
Towards a Framework for Better Understanding of Quiescent Cancer Cells

 ,
,  , ,
, ,  ,
,
Abstract
:1. Introduction
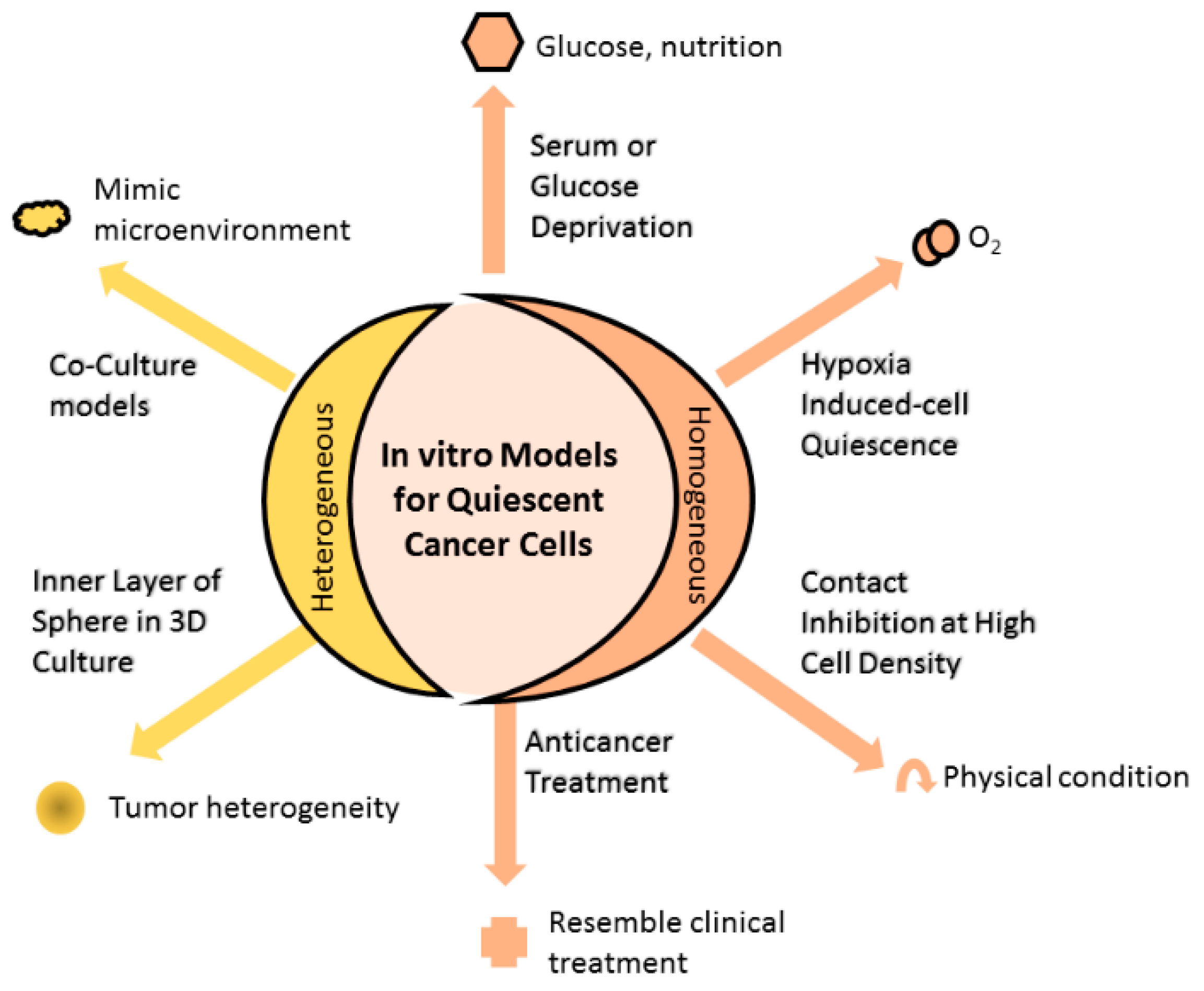
2. Models Mimicking Quiescent Cancer Cells
2.1. Homogenous In Vitro Models of QCC
2.1.1. Serum or Glucose Deprivation
2.1.2. Hypoxia-Induced Cell Quiescence
2.1.3. Contact Inhibition at High Cell Density
2.1.4. Anticancer Treatment Increase Proportion of QCC
2.2. Heterogenous In Vitro Models of QCC
2.2.1. Inner Layer of Spheres in Three-Dimensional (3D) Culture
2.2.2. Coculture Models
2.3. In Vivo Quiescent Models
2.4. Discussion
3. Measurement of Quiescent Cancer Cells
3.1. Ki-67
3.2. RNA and DNA Content
3.3. Fluorescence Ubiquitin Cell Cycle Indicator (FUCCI)
3.4. Potential QCC Molecular Markers
3.5. Pulse-Chase Identification
3.6. Discussion
4. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AO | Acridine orange |
| BME | Basement membrane extract |
| BrdU | 5-bromo-2′-deoxyuridine |
| CFSE | Carboxyfluorescein succinimidyl ester |
| CoCl2 | Cobalt chloride |
| cPLA2α | Cytosolic phospholipase A2α |
| DAPI | 4′,6′-diamino-2-phenylindole |
| DiD | 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindo-dicarbocyanine |
| Dil | 1,1′-dioctadecyl-3,3,3′3′-tetramethylindocarbocyanine perchlorate |
| DYRK1B | Dual specificity tyrosine phosphorylation-regulated kinase 1B |
| DTCs | Disseminating tumor cells |
| EdU | 5-ethynyl-2′-deoxyuridine |
| FUCCI | Fluorescence ubiquitin cell cycle indicator |
| GOS2 | G0/G1 switch gene 2 |
| HIF-1 | Hypoxia-inducible factor-1 |
| HUVECs | Human umbilical vein endothelial cells |
| IGF-1 | Insulin-like growth factor |
| miRNAs | MicroRNAs |
| MMC | Mouse mammary carcinoma |
| NR2F1 | Nuclear receptor subfamily 2 group F member 1 |
| PDGF | Platelet-derived growth factor |
| PI | Propidium iodide |
| QCCs | Quiescent cancer cells |
| Rb | Retinoblastoma protein |
| ROS | Reactive oxygen species |
| SFM | Serum- and cytokine-free medium |
| TBK1 | TANK binding kinase 1 |
| TSP-1 | Thrombospondin-1 |
References
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Comprehensive Network (NCCN). Breast Cancer, Version 4.2020, NCCN Clinical Practice Guidelines in Oncology. Available online: https://www.nccn.org/professionals/physician_gls/pdf/breast.pdf (accessed on 16 June 2020).
- Yao, G. Modelling mammalian cellular quiescence. Interface Focus 2014, 4, 34458–34474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci. 2019, 110, 474–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pack, L.R.; Daigh, L.H.; Meyer, T. Putting the brakes on the cell cycle: Mechanisms of cellular growth arrest. Curr. Opin. Cell Biol. 2019, 60, 106–113. [Google Scholar] [CrossRef]
