
Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs


Abstract
:1. Introduction
Hepatocellular Carcinoma
2. Notch Genes
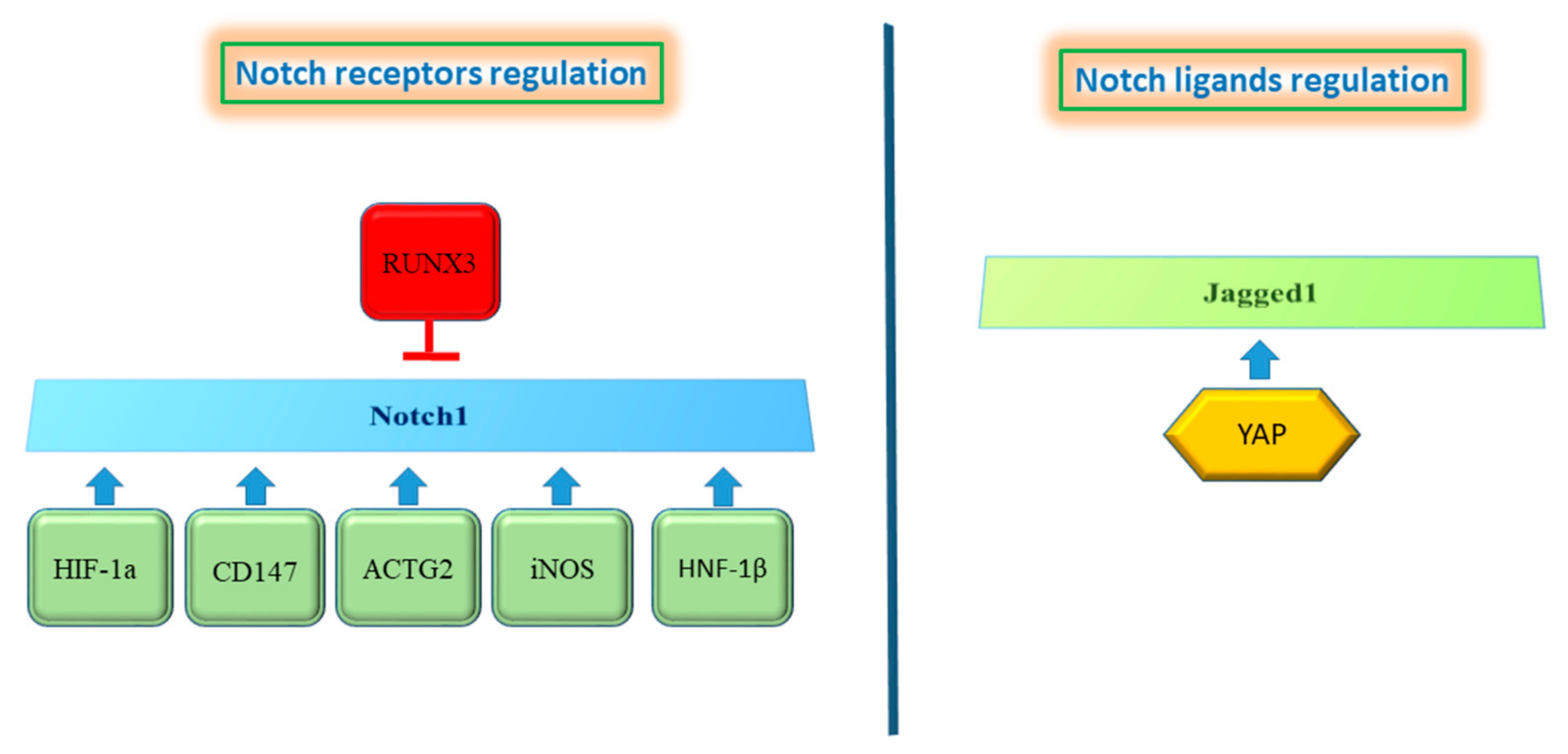
Notch Signaling in HCC
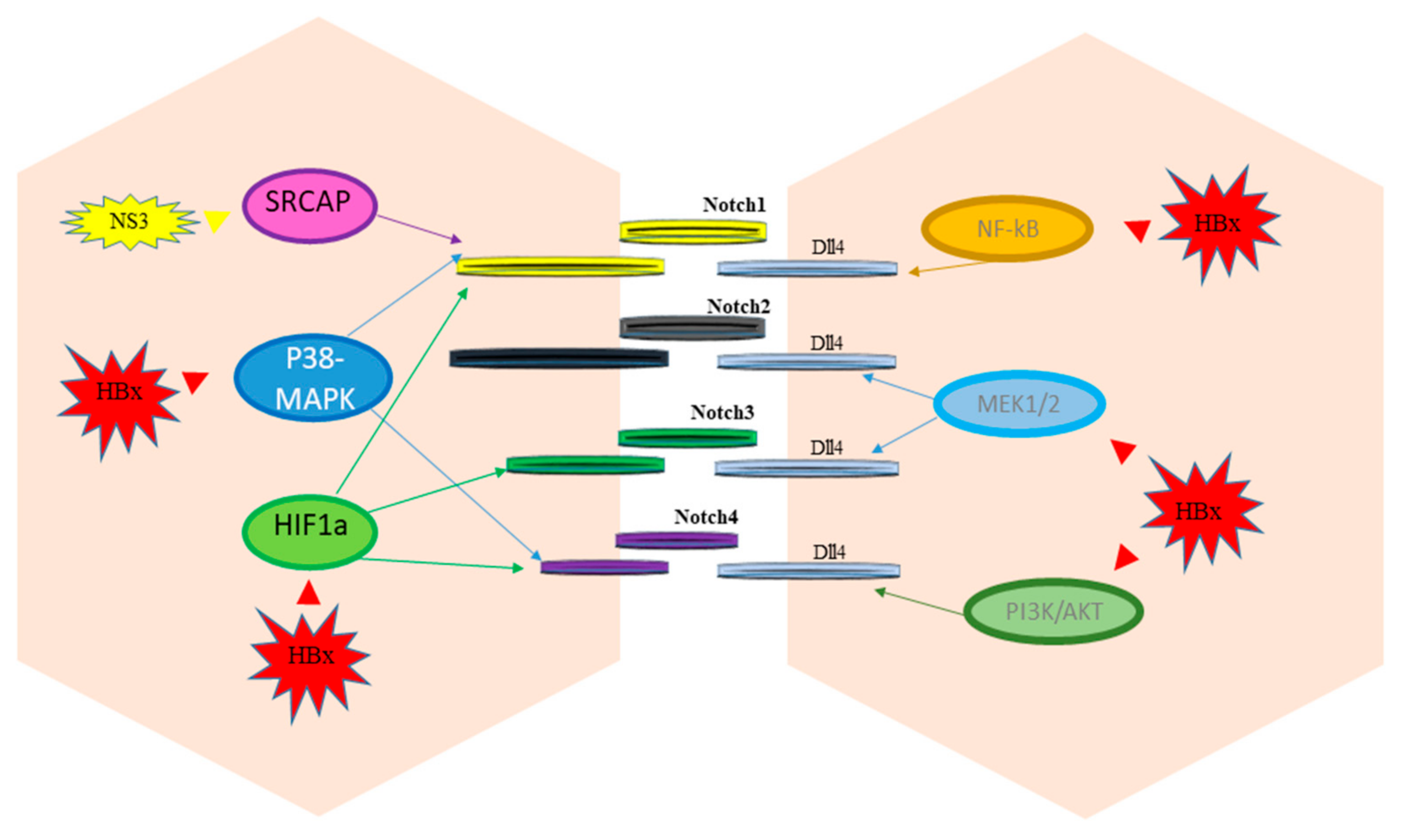
3. Notch and Hepatitis Viruses
4. Crosstalk of Notch Signaling with Other Signaling Pathways in HCC
5. Regulation of Notch Signaling by Non-Coding RNAs in HCC
6. Concluding Remarks
Funding
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef]
- Kulik, L.; El-Serag, H.B. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019, 156, 477–491.e1. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. Electronic Address EEE, European Association for the Study of the L. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J. Hepatol. 2018, 69, 182–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J.M.; Lynch, M. Preclinical Overview of Sorafenib, a Multikinase Inhibitor That Targets Both Raf and VEGF and PDGF Receptor Tyrosine Kinase Signaling. Mol. Cancer Ther. 2008, 7, 3129–3140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [Green Version]
- Da Fonseca, L.G.; Reig, M.; Bruix, J. Tyrosine Kinase Inhibitors and Hepatocellular Carcinoma. Clin. Liver Dis. 2020, 24, 719–737. [Google Scholar] [CrossRef]
- Constantinidou, A.; Alifieris, C.; Trafalis, D.T. Targeting Programmed Cell Death-1 (PD-1) and Ligand (PD-L1): A New Era in Cancer Active Immunotherapy. Pharmacol. Ther. 2019, 194, 84–106. [Google Scholar] [CrossRef]
- Duffy, A.G.; Ulahannan, S.V.; Makorova-Rusher, O.; Rahma, O.; Wedemeyer, H.; Pratt, D.; Davis, J.L.; Hughes, M.S.; Heller, T.; ElGindi, M.; et al. Tremelimumab in Combination with Ablation in Patients with Advanced Hepatocellular Carcinoma. J. Hepatol. 2017, 66, 545–551. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.-Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N. Engl. J. Med. 2020, 382, 1894–1905. [Google Scholar] [CrossRef]
- Moeini, A.; Cornella, H.; Villanueva, A. Emerging Signaling Pathways in Hepatocellular Carcinoma. Liver Cancer 2012, 1, 83–93. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.; Fasnacht, N.; MacDonald, H.R. Notch Signaling in the Immune System. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, T.H. The Origin of Nine Wing Mutations in Drosophila. Science 1911, 33, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide Sequence from the Neurogenic Locus Notch Implies a Gene Product That Shares Homology with Proteins Containing EGF-Like Repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Miller, A.C.; Lyons, E.L.; Herman, T.G. Cis-Inhibition of Notch by Endogenous Delta Biases the Outcome of Lateral Inhibition. Curr. Biol. 2009, 19, 1378–1383. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Haltiwanger, R.S. O-Fucosylation of Notch Occurs in the Endoplasmic Reticulum. J. Biol. Chem. 2005, 280, 11289–11294. [Google Scholar] [CrossRef] [Green Version]
- Blaumueller, C.M.; Qi, H.; Zagouras, P.; Artavanis-Tsakonas, S. Intracellular Cleavage of Notch Leads to a Heterodimeric Receptor on the Plasma Membrane. Cell 1997, 90, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R. Notch Signaling. Cold Spring Harb. Perspect. Biol. 2012, 4, a011213. [Google Scholar] [CrossRef] [Green Version]
- Borggrefe, T.; Oswald, F. The Notch Signaling Pathway: Transcriptional Regulation at Notch Target Genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 Signalling Requires Ligand-Induced Proteolytic Release of Intracellular Domain. Nat. Cell Biol. 1998, 393, 382–386. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef] [Green Version]
- Gil-García, B.; Baladrón, V. The Complex Role of NOTCH Receptors and Their Ligands in the Development of Hepatoblastoma, Cholangiocarcinoma and Hepa-Tocellular Carcinoma. Biol. Cell 2015, 108, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Rauff, B.; Malik, A.; Bhatti, Y.A.; Chudhary, S.A.; Qadri, I.; Rafiq, S. Notch Signalling Pathway in Development of Cholangiocarcinoma. World J. Gastrointest. Oncol. 2020, 12, 957–974. [Google Scholar] [CrossRef] [PubMed]
- Huntzicker, E.G.; Hötzel, K.; Choy, L.; Che, L.; Ross, J.; Pau, G.; Sharma, N.; Siebel, C.W.; Chen, X.; French, D.M. Differential Effects of Targeting Notch Receptors in a Mouse Model of Liver Cancer. Hepatology 2015, 61, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, C.; Hong, L.; Wang, J.; Du, Y.; Song, J.; Shao, X.; Zhang, J.; Han, H.; Liu, J.; et al. Expression of Jagged1 and its Association with Hepatitis B Virus X Protein in Hepatocellular Carcinoma. Biochem. Biophys. Res. Commun. 2007, 356, 341–347. [Google Scholar] [CrossRef]
