; Carrì, M.T. Astroglial Inhibition of NF-κB Does Not Ameliorate Disease Onset and Progression in a Mouse Model for Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2011, 6, e17187. [Google Scholar] [CrossRef] [Green Version]
- Alami, N.O.; Schurr, C.; Heuvel, F.O.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Afonso, V.; Santos, G.; Collin, P.; Khatib, A.-M.; Mitrovic, D.R.; Lomri, N.; Leitman, D.C.; Lomri, A. Tumor necrosis factor-α down-regulates human Cu/Zn superoxide dismutase 1 promoter via JNK/AP-1 signaling pathway. Free. Radic. Biol. Med. 2006, 41, 709–721. [Google Scholar] [CrossRef]
- Semmler, S.; Gagné, M.; Garg, P.; Pickles, S.R.; Baudouin, C.; Hamon-Keromen, E.; Destroismaisons, L.; Khalfallah, Y.; Chaineau, M.; Caron, E.; et al. TNF receptor–associated factor 6 interacts with ALS-linked misfolded superoxide dismutase 1 and promotes aggregation. J. Biol. Chem. 2020, 295, 3808–3825. [Google Scholar] [CrossRef] [PubMed]
- Vitkovic, L.; Bockaert, J.; Jacque, C. “Inflammatory” Cytokines. J. Neurochem. 2001, 74, 457–471. [Google Scholar] [CrossRef]
- Cearley, C.; Churchill, L.; Krueger, J.M. Time of day differences in IL1β and TNFα mRNA levels in specific regions of the rat brain. Neurosci. Lett. 2003, 352, 61–63. [Google Scholar] [CrossRef]
- Iosif, R.E.; Ekdahl, C.T.; Ahlenius, H.; Pronk, C.J.H.; Bonde, S.; Kokaia, Z.; Jacobsen, S.-E.W.; Lindvall, O. Tumor Necrosis Factor Receptor 1 Is a Negative Regulator of Progenitor Proliferation in Adult Hippocampal Neurogenesis. J. Neurosci. 2006, 26, 9703–9712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, M.A.; Heffner, D.L.; Kim, S.; Espy, S.M.; Spano, A.J.; Cleland, C.L.; Deppmann, C.D. TNF-α/TNFR1 Signaling Is Required for the Development and Function of Primary Nociceptors. Neuron 2014, 82, 587–602. [Google Scholar] [CrossRef] [Green Version]
- Santello, M.; Volterra, A. TNFα in synaptic function: Switching gears. Trends Neurosci. 2012, 35, 638–647. [Google Scholar] [CrossRef]
- Beattie, E.C.; Stellwagen, D.; Morishita, W.; Bresnahan, J.C.; Ha, B.K.; Von Zastrow, M.; Beattie, M.S.; Malenka, R.C. Control of Synaptic Strength by Glial TNFalpha. Science 2002, 295, 2282–2285. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, D.; Beattie, E.C.; Seo, J.Y.; Malenka, R.C. Differential Regulation of AMPA Receptor and GABA Receptor Trafficking by Tumor Necrosis Factor. J. Neurosci. 2005, 25, 3219–3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellwagen, D.; Malenka, R.C. Synaptic scaling mediated by glial TNF-α. Nat. Cell Biol. 2006, 440, 1054–1059. [Google Scholar] [CrossRef]
- Santello, M.; Bezzi, P.; Volterra, A. TNFα Controls Glutamatergic Gliotransmission in the Hippocampal Dentate Gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Taoufik, E.; Petit, E.; Divoux, D.; Tseveleki, V.; Mengozzi, M.; Roberts, M.L.; Valable, S.; Ghezzi, P.; Quackenbush, J.; Brines, M.; et al. TNF receptor I sensitizes neurons to erythropoietin- and VEGF-mediated neuroprotection after ischemic and excitotoxic injury. Proc. Natl. Acad. Sci. USA 2008, 105, 6185–6190. [Google Scholar] [CrossRef] [Green Version]