- Terzi, M.Y.; Izmirli, M.; Gogebakan, B. The cell fate: Senescence or quiescence. Mol. Biol. Rep. 2016, 43, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of cancer cell dormancy–another hallmark of cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kida, A.; Kahn, M. Hypoxia selects for a quiescent, CML stem/leukemia initiating-like population dependent on CBP/catenin transcription. Curr. Mol. Pharmacol. 2013, 6, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Ruch, R.J. Cell-cell communication in carcinogenesis. Front. Biosci. 1998, 3, d208–d236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; de Milito, A.; Olofsson, M.H.; Gullbo, J.; D’Arcy, P.; Linder, S. Targeting mitochondrial function to treat quiescent tumor cells in solid tumors. Int. J. Mol. Sci. 2015, 16, 27313–27326. [Google Scholar] [CrossRef] [Green Version]
- Jin, K.; Ewton, D.Z.; Park, S.; Hu, J.; Friedman, E. Mirk regulates the exit of colon cancer cells from quiescence. J. Biol. Chem. 2009, 284, 22916–22925. [Google Scholar] [CrossRef] [Green Version]
- Xi, Z.; Yao, M.; Li, Y.; Xie, C.; Holst, J.; Liu, T.; Cai, S.; Lao, Y.; Tan, H.; Xu, H.-X.; et al. Guttiferone K impedes cell cycle re-entry of quiescent prostate cancer cells via stabilization of FBXW7 and subsequent c-MYC degradation. Cell Death Dis. 2016, 7, e2252. [Google Scholar] [CrossRef] [Green Version]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-cycle regulation accounts for variability in Ki-67 expression levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Sederstrom, J.M. Assaying cell cycle status using flow cytometry. Curr. Protoc. Mol. Biol. 2015, 111, 28.6.1–28.6.11. [Google Scholar] [CrossRef]
- Lee, H.R.; Leslie, F.; Azarin, S.M. A facile in vitro platform to study cancer cell dormancy under hypoxic microenvironments using CoCl2. J. Biol. Eng. 2018, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Xie, C.; Kiang, M.-Y.; Teng, Y.; Harman, D.; Tiffen, J.; Wang, Q.; Sved, P.; Bao, S.; Witting, P.; et al. Targeting of cytosolic phospholipase A2 alpha impedes cell cycle re-entry of quiescent prostate cancer cells. Oncotarget 2015, 6, 34458–34474. [Google Scholar] [CrossRef] [Green Version]
- Yano, S.; Takehara, K.; Tazawa, H.; Kishimoto, H.; Urata, Y.; Kagawa, S.; Fujiwara, T.; Hoffman, R.M. Cell-cycle-dependent drug-resistant quiescent cancer cells induce tumor angiogenesis after chemotherapy as visualized by real-time FUCCI imaging. Cell Cycle 2017, 16, 406–414. [Google Scholar] [CrossRef]
- Klein, C.A. Cancer progression and the invisible phase of metastatic colonization. Nat. Rev. Cancer 2020, 20, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Nakhla, H.; Friedman, E. Transient arrest in a quiescent state allows ovarian cancer cells to survive suboptimal growth conditions and is mediated by both Mirk/dyrk1b and p130/RB2. Int. J. Cancer 2011, 129, 307–318. [Google Scholar] [CrossRef]