- Paranjpe, S.; Bowen, W.C.; Bell, A.W.; Nejak-Bowen, K.; Luo, J.-H.; Michalopoulos, G.K. Cell Cycle Effects Resulting from Inhibition of Hepatocyte Growth Factor and Its Receptor C-Met in Regenerating Rat Livers by RNA Interference. Hepatology 2007, 45, 1471–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, K.; Honda, M.; Yamashita, T.; Okada, H.; Shirasaki, T.; Nishikawa, M.; Nio, K.; Arai, K.; Sakai, Y.; Yamashita, T.; et al. Jagged1 DNA Copy Number Variation Is Associated with Poor Outcome in Liver Cancer. Am. J. Pathol. 2016, 186, 2055–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.; Xue, L.; Cao, Q.; Lin, Y.; Ding, Y.; Yang, P.; Che, L. Expression of Notch1, Jagged1 and β-Catenin and Their Clinicopathological Significance in Hepatocellular Carcinoma. Neoplasma 2009, 56, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A.; Alsinet, C.; Yanger, K.; Hoshida, Y.; Zong, Y.; Toffanin, S.; Rodriguez–Carunchio, L.; Solé, M.; Thung, S.; Stanger, B.Z.; et al. Notch Signaling Is Activated in Human Hepatocellular Carcinoma and Induces Tumor Formation in Mice. Gastroenterology 2012, 143, 1660–1669.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tovar, V.; Alsinet, C.; Villanueva, A.; Hoshida, Y.; Chiang, D.Y.; Solé, M.; Thung, S.; Moyano, S.; Toffanin, S.; Mínguez, B.; et al. IGF Activation in a Molecular Subclass of Hepatocellular Carcinoma and Pre-Clinical Efficacy of IGF-1R Blockage. J. Hepatol. 2010, 52, 550–559. [Google Scholar] [CrossRef] [Green Version]
- Giovannini, C.; Gramantieri, L.; Chieco, P.; Minguzzi, M.; Lago, F.; Pianetti, S.; Ramazzotti, E.; Marcu, K.B.; Bolondi, L. Selective Ablation of Notch3 in HCC Enhances Doxorubicin’s Death Promoting Effect by a p53 Dependent Mechanism. J. Hepatol. 2009, 50, 969–979. [Google Scholar] [CrossRef]
- Gramantieri, L.; Giovannini, C.; Lanzi, A.; Chieco, P.; Ravaioli, M.; Venturi, A.; Grazi, G.L.; Bolondi, L. Aberrant Notch3 and Notch4 Expression in Human Hepatocellular Carcinoma. Liver Int. 2007, 27, 997–1007. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lu, C.; Fang, T.; Wang, Y.; Hu, W.; Qiao, J.; Liu, B.; Liu, J.; Chen, N.; Li, M.; et al. Notch3 Functions as a Regulator of Cell Self-Renewal by Interacting with the β-Catenin Pathway in Hepatocellular Carcinoma. Oncotarget 2015, 6, 3669–3679. [Google Scholar] [CrossRef] [Green Version]
- Moore, G.; Annett, S.; McClements, L.; Robson, T. Top Notch Targeting Strategies in Cancer: A Detailed Overview of Recent Insights and Current Perspectives. Cells 2020, 9, 1503. [Google Scholar] [CrossRef]
- Morell, C.M.; Fiorotto, R.; Fabris, L.; Strazzabosco, M. Notch Signalling beyond Liver Development: Emerging Concepts in Liver Repair and Oncogenesis. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Li, J.; Zheng, J.; Wei, A. The Carcinogenic Role of the Notch Signaling Pathway in the Development of Hepatocellular Carcinoma. J. Cancer 2019, 10, 1570–1579. [Google Scholar] [CrossRef] [Green Version]
- Giovannini, C.; Bolondi, L.; Gramantieri, L. Targeting Notch3 in Hepatocellular Carcinoma: Molecular Mechanisms and Therapeutic Perspectives. Int. J. Mol. Sci. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Benhenda, S.; Cougot, D.; Buendia, M.-A.; Neuveut, C. Chapter 4 Hepatitis B Virus X Protein. Adv. Cancer Res. 2009, 103, 75–109. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Xiong, Y.; Wang, Y.; Wang, Y.; Zheng, G.; Xu, H. Hepatitis B virus X Protein Activates Notch Signaling by Its Effects on Notch1 and Notch4 in Human Hepatocellular Carcinoma. Int. J. Oncol. 2015, 48, 329–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.-L.; Ren, Q.-G.; Zhang, T.; Pan, X.; Wen, L.; Hu, J.-L.; Yu, C.; He, Q.-J. Hepatitis B Virus X Protein and Hypoxia-Inducible Factor-1α Stimulate Notch Gene Expression in Liver Cancer Cells. Oncol. Rep. 2016, 37, 348–356. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Wang, R.; Luo, J.; Wang, P.; Xiong, S.; Liu, M.; Cheng, B. Notch1 Promotes Hepatitis B Virus X Protein-Induced Hepatocarcinogenesis via Wnt/β-Catenin Pathway. Int. J. Oncol. 2014, 45, 1638–1648. [Google Scholar] [CrossRef] [Green Version]
- Kongkavitoon, P.; Tangkijvanich, P.; Hirankarn, N.; Palaga, T. Hepatitis B Virus HBx Activates Notch Signaling via Delta-Like 4/Notch1 in Hepatocellular Carcinoma. PLoS ONE 2016, 11, e0146696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, A.; Takegami, T.; Shiozaki, T.; Miyazaki, T. Hepatitis C Virus NS3 Protein Can Activate the Notch-Signaling Pathway through Binding to a Transcription Factor, SRCAP. PLoS ONE 2011, 6, e20718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, N.J.; Tiriveedhi, V.; Subramanian, V.; Shenoy, S.; Crippin, J.S.; Chapman, W.C.; Mohanakumar, T. Hepatitis C Virus Mediated Changes in miRNA-449a Modulates Inflammatory Biomarker YKL40 through Components of the NOTCH Signaling Pathway. PLoS ONE 2012, 7, e50826. [Google Scholar] [CrossRef] [PubMed]