- Ngo, S.; Steyn, F.; Huang, L.; Mantovani, S.; Pfluger, C.; Woodruff, T.M.; Osullivan, J.D.; Henderson, R.; A McCombe, P. Altered expression of metabolic proteins and adipokines in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2015, 357, 22–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.-H.; Allen, K.; Oei, F.; Leoni, E.; Kuhle, J.; Tree, T.; Fratta, P.; Sharma, N.; Sidle, K.; Howard, R.; et al. Systemic inflammatory response and neuromuscular involvement in amyotrophic lateral sclerosis. Neurol.-Neuroimmunol. Neuroinflamm. 2016, 3, e244. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, T.; Yamasaki, R.; Tanaka, M.; Matsushita, T.; Kikuchi, H.; Isobe, N.; Ohyagi, Y.; Kira, J.-I. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J. Neuroimmunol. 2010, 222, 76–81. [Google Scholar] [CrossRef]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [Green Version]
- Babu, G.N.; Kumar, A.; Chandra, R.; Puri, S.K.; Kalita, J.; Misra, U.K. Elevated Inflammatory Markers in a Group of Amyotrophic Lateral Sclerosis Patients from Northern India. Neurochem. Res. 2008, 33, 1145–1149. [Google Scholar] [CrossRef]
- Fukazawa, H.; Tsukie, T.; Higashida, K.; Fujikura, M.; Ono, S. An immunohistochemical study of increased tumor necrosis factor-α in the skin of patients with amyotrophic lateral sclerosis. J. Clin. Neurosci. 2013, 20, 1371–1376. [Google Scholar] [CrossRef]
- Kiaei, M.; Petri, S.; Kipiani, K.; Gardian, G.; Choi, D.-K.; Chen, J.; Calingasan, N.Y.; Schafer, P.; Muller, G.W.; Stewart, C.; et al. Thalidomide and Lenalidomide Extend Survival in a Transgenic Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurosci. 2006, 26, 2467–2473. [Google Scholar] [CrossRef] [Green Version]
- Brohawn, D.G.; O’Brien, L.C.; Bennett, J.P. RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PLoS ONE 2016, 11, e0160520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hensley, K.; Floyd, R.A.; Gordon, B.; Mou, S.; Pye, Q.N.; Stewart, C.; West, M.; Williamson, K. Temporal patterns of cytokine and apoptosis-related gene expression in spinal cords of the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2002, 82, 365–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, T.; Ishigaki, S.; Yamamoto, M.; Liang, Y.; Niwa, J.-I.; Takeuchi, H.; Doyu, M.; Sobue, G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 80, 158–167. [Google Scholar] [CrossRef]
- Veglianese, P.; Coco, D.L.; Cutrona, M.B.; Magnoni, R.; Pennacchini, D.; Pozzi, B.; Gowing, G.; Julien, J.; Tortarolo, M.; Bendotti, C. Activation of the p38MAPK cascade is associated with upregulation of TNF alpha receptors in the spinal motor neurons of mouse models of familial ALS. Mol. Cell. Neurosci. 2006, 31, 218–231. [Google Scholar] [CrossRef]
- Tolosa, L.; Caraballo-Miralles, V.; Olmos, G.; Llado, J. TNF-α potentiates glutamate-induced spinal cord motoneuron death via NF-κB. Mol. Cell. Neurosci. 2011, 46, 176–186. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sullivan, J.S.; Henkel, J.S.; Appel, S.H.; Zlokovic, B.V. Blood–spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 2012, 125, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef] [Green Version]
- Jablonski, M.R.; Jacob, D.A.; Campos, C.; Miller, D.S.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Selective increase of two ABC drug efflux transporters at the blood–spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 2012, 47, 194–200. [Google Scholar] [CrossRef] [Green Version]
- Qosa, H.; Lichter, J.; Sarlo, M.; Markandaiah, S.S.; McAvoy, K.; Richard, J.-P.; Jablonski, M.R.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Astrocytes drive upregulation of the multidrug resistance transporter ABCB1 (P-Glycoprotein) in endothelial cells of the blood-brain barrier in mutant superoxide dismutase 1-linked amyotrophic lateral sclerosis. Glia 2016, 64, 1298–1313. [Google Scholar] [CrossRef] [Green Version]