- Coller, H.A.; Sang, L.; Roberts, J.M. A new description of cellular quiescence. PLoS Biol. 2006, 4, e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, X.Y.; Dai, Y.; Youssefian, L.E.; Chen, S.; Bodie, W.W.; Takabatake, Y.; Felthousen, J.; Almenara, J.A.; Kramer, L.B.; Dent, P.; et al. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood 2011, 118, 5189–5200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, L.; Xie, C.; Yao, M.; Hnit, S.S.T.; Vignarajan, S.; Wang, Y.; Wang, Q.; Xi, Z.; Xu, H.; Li, Z.; et al. The histone chaperone complex FACT promotes proliferative switch of G0 cancer cells. Int. J. Cancer 2019, 145, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Ewton, D.Z.; Hu, J.; Vilenchik, M.; Deng, X.; Luk, K.-C.; Polonskaia, A.; Hoffman, A.F.; Zipf, K.; Boylan, J.F.; Friedman, E.A. Inactivation of mirk/dyrk1b kinase targets quiescent pancreatic cancer cells. Mol. Cancer Ther. 2011, 10, 2104–2114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pause, A.; Lee, S.; Lonergan, K.M.; Klausner, R.D. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc. Natl. Acad. Sci. USA 1998, 95, 993–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avril, P.; Duteille, F.; Ridel, P.; Heymann, M.-F.; De Pinieux, G.; Rédini, F.; Blanchard, F.; Heymann, D.; Trichet, V.; Perrot, P. Opposite Effects of Soluble Factors Secreted by Adipose Tissue on Proliferating and Quiescent Osteosarcoma Cells. Plast. Reconstr. Surg. 2016, 137, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Forbes, N.S. Single-cell analysis demonstrates how nutrient deprivation creates apoptotic and quiescent cell populations in tumor cylindroids. Biotechnol. Bioeng. 2008, 101, 797–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, H.; Okuyama, H.; Ohue, M.; Inoue, M. Dormancy of cancer cells with suppression of AKT activity contributes to survival in chronic hypoxia. PLoS ONE 2014, 9, e98858. [Google Scholar] [CrossRef]
- Gade, T.P.F.; Tucker, E.; Nakazawa, M.S.; Hunt, S.J.; Wong, W.; Krock, B.; Weber, C.N.; Nadolski, G.J.; Clark, T.W.I.; Soulen, M.C.; et al. Ischemia Induces Quiescence and Autophagy Dependence in Hepatocellular Carcinoma. Radiology 2017, 283, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Gos, M.; Miloszewska, J.; Swoboda, P.; Trembacz, H.; Skierski, J.; Janik, P. Cellular quiescence induced by contact inhibition or serum withdrawal in C3H10T1/2 cells. Cell Prolif. 2005, 38, 107–116. [Google Scholar] [CrossRef]
- Fuse, T.; Tanikawa, M.; Nakanishi, M.; Ikeda, K.; Tada, T.; Inagaki, H.; Asai, K.; Kato, T.; Yamada, K. p27Kip1 expression by contact inhibition as a prognostic index of human glioma. J. Neurochem. 2000, 74, 1393–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onozato, Y.; Kaida, A.; Harada, H.; Miura, M. Radiosensitivity of quiescent and proliferating cells grown as multicellular tumor spheroids. Cancer Sci. 2017, 108, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellor, H.R.; Ferguson, D.J.P.; Callaghan, R. A model of quiescent tumour microregions for evaluating multicellular resistance to chemotherapeutic drugs. Br. J. Cancer 2005, 93, 302–309. [Google Scholar] [CrossRef] [Green Version]