- Papic, N.; Maxwell, C.I.; Delker, D.A.; Liu, S.; Heale, B.S.E.; Hagedorn, C.H. RNA-Sequencing Analysis of 5′ Capped RNAs Identifies Many New Differentially Expressed Genes in Acute Hepatitis C Virus Infection. Viruses 2012, 4, 581–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.-C.; Liu, X.; Liu, X.-H.; Li, Z.-S.-N.; Zhu, G.-Z. Notch Signaling Regulates Circulating T Helper 22 Cells in Patients with Chronic Hepatitis, C. Viral Immunol. 2017, 30, 522–532. [Google Scholar] [CrossRef]
- Qin, L.; Zhou, Y.-C.; Wu, H.-J.; Zhuo, Y.; Wang, Y.-P.; Si, C.-Y.; Qin, Y.-M. Notch Signaling Modulates the Balance of Regulatory T Cells and T Helper 17 Cells in Patients with Chronic Hepatitis, C. DNA Cell Biol. 2017, 36, 311–320. [Google Scholar] [CrossRef]
- Guo, Y.; Xiao, Z.; Yang, L.; Gao, Y.; Zhu, Q.; Hu, L.; Huang, D.; Xu, Q. Hypoxia Inducible Factors in Hepatocellular Carcinoma (Review). Oncol. Rep. 2019, 43, 3–15. [Google Scholar] [CrossRef]
- Qin, Y.; Liu, H.-J.; Li, M.; Zhai, D.-H.; Tang, Y.-H.; Yang, L.; Qiao, K.-L.; Yang, J.-H.; Zhong, W.-L.; Zhang, Q.; et al. Salidroside Improves the Hypoxic Tumor Microenvironment and Reverses the Drug Resistance of Platinum Drugs via HIF-1α Signaling Pathway. EBioMedicine 2018, 38, 25–36. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Duan, W.; Xu, Q.; Li, X.; Han, L.; Li, W.; Zhang, D.; Wang, Z.; Lei, J. Curcumin Suppresses Hepatic Stellate Cell-Induced Hepatocarcinoma Angiogenesis and Invasion through Downregulating CTGF. Oxidative Med. Cell. Longev. 2019, 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yong, Y.; Zhang, R.; Liu, Z.; Wei, D.; Shang, Y.; Wu, J.; Zhang, Z.; Li, C.; Chen, Z.; Bian, H. Gamma-Secretase Complex-Dependent Intramembrane Proteolysis of CD147 Regulates the Notch1 Signaling Pathway in Hepatocellular Carcinoma. J. Pathol. 2019, 249, 255–267. [Google Scholar] [CrossRef]
- Tanaka, S.; Shiraha, H.; Nakanishi, Y.; Nishina, S.-I.; Matsubara, M.; Horiguchi, S.; Takaoka, N.; Iwamuro, M.; Kataoka, J.; Kuwaki, K.; et al. Runt-Related Transcription Factor 3 Reverses Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma. Int. J. Cancer 2012, 131, 2537–2546. [Google Scholar] [CrossRef] [Green Version]
- Gou, Y.; Zhai, F.; Zhang, L.; Cui, L. RUNX3 Regulates Hepatocellular Carcinoma Cell Metastasis via Targeting miR-186/E-Cadherin/EMT Pathway. Oncotarget 2017, 8, 61475–61486. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Chen, Y.; Wu, K.-C.; Liu, J.; Zhao, Y.-Q.; Pan, Y.-L.; Du, R.; Zheng, G.-R.; Xiong, Y.-M.; Xu, H.-L.; et al. RUNX3 Directly Interacts with Intracellular Domain of Notch1 and Suppresses Notch Signaling in Hepatocellular Carcinoma Cells. Exp. Cell Res. 2010, 316, 149–157. [Google Scholar] [CrossRef]
- Wang, R.; Geller, D.A.; Wink, D.A.; Cheng, B.; Billiar, T.R. NO and Hepatocellular Cancer. Br. J. Pharmacol. 2020, 177, 5459–5466. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Y.; Tsung, A.; Huang, H.; Du, Q.; Yang, M.; Deng, M.; Xiong, S.; Wang, X.; Zhang, L.; et al. iNOS promotes CD24+CD133+Liver Cancer Stem Cell Phenotype through a TACE/ADAM17-Dependent Notch Signaling Pathway. Proc. Natl. Acad. Sci. USA 2018, 115, E10127–E10136. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Liu, Z.G.; Shi, M.Q.; Yu, H.Z.; Jiang, X.Y.; Yang, A.H.; Fu, X.S.; Xu, Y.; Yang, S.; Ni, H.; et al. Identification of ACTG2 Functions as a Promoter Gene in Hepatocellular Carcinoma Cells Migration and Tumor Metastasis. Biochem. Biophys. Res. Commun. 2017, 491, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.-D.; Jing, Y.-Y.; Guo, S.-W.; Ye, F.; Lu, W.; Li, Q.; Dong, Y.-L.; Gao, L.; Yang, Y.-T.; Yang, Y.; et al. Overexpression of Hepatocyte Nuclear Factor-1beta Predicting Poor Prognosis Is Associated with Biliary Phenotype In Patients With Hepatocellular Carcinoma. Sci. Rep. 2015, 5, 13319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.-N.; Jiang, L.; Jiang, J.-H.; Yang, X.; Li, X.-Y.; Zeng, J.-X.; Shi, R.-Y.; Shi, Y.; Pan, X.-R.; Han, Z.-P.; et al. Hepatocyte Nuclear Factor-1beta Enhances the Stemness of Hepatocellular Carcinoma Cells through Activation of the Notch Pathway. Sci. Rep. 2017, 7, 4793. [Google Scholar] [CrossRef] [Green Version]
- Abylkassov, R.; Xie, Y. Role of Yes-Associated Protein in Cancer: An Update. Oncol. Lett. 2016, 12, 2277–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laquaglia, M.J.; Grijalva, J.L.; Mueller, K.A.; Perez-Atayde, A.R.; Kim, H.B.; Sadri-Vakili, G.; Vakili, K. YAP Subcellular Localization and Hippo Pathway Transcriptome Analysis in Pediatric Hepatocellular Carcinoma. Sci. Rep. 2016, 6, 30238. [Google Scholar] [CrossRef]