- Milane, A.; Fernandez, C.; Vautier, S.; Bensimon, G.; Meininger, V.; Farinotti, R. Minocycline and riluzole brain disposition: Interactions with p-glycoprotein at the blood?brain barrier. J. Neurochem. 2007, 103, 164–173. [Google Scholar] [CrossRef]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes expressing ALS-linked mutant FUS induce motor neuron death through release of tumor necrosis factor-alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Van Dyke, J.M.; Smit-Oistad, I.M.; Macrander, C.; Krakora, D.; Meyer, M.G.; Suzuki, M. Macrophage-mediated inflammation and glial response in the skeletal muscle of a rat model of familial amyotrophic lateral sclerosis (ALS). Exp. Neurol. 2016, 277, 275–282. [Google Scholar] [CrossRef] [Green Version]
- García-Martínez, C.; Agell, N.; Llovera, M.; López-Soriano, F.J.; Argilés, J.M. Tumour necrosis factor-α increases the ubiquitinization of rat skeletal muscle proteins. FEBS Lett. 1993, 323, 211–214. [Google Scholar] [CrossRef] [Green Version]
- Stommel, E.W.; Cohen, J.A.; Fadul, C.E.; Cogbill, C.H.; Graber, D.J.; Kingman, L.; Mackenzie, T.; Channon Smith, J.Y.; Harris, B.T. Efficacy of thalidomide for the treatment of amyotrophic lateral sclerosis: A phase II open label clinical trial. Amyotroph. Lateral. Scler. 2009, 10, 393–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gowing, G.; Dequen, F.; Soucy, G.; Julien, J.-P. Absence of Tumor Necrosis Factor- Does Not Affect Motor Neuron Disease Caused by Superoxide Dismutase 1 Mutations. J. Neurosci. 2006, 26, 11397–11402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tortarolo, M.; Vallarola, A.; Lidonnici, D.; Battaglia, E.; Gensano, F.; Spaltro, G.; Fiordaliso, F.; Corbelli, A.; Garetto, S.; Martini, E.; et al. Lack of TNF-alpha receptor type 2 protects motor neurons in a cellular model of amyotrophic lateral sclerosis and in mutant SOD1 mice but does not affect disease progression. J. Neurochem. 2015, 135, 109–124. [Google Scholar] [CrossRef] [Green Version]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 domain of Bcl-X(L) rescues astrocyte degeneration in amyotrophic lateral sclerosis by modulating intracellular calcium signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef] [Green Version]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Petitpain, N.; Devos, D.; Bagheri, H.; Rocher, F.; Gouraud, A.; Masmoudi, K.; Coquerel, A. Is TNF inhibitor exposure a risk factor for amyotrophic lateral sclerosis? Fundam. Clin. Pharmacol. 2019, 33, 689–694. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guidotti, G.; Scarlata, C.; Brambilla, L.; Rossi, D. Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells 2021, 10, 518. https://doi.org/10.3390/cells10030518
Guidotti G, Scarlata C, Brambilla L, Rossi D. Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells. 2021; 10(3):518. https://doi.org/10.3390/cells10030518
Chicago/Turabian StyleGuidotti, Giulia, Chiara Scarlata, Liliana Brambilla, and Daniela Rossi. 2021. "Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe?" Cells 10, no. 3: 518. https://doi.org/10.3390/cells10030518
APA StyleGuidotti, G., Scarlata, C., Brambilla, L., & Rossi, D. (2021). Tumor Necrosis Factor Alpha in Amyotrophic Lateral Sclerosis: Friend or Foe? Cells, 10(3), 518. https://doi.org/10.3390/cells10030518