- Martins-Neves, S.R.; Corver, W.E.; Paiva-Oliveira, D.I.; Akker, B.E.W.M.V.D.; Briaire-De-Bruijn, I.H.; Bovée, J.V.M.G.; Gomes, C.M.F.; Cleton-Jansen, A.-M. Osteosarcoma Stem Cells Have Active Wnt/β-catenin and Overexpress SOX2 and KLF4. J. Cell Physiol. 2016, 231, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, R.; Broglie, J.J.; Adcock, A.F.; Yang, L. Three-dimensional cell culture systems and their applications in drug discovery and cell-based biosensors. Assay Drug Dev. Technol. 2014, 12, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sant, S.; Johnston, P.A. The production of 3D tumor spheroids for cancer drug discovery. Drug Discov. Today Technol. 2017, 23, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Alves, C.P.; Dey-Guha, I.; Kabraji, S.; Yeh, A.C.; Talele, N.P.; Solé, X.; Chowdhury, J.; Mino-Kenudson, M.; Loda, M.; Sgroi, D.; et al. AKT1(low) Quiescent Cancer Cells Promote Solid Tumor Growth. Mol. Cancer Ther. 2018, 17, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.-H.; Mu, L.; Li, X.-L.; Hu, Y.-B.; Liu, H.; Han, L.-T.; Gong, J.-P. Characterization and functional analysis of a slow-cycling subpopulation in colorectal cancer enriched by cell cycle inducer combined chemotherapy. Oncotarget 2017, 8, 78466–78479. [Google Scholar] [CrossRef] [PubMed]
- Touil, Y.; Igoudjil, W.; Corvaisier, M.; Dessein, A.-F.; Vandomme, J.; Monté, D.; Stechly, L.; Skrypek, N.; Langlois, C.; Grard, G.; et al. Colon cancer cells escape 5FU chemotherapy-induced cell death by entering stemness and quiescence associated with the c-Yes/YAP axis. Clin. Cancer Res. 2014, 20, 837–846. [Google Scholar] [CrossRef] [Green Version]
- Aqbi, H.F.; Tyutyunyk-Massey, L.; Keim, R.C.; Butler, S.E.; Thekkudan, T.; Joshi, S.; Smith, T.M.; Bandyopadhyay, D.; Idowu, M.O.; Bear, H.D.; et al. Autophagy-deficient breast cancer shows early tumor recurrence and escape from dormancy. Oncotarget 2018, 9, 22113–22122. [Google Scholar] [CrossRef] [Green Version]
- Eckers, J.C.; Kalen, A.L.; Sarsour, E.H.; Tompkins, V.S.; Janz, S.; Son, J.M.; Doskey, C.M.; Buettner, G.R.; Goswami, P.C. Forkhead box M1 regulates quiescence-associated radioresistance of human head and neck squamous carcinoma cells. Radiat. Res. 2014, 182, 420–429. [Google Scholar] [CrossRef] [Green Version]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; De Almeida, D.L.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.R.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- Kim, J.K.; Jung, Y.; Wang, J.; Joseph, J.; Mishra, A.; Hill, E.E.; Krebsbach, P.H.; Pienta, K.J.; Shiozawa, Y.; Taichman, R.S. TBK1 regulates prostate cancer dormancy through mTOR inhibition. Neoplasia 2013, 15, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-U.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Yumoto, K.; Eber, M.R.; Wang, J.; Cackowski, F.C.; Decker, A.M.; Lee, E.; Nobre, A.R.; Aguirre-Ghiso, J.A.; Jung, Y.; Taichman, R.S.; et al. Axl is required for TGF-beta2-induced dormancy of prostate cancer cells in the bone marrow. Sci. Rep. 2016, 6, 36520. [Google Scholar] [CrossRef] [PubMed]