- Tschaharganeh, D.F.; Chen, X.; Latzko, P.; Malz, M.; Gaida, M.M.; Felix, K.; Ladu, S.; Singer, S.; Pinna, F.; Gretz, N.; et al. Yes-Associated Protein Up-Regulates Jagged-1 and Activates the NOTCH Pathway in Human Hepatocellular Carcinoma. Gastroenterology 2013, 144, 1530–1542. [Google Scholar] [CrossRef] [Green Version]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.-J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and Pharmacological Disruption of the TEAD-YAP Complex Suppresses the Oncogenic Activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, V.R.; Mazack, V.; Feng, W.; Nash, J.; Carey, D.J.; Gogoi, R. Verteporfin Exhibits YAP-Independent Anti-proliferative and Cytotoxic Effects in Endometrial Cancer Cells. Oncotarget 2017, 8, 28628–28640. [Google Scholar] [CrossRef] [Green Version]
- Tay, Y.; Rinn, J.L.; Pandolfi, P.P. The Multilayered Complexity of ceRNA Crosstalk and Competition. Nat. Cell Biol. 2014, 505, 344–352. [Google Scholar] [CrossRef] [Green Version]
- Volinia, S.; Calin, G.A.; Liu, C.-G.; Ambs, S.; Cimmino, A.; Petrocca, F.; Visone, R.; Iorio, M.; Roldo, C.; Ferracin, M.; et al. A microRNA Expression Signature of Human Solid Tumors Defines Cancer Gene Targets. Proc. Natl. Acad. Sci. USA 2006, 103, 2257–2261. [Google Scholar] [CrossRef] [Green Version]
- Gramantieri, L.; Ferracin, M.; Fornari, F.; Veronese, A.; Sabbioni, S.; Liu, C.-G.; Calin, G.A.; Giovannini, C.; Ferrazzi, E.; Grazi, G.L.; et al. Cyclin G1 Is a Target of miR-122a, a MicroRNA Frequently Down-regulated in Human Hepatocellular Carcinoma. Cancer Res. 2007, 67, 6092–6099. [Google Scholar] [CrossRef] [Green Version]
- Gramantieri, L.; Fornari, F.; Callegari, E.; Sabbioni, S.; Lanza, G.; Croce, C.M.; Bolondi, L.; Negrini, M. MicroRNA Involvement in Hepatocellular Carcinoma. J. Cell. Mol. Med. 2008, 12, 2189–2204. [Google Scholar] [CrossRef] [Green Version]
- Murakami, Y.; Yasuda, T.; Saigo, K.; Urashima, T.; Toyoda, H.; Okanoue, T.; Shimotohno, K. Comprehensive Analysis of MicroRNA Expression Patterns in Hepatocellular Carcinoma and Non-Tumorous Tissues. Oncogene 2005, 25, 2537–2545. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Gramantieri, L.; Giovannini, C.; Veronese, A.; Ferracin, M.; Sabbioni, S.; Calin, G.A.; Grazi, G.L.; Croce, C.M.; Tavolari, S.; et al. MiR-122/Cyclin G1 Interaction Modulates p53 Activity and Affects Doxorubicin Sensitivity of Human Hepatocarcinoma Cells. Cancer Res. 2009, 69, 5761–5767. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Gardini, A.C.; Marisi, G.; Toaldo, M.B.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3–Mediated Apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, C.; Fornari, F.; Dallo, R.; Gagliardi, M.; Nipoti, E.; Vasuri, F.; Coadă, C.A.; Ravaioli, M.; Bolondi, L.; Gramantieri, L. MiR-199-3p Replacement Affects E-Cadherin Expression through Notch1 Targeting in Hepatocellular Carcinoma. Acta Histochem. 2018, 120, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Jia, H.-L.; Forgues, M.; Liu, C.-G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.-H.; Qin, L.-X.; Croce, C.M.; et al. Identification of Metastasis-Related microRNAs in Hepatocellular Carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Henson, R.; Wehbe–Janek, H.; Ghoshal, K.; Jacob, S.T.; Patel, T. MicroRNA-21 Regulates Expression of the PTEN Tumor Suppressor Gene in Human Hepatocellular Cancer. Gastroenterology 2007, 133, 647–658. [Google Scholar] [CrossRef] [Green Version]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.S.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs In Vivo with ‘Antagomirs’. Nat. Cell Biol. 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-Mediated microRNA Silencing in Non-Human Primates. Nat. Cell Biol. 2008, 452, 896–899. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [Green Version]
- Callegari, E.; Domenicali, M.; Shankaraiah, R.C.; D’Abundo, L.; Guerriero, P.; Giannone, F.; Baldassarre, M.; Bassi, C.; Elamin, B.K.; Zagatti, B.; et al. MicroRNA-Based Prophylaxis in a Mouse Model of Cirrhosis and Liver Cancer. Mol. Ther. Nucleic Acids 2019, 14, 239–250. [Google Scholar] [CrossRef] [Green Version]
- Callegari, E.; Elamin, B.K.; Giannone, F.; Milazzo, M.; Altavilla, G.; Fornari, F.; Giacomelli, L.; D’Abundo, L.; Ferracin, M.; Bassi, C.; et al. Liver Tumorigenicity Promoted by microRNA-221 in a Mouse Transgenic Model. Hepatology 2012, 56, 1025–1033. [Google Scholar] [CrossRef]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; Van Der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [Green Version]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and Activity of MicroRNA-Loaded Minicells in Patients with Recurrent Malignant Pleural Mesothelioma: A First-in-Man, Phase 1, Open-Label, Dose-Escalation Study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.-C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239. [Google Scholar] [CrossRef] [Green Version]