- Khoo, W.H.; Ledergor, G.; Weiner, A.; Roden, D.L.; Terry, R.L.; McDonald, M.M.; Chai, R.C.; De Veirman, K.; Owen, K.L.; Opperman, K.S.; et al. A niche-dependent myeloid transcriptome signature defines dormant myeloma cells. Blood Cells 2019, 134, 30–43. [Google Scholar] [CrossRef]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.-Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BitMansour, A.; Pop, L.M.; Vitetta, E.S. The Role of Regulatory B Cell-Like Malignant Cells and Treg Cells in the Mouse Model of BCL1 Tumor Dormancy. PLoS ONE 2016, 11, e0167618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlow, R.; Honeth, G.; Lombardi, S.; Cariati, M.; Hessey, S.M.; Pipili, A.; Mariotti, V.; Buchupalli, B.; Foster, K.; Bonnet, D.; et al. A novel model of dormancy for bone metastatic breast cancer cells. Cancer Res. 2013, 73, 6886–6899. [Google Scholar] [CrossRef] [Green Version]
- Barkan, D.; El Touny, L.H.; Michalowski, A.M.; Smith, J.A.; Chu, I.; Davis, A.S.; Webster, J.D.; Hoover, S.; Simpson, R.M.; Gauldie, J.; et al. Metastatic growth from dormant cells induced by a col-I-enriched fibrotic environment. Cancer Res. 2010, 70, 5706–5716. [Google Scholar] [CrossRef] [Green Version]
- Payne, K.K.; Keim, R.C.; Graham, L.; Idowu, M.O.; Wan, W.; Wang, X.-Y.; Toor, A.A.; Bear, H.D.; Manjili, M.H. Tumor-reactive immune cells protect against metastatic tumor and induce immunoediting of indolent but not quiescent tumor cells. J. Leukoc. Biol. 2016, 100, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Fryknäs, M.; Hernlund, E.; Fayad, W.; De Milito, A.; Olofsson, M.H.; Gogvadze, V.; Dang, L.; Påhlman, S.; Schughart, L.A.K.; et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat. Commun. 2014, 5, 3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, J.; Eder, W.; Castaneda, J.; Doss, M.; Huber, E.; Ebner, R.; Kunz-Schughart, L.A. A reliable tool to determine cell viability in complex 3-d culture: The acid phosphatase assay. J. Biomol. Screen. 2007, 12, 925–937. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Aulestia, F.J.; Kahn, S.A.; Zeniou, M.; Dubois, L.G.; El-Habr, E.A.; Daubeuf, F.; Tounsi, N.; Cheshier, S.H.; Frossard, N.; et al. Bisacodyl and its cytotoxic activity on human glioblastoma stem-like cells. Implication of inositol 1,4,5-triphosphate receptor dependent calcium signaling. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Weidenfeld, K.; Schif-Zuck, S.; Abu-Tayeh, H.; Kang, K.; Kessler, O.; Weissmann, M.; Neufeld, G.; Barkan, D. Dormant tumor cells expressing LOXL2 acquire a stem-like phenotype mediating their transition to proliferative growth. Oncotarget 2016, 7, 71362–71377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uhr, J.W.; Marches, R. Dormancy in a model of murine B cell lymphoma. Semin. Cancer Biol. 2001, 11, 277–283. [Google Scholar] [CrossRef]
- Ottewell, P.D.; Quayle, L.A. Tumor dormancy in the bone. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2019; pp. 1–14. [Google Scholar]