- Pollutri, D.; Gramantieri, L.; Bolondi, L.; Fornari, F. TP53/MicroRNA Interplay in Hepatocellular Carcinoma. Int. J. Mol. Sci. 2016, 17, 2029. [Google Scholar] [CrossRef] [Green Version]
- Fornari, F.; Milazzo, M.; Galassi, M.; Callegari, E.; Veronese, A.; Miyaaki, H.; Sabbioni, S.; Mantovani, V.; Marasco, E.; Chieco, P.; et al. p53/mdm2 Feedback Loop Sustains miR-221 Expression and Dictates the Response to Anticancer Treatments in Hepatocellular Carcinoma. Mol. Cancer Res. 2014, 12, 203–216. [Google Scholar] [CrossRef] [Green Version]
- Giovannini, C.; Minguzzi, M.; Baglioni, M.; Fornari, F.; Giannone, F.; Ravaioli, M.; Cescon, M.; Chieco, P.; Bolondi, L.; Gramantieri, L. Suppression of p53 by Notch3 is mediated by Cyclin G1 and sustained by MDM2 and miR-221 axis in hepatocellular carcinoma. Oncotarget 2014, 5, 10607–10620. [Google Scholar] [CrossRef] [Green Version]
- Giovannini, C.; Gramantieri, L.; Minguzzi, M.; Fornari, F.; Chieco, P.; Grazi, G.L.; Bolondi, L. CDKN1C/P57 Is Regulated by the Notch Target Gene Hes1 and Induces Senescence in Human Hepatocellular Carcinoma. Am. J. Pathol. 2012, 181, 413–422. [Google Scholar] [CrossRef]
- Peng, J.-M.; Bera, R.; Chiou, C.-Y.; Chih-Yung, C.; Chen, T.-C.; Chen, C.-W.; Wang, T.-R.; Chiang, W.-L.; Chai, S.-P.; Chia-Wei, C.; et al. Actin Cytoskeleton Remodeling Drives Epithelial-Mesenchymal Transition for Hepatoma Invasion and Metastasis in Mice. Hepatology 2018, 67, 2226–2243. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Chang, R.-M.; Yang, M.-Y.; Lei, X.; Liu, X.; Gao, W.-B.; Xiao, J.-L.; Yang, L.-Y. Actin-like 6A Predicts Poor Prognosis of Hepatocellular Carcinoma and Promotes Metastasis and Epithelial-Mesenchymal Transition. Hepatology 2016, 63, 1256–1271. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.; Veronese, A.; Pichiorri, F.; Lee, T.J.; Jeon, Y.-J.; Volinia, S.; Pineau, P.; Marchio, A.; Palatini, J.; Suh, S.-S.; et al. p53 Regulates Epithelial–Mesenchymal Transition Through microRNAs Targeting ZEB1 and ZEB2. J. Exp. Med. 2011, 208, 875–883. [Google Scholar] [CrossRef]
- Deng, L.; Tang, J.; Yang, H.; Cheng, C.; Lu, S.; Jiang, R.; Sun, B. MTA1 Modulated by miR-30e Contributes to Epithelial-to-Mesenchymal Transition in Hepatocellular Carcinoma through an ErbB2-Dependent Pathway. Oncogene 2017, 36, 3976–3985. [Google Scholar] [CrossRef]
- Natsuizaka, M.; Whelan, K.A.; Kagawa, S.; Tanaka, K.; Giroux, V.; Chandramouleeswaran, P.M.; Long, A.; Sahu, V.; Darling, D.S.; Veronique, G.; et al. Interplay between Notch1 and Notch3 Promotes EMT and Tumor Initiation in Squamous Cell Carcinoma. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Wang, Y.; Peng, B.; Liang, L.; Li, J. The Roles of Notch1 Expression in the Migration of Intrahepatic Cholangiocarcinoma. BMC Cancer 2013, 13, 244. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Ooi, L.L.P.; Hui, K.M. MicroRNA-216a/217-Induced Epithelial-Mesenchymal Transition Targets PTEN and SMAD7 to Promote Drug Resistance and Recurrence of Liver Cancer. Hepatology 2013, 58, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Tu, K.; Liu, Q. Effects of microRNA-30a on Migration, Invasion and Prognosis of Hepatocellular Carcinoma. FEBS Lett. 2014, 588, 3089–3097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, C.; Minguzzi, M.; Genovese, F.; Baglioni, M.; Gualandi, A.; Ravaioli, M.; Milazzo, M.; Tavolari, S.; Bolondi, L.; Gramantieri, L. Molecular and Proteomic Insight into Notch1 Characterization in Hepatocellular Carcinoma. Oncotarget 2016, 7, 39609–39626. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Lin, L.; Zhou, W.; Wang, Z.; Ding, G.; Dong, Q.; Qin, L.; Wu, X.; Zheng, Y.; Yang, Y.; et al. Identification of miRNomes in Human Liver and Hepatocellular Carcinoma Reveals miR-199a/b-3p as Therapeutic Target for Hepatocellular Carcinoma. Cancer Cell 2011, 19, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callegari, E.; D’Abundo, L.; Guerriero, P.; Simioni, C.; Elamin, B.K.; Russo, M.; Cani, A.; Bassi, C.; Zagatti, B.; Giacomelli, L.; et al. miR-199a-3p Modulates MTOR and PAK4 Pathways and Inhibits Tumor Growth in a Hepatocellular Carcinoma Transgenic Mouse Model. Mol. Ther. Nucleic Acids 2018, 11, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I Study of MRX34, a Liposomal miR-34a Mimic, Administered Twice Weekly in Patients with Advanced Solid Tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Forterre, A.; Komuro, H.; Aminova, S.; Harada, M. A Comprehensive Review of Cancer MicroRNA Therapeutic Delivery Strategies. Cancers 2020, 12, 1852. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.-J.; Deng, Y.-L.; Liang, H.-F.; Jaoude, J.C.; Liu, F.-Y. Hepatitis B virus X Protein Promotes CREB-Mediated Activation of miR-3188 and Notch Signaling in Hepatocellular Carcinoma. Cell Death Differ. 2017, 24, 1577–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Zheng, Y.; Wang, L.; Wang, H.; Du, J.; Ye, F.; Sun, T.; Zhang, L. A Novel microRNA Signature Predicts Vascular Invasion in Hepatocellular Carcinoma. J. Cell. Physiol. 2019, 234, 20859–20868. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Li, M.; Wang, T.; Linghu, E.; Wu, B. MicroRNA-137 Represses FBI-1 to Inhibit Proliferation and in Vitro Invasion and Migration of Hepatocellular Carcinoma Cells. Tumor Biol. 2016, 37, 13995–14008. [Google Scholar] [CrossRef]
- El–Serag, H.B.; Rudolph, K.L. Hepatocellular Carcinoma: Epidemiology and Molecular Carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef] [PubMed]
- Jue, C.; Lin, C.; Zhisheng, Z.; Yayun, Q.; Feng, J.; Min, Z.; Haibo, W.; Youyang, S.; Hisamitsu, T.; Shintaro, I.; et al. Notch1 Inhibit Proliferation and in Vitro Invasion and Migration of Hepatocellular Carcinoma Cellspromotes Vasculogenic Mimicry in Hepatocellular Carcinoma by Inducing EMT Signaling. Oncotarget 2016, 8, 2501–2513. [Google Scholar] [CrossRef] [PubMed]
- Xue, T.-C.; Zou, J.-H.; Chen, R.-X.; Cui, J.-F.; Tang, Z.-Y.; Ye, S.-L. Spatial Localization of the JAG1/Notch1/Osteopontin Cascade Modulates Extrahepatic Metastasis in Hepatocellular Carcinoma. Int. J. Oncol. 2014, 45, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Wang, C.; Xie, H.; Wang, Y.; Huang, J.; Rong, Y.; Zhang, H.; Kong, H.; Yang, Y.; Lu, Y. MicroRNA-3163 Targets ADAM-17 and Enhances the Sensitivity of Hepatocellular Carcinoma Cells to Molecular Targeted Agents. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, C.; Baglioni, M.; Toaldo, M.B.; Ventrucci, C.; D’Adamo, S.; Cipone, M.; Chieco, P.; Gramantieri, L.; Bolondi, L. Notch3 Inhibition Enhances Sorafenib Cytotoxic Efficacy by Promoting GSK3β Phosphorylation and p21 Down-Regulation in Hepato-Cellular Carcinoma. Oncotarget 2013, 4, 1618–1631. [Google Scholar] [CrossRef] [Green Version]
- Jung, K.H.; Zhang, J.; Zhou, C.; Shen, H.; Gagea, M.; Rodriguez-Aguayo, C.; Lopez-Berestein, G.; Sood, A.K.; Beretta, L. Differentiation therapy for hepatocellular carcinoma: Multifaceted effects of miR-148a on tumor growth and phenotype and liver fibrosis. Hepatology 2016, 63, 864–879. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, Y.; Guo, Y.; Liu, B.; Zhao, Y.; Li, P.; Song, F.; Zheng, H.; Yu, J.; Song, T.; et al. Regulatory MiR-148a-ACVR1/BMP Circuit Defines a Cancer Stem Cell-like Aggressive Subtype of Hepatocellular Carcinoma. Hepatology 2015, 61, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Jiao, J.; Cermelli, S.; Muir, K.; Jung, K.H.; Zou, R.; Rashid, A.; Gagea, M.; Zabludoff, S.; Kalluri, R.; et al. miR-21 Inhibition Reduces Liver Fibrosis and Prevents Tumor Development by Inducing Apoptosis of CD24+ Progenitor Cells. Cancer Res. 2015, 75, 1859–1867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selaru, F.M.; Olaru, A.V.; Kan, T.; David, S.; Cheng, Y.; Mori, Y.; Yang, J.; Paun, B.; Jin, Z.; Agarwal, R.; et al. MicroRNA-21 is Overexpressed in Human Cholangiocarcinoma and Regulates Programmed Cell Death 4 and Tissue Inhibitor of Metalloproteinase 3. Hepatology 2009, 49, 1595–1601. [Google Scholar] [CrossRef]
- Jeliazkova, P.; Jörs, S.; Lee, M.; Zimber-Strobl, U.; Ferrer, J.; Schmid, R.M.; Siveke, J.T.; Geisler, F. Canonical Notch2 Signaling Determines Biliary Cell Fates of Embryonic Hepatoblasts and Adult Hepatocytes Independent of Hes1. Hepatology 2013, 57, 2469–2479. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gores, G.J. Notch-Driven Carcinogenesis: The Merging of Hepatocellular Cancer and Cholangiocarcinoma into a Common Molecular Liver Cancer Subtype. J. Hepatol. 2013, 58, 1244–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; He, Y.; Mackowiak, B.; Gao, B. MicroRNAs as Regulators, Biomarkers and Therapeutic Targets in Liver Diseases. Gut 2020, 10, 1136. [Google Scholar] [CrossRef]
- Tomimaru, Y.; Eguchi, H.; Nagano, H.; Wada, H.; Kobayashi, S.; Marubashi, S.; Tanemura, M.; Tomokuni, A.; Takemasa, I.; Umeshita, K.; et al. Circulating microRNA-21 as a Novel Biomarker for Hepatocellular Carcinoma. J. Hepatol. 2012, 56, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Chen, X.; Liang, C.; Ling, Y.; Yang, X.; Ye, X.; Zhang, H.; Yang, P.; Cui, X.; Ren, Y.; et al. A Noncoding Regulatory RNAs Network Driven by Circ-CDYL Acts Specifically in the Early Stages Hepatocellular Carcinoma. Hepatology 2020, 71, 130–147. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Engelman, D.M.; Slack, F.J. OncomiR or Tumor Suppressor? The Duplicity of MicroRNAs in Cancer. Cancer Res. 2016, 76, 3666–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramantieri, L.; Pollutri, D.; Gagliardi, M.; Giovannini, C.; Quarta, S.; Ferracin, M.; Casadei-Gardini, A.; Callegari, E.; De Carolis, S.; Marinelli, S.; et al. MiR-30e-3p Influences Tumor Phenotype through MDM2/TP53 Axis and Predicts Sorafenib Resistance in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 1720–1734. [Google Scholar] [CrossRef]
- Lobry, C.; Oh, P.; Mansour, M.R.; Look, A.T.; Aifantis, I. Notch Signaling: Switching an Oncogene to a Tumor Suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radtke, F.; Raj, K. The Role of Notch in Tumorigenesis: Oncogene or Tumour Suppressor? Nat. Rev. Cancer 2003, 3, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.Q.; Nolasco, S.; Soares, H. Non-Coding RNAs: Multi-Tasking Molecules in the Cell. Int. J. Mol. Sci. 2013, 14, 16010–16039. [Google Scholar] [CrossRef]
- Gutschner, T.; Diederichs, S. The Hallmarks of Cancer. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramantieri, L.; Baglioni, M.; Fornari, F.; Laginestra, M.A.; Ferracin, M.; Indio, V.; Ravaioli, M.; Cescon, M.; De Pace, V.; Leoni, S.; et al. LncRNAs as Novel Players in Hepatocellular Carcinoma Recurrence. Oncotarget 2018, 9, 35085–35099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, D.; Chen, Y.; Li, X.; Li, J.; Zhao, Y.; Shen, J.; Du, F.; Kaboli, P.J.; Li, M.; Wu, X.; et al. Long Non-Coding RNAs: Potential Biomarkers and Targets for Hepatocellular Carcinoma Therapy and Diagnosis. Int. J. Biol. Sci. 2021, 17, 220–235. [Google Scholar] [CrossRef]
- Zhang, H.-F.; Li, W.; Han, Y.-D. LINC00261 Suppresses Cell Proliferation, Invasion and Notch Signaling Pathway in Hepatocellular Carcinoma. Cancer Biomark. 2018, 21, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liu, J.-Z.; Hu, G.-J.; Shi, L.-L.; Lan, T. Promotion of Proliferation and Metastasis of Hepatocellular Carcinoma by LncRNA00673 Based on the Targeted-Regulation of Notch Signaling Pathway. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3412–3420. [Google Scholar] [PubMed]
- Yoon, J.-H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 Suppresses Target mRNA Translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Jia, M.; Jiang, L.; Wang, Y.-D.; Huang, J.-Z.; Yu, M.; Xue, H.-Z. LincRNA-p21 Inhibits Invasion and Metastasis of Hepatocellular Carcinoma through Notch Signaling-Induced Epithelial-Mesenchymal Tran-Sition. Hepatol. Res. 2016, 46, 1137–1144. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, H.; Wan, X.; Yang, X.; Zhu, C.; Wang, A.; He, L.; Miao, R.; Chen, S.; Zhao, H. Long Noncoding RNA Plays a Key Role in Metastasis and Prognosis of Hepatocellular Carcinoma. BioMed Res. Int. 2014, 2014, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Abbastabar, M.; Sarfi, M.; Golestani, A.; Khalili, E. lncRNA Involvement in Hepatocellular Carcinoma Metastasis and Prognosis. EXCLI J. 2018, 17, 900–913. [Google Scholar] [PubMed]
- Sonohara, F.; Inokawa, Y.; Hayashi, M.; Yamada, S.; Sugimoto, H.; Fujii, T.; Kodera, Y.; Nomoto, S. Prognostic Value of Long Non-Coding RNA HULC and MALAT1 Following the Curative Resection of Hepatocellular Carcinoma. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.J.; Xie, S.L.; Li, Q.; Ma, J.; Wang, G.Y. Large Intervening Non-Coding RNA HOTAIR is Associated with Hepatocellular Carcinoma Progression. J. Int. Med. Res. 2011, 39, 2119–2128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, H.; Li, B.; He, J.; Zeng, Y.; Zhang, Y.; He, F. lncRNA HULC Promotes the Growth of Hepatocellular Carcinoma Cells via Stabilizing COX-2 Protein. Biochem. Biophys. Res. Commun. 2017, 490, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Quagliata, L.; Matter, M.S.; Piscuoglio, S.; Arabi, L.; Ruiz, C.; Procino, A.; Kovac, M.; Moretti, F.; Makowska, Z.; Boldanova, T.; et al. Long Noncoding RNA HOTTIP/HOXA13 Expression Is Associated with Disease Progression and Predicts Outcome in Hepatocellular Carcinoma Patients. Hepatology 2014, 59, 911–923. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Yang, Z.; Zhang, J.; Xie, H.; Zhou, L.; Zheng, S. Long Noncoding RNA HOTTIP Expression Predicts Tumor Recurrence in Hepatocellular Carcinoma Patients Following Liver Transplantation. Hepatobiliary Surg. Nutr. 2018, 7, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Fan, R.-G.; Qin, C.-L.; Jia, J.; Wu, X.-D.; Zha, W.-Z. LncRNA-H19 Activates CDC42/PAK1 Pathway to Promote Cell Proliferation, Migration and Invasion by Targeting miR-15b in Hepatocellular Carcinoma. Genomes 2019, 111, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Gamaev, L.; Mizrahi, L.; Friehmann, T.; Rosenberg, N.; Pappo, O.; Olam, D.; Zeira, E.; Halpern, K.B.; Caruso, S.; Zucman-Rossi, J.; et al. The Pro-Oncogenic Effect of the lncRNA H19 in The Development of Chronic Inflammation-Mediated Hepatocellular Carcinoma. Oncogene 2021, 40, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, J.; Kan, A.; Hao, T.; Huang, L. SNHG and UCA1 As Prognostic Molecular Biomarkers in Hepatocellular Carcinoma: Recent Research and Meta-Analysis. Minerva Med. 2017, 108, 568–574. [Google Scholar] [PubMed]
- Zhou, Y.; Li, Y.; Wang, N.; Li, X.; Zheng, J.; Ge, L. UPF1 Inhibits the Hepatocellular Carcinoma Progression by Targeting Long Non-coding RNA UCA1. Sci. Rep. 2019, 9, 6652. [Google Scholar] [CrossRef]
- Masiero, M.; Li, D.; Whiteman, P.; Bentley, C.; Greig, J.; Hassanali, T.; Watts, S.; Stribbling, S.; Yates, J.; Bealing, E.; et al. Development of Therapeutic Anti-JAGGED1 Antibodies for Cancer Therapy. Mol. Cancer Ther. 2019, 18, 2030–2042. [Google Scholar] [CrossRef] [Green Version]
- Thurber, G.M.; Zajic, S.C.; Wittrup, K.D. Theoretic Criteria for Antibody Penetration into Solid Tumors and Micrometastases. J. Nucl. Med. 2007, 48, 995–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurber, G.M.; Schmidt, M.M.; Wittrup, K.D. Factors Determining Antibody Distribution in Tumors. Trends Pharmacol. Sci. 2008, 29, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Tannock, I.F. The Distribution of the Therapeutic Monoclonal Antibodies Cetuximab and Trastuzumab within Solid Tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic Antibodies: Successes, Limitations and Hopes for the Future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Tsukumo, S.-I.; Yasutomo, K. Regulation of CD8+ T Cells and Antitumor Immunity by Notch Signaling. Front. Immunol. 2018, 9, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sierra, R.A.; Thevenot, P.; Raber, P.L.; Cui, Y.; Parsons, C.; Ochoa, A.C.; Trillo-Tinoco, J.; Del Valle, L.; Rodriguez, P.C. Rescue of Notch-1 Signaling in Antigen-Specific CD8+ T Cells Overcomes Tumor-Induced T-cell Suppression and Enhances Immunotherapy in Cancer. Cancer Immunol. Res. 2014, 2, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roti, G.; Sorrentino, C.; Cuneo, A. Therapeutic Targeting of Notch Signaling Pathway in Hematological Malignancies. Mediterr. J. Hematol. Infect. Dis. 2019, 11, e2019037. [Google Scholar] [CrossRef] [PubMed]
- Nolin, E.; Gans, S.; Llamas, L.; Bandyopadhyay, S.; Brittain, S.M.; Bernasconi-Elias, P.; Carter, K.P.; Loureiro, J.J.; Thomas, J.R.; Schirle, M.; et al. Discovery of a ZIP7 Inhibitor from a Notch Pathway Screen. Nat. Chem. Biol. 2019, 15, 179–188. [Google Scholar] [CrossRef]
- Fortini, M.E. Notch Signaling: The Core Pathway and Its Posttranslational Regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Pagliaro, L.; Marchesini, M.; Roti, G. Targeting Oncogenic Notch Signaling with SERCA Inhibitors. J. Hematol. Oncol. 2021, 14, 1–17. [Google Scholar] [CrossRef]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef] [PubMed]
- Arun, G.; Diermeier, S.; Akerman, M.; Chang, K.-C.; Wilkinson, J.E.; Hearn, S.; Kim, Y.; MacLeod, A.R.; Krainer, A.R.; Norton, L.; et al. Differentiation of Mammary Tumors and Reduction in Metastasis UponMalat1lncRNA Loss. Genes Dev. 2016, 30, 34–51. [Google Scholar] [CrossRef] [Green Version]
- Gong, N.; Teng, X.; Li, J.; Liang, X.-J. Antisense Oligonucleotide-Conjugated Nanostructure-Targeting lncRNA MALAT1 Inhibits Cancer Metastasis. ACS Appl. Mater. Interfaces 2018, 11, 37–42. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Size (bp) | Deregulation in HCC | Role in HCC | Ref |
|---|---|---|---|---|
| MALAT1 | 8708 | Upregulated | Tumor metastasis and recurrence | [130,131,132] |
| HOTAIR | 12649 | Upregulated | Associated with invasion | [130,133] |
| HULC | 1638 | Upregulated | Associated with tumor growth | [131,134] |
| HOTTIP | 6839 | Upregulated | Associated with tumor progression | [135,136] |
| H19 | 2660 | Upregulated | Promotes cell proliferation | [137,138] |
| UCA1 | 7375 | Upregulated | Associated with disease outcome | [139,140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giovannini, C.; Fornari, F.; Piscaglia, F.; Gramantieri, L. Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs. Cells 2021, 10, 521. https://doi.org/10.3390/cells10030521
Giovannini C, Fornari F, Piscaglia F, Gramantieri L. Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs. Cells. 2021; 10(3):521. https://doi.org/10.3390/cells10030521
Chicago/Turabian StyleGiovannini, Catia, Francesca Fornari, Fabio Piscaglia, and Laura Gramantieri. 2021. "Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs" Cells 10, no. 3: 521. https://doi.org/10.3390/cells10030521
APA StyleGiovannini, C., Fornari, F., Piscaglia, F., & Gramantieri, L. (2021). Notch Signaling Regulation in HCC: From Hepatitis Virus to Non-Coding RNAs. Cells, 10(3), 521. https://doi.org/10.3390/cells10030521