- García-Gutiérrez, L.; Delgado, M.D.; León, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, L.B.; Luengo, A.; Danai, L.V.; Bush, L.N.; Diehl, F.F.; Hosios, A.M.; Lau, A.N.; Elmiligy, S.; Malstrom, S.; Lewis, C.A.; et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat. Cell Biol. 2018, 20, 782–788. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Gala, H.P.; Mishra, R.K.; Dhawan, J. Distinguishing states of arrest: Genome-wide descriptions of cellular quiescence using ChIP-Seq and RNA-Seq analysis. Methods Mol. Biol. 2018, 1686, 215–239. [Google Scholar] [PubMed]
- So, W.K.; Cheung, T.H. Molecular regulation of cellular quiescence: A perspective from adult stem cells and its niches. In Cellular Quiescence. Methods in Molecular Biology; Daniel Lacorazza, H., Ed.; Humana Press: New York, NY, USA, 2018; pp. 1–25. [Google Scholar]
- Van Staveren, W.C.; Solís, D.Y.; Hébrant, A.; Detours, V.; Dumont, J.E.; Maenhaut, C. Human cancer cell lines: Experimental models for cancer cells in situ? For cancer stem cells? Biochim. Biophys. Acta 2009, 1795, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Darzynkiewicz, Z. Differential staining of DNA and RNA in intact cells and isolated cell nuclei with acridine orange. Methods Cell Biol. 1990, 33, 285–298. [Google Scholar]
- Oh, M.; Tanaka, T.; Kobayashi, M.; Furukawa, T.; Mori, T.; Kudo, T.; Fujieda, S.; Fujibayashi, Y. Radio-copper-labeled Cu-ATSM: An indicator of quiescent but clonogenic cells under mild hypoxia in a Lewis lung carcinoma model. Nucl. Med. Biol. 2009, 36, 419–426. [Google Scholar] [CrossRef]
- Eddaoudi, A.; Canning, S.L.; Kato, I. Flow cytometric detection of G0 in live cells by Hoechst 33342 and Pyronin Y staining. Methods Mol. Biol. 2018, 1686, 49–57. [Google Scholar]
- Byvaltsev, V.A.; Bardonova, L.A.; Onaka, N.R.; Polkin, R.A.; Ochkal, S.V.; Shepelev, V.V.; Aliyev, M.A.; Potapov, A.A. Acridine Orange: A Review of Novel Applications for Surgical Cancer Imaging and Therapy. Front. Oncol. 2019, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Rothaeusler, K.; Baumgarth, N. Assessment of cell proliferation by 5-bromodeoxyuridine (BrdU) labeling for multicolor flow cytometry. Curr. Protoc. Cytom. 2007, 40, 7–31. [Google Scholar] [CrossRef] [PubMed]
- Tomura, M.; Sakaue-Sawano, A.; Mori, Y.; Takase-Utsugi, M.; Hata, A.; Ohtawa, K.; Kanagawa, O.; Miyawaki, A. Contrasting quiescent G0 phase with mitotic cell cycling in the mouse immune system. PLoS ONE 2013, 8, e73801. [Google Scholar] [CrossRef] [PubMed]
- Oki, T.; Nishimura, K.; Kitaura, J.; Togami, K.; Maehara, A.; Izawa, K.; Sakaue-Sawano, A.; Niida, A.; Miyano, S.; Aburatani, H.; et al. A novel cell-cycle-indicator, mVenus-p27K-, identifies quiescent cells and visualizes G0–G1 transition. Sci. Rep. 2014, 4, 4012. [Google Scholar] [CrossRef] [Green Version]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARbeta-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [Green Version]
- Dey-Guha, I.; Wolfer, A.; Yeh, A.C.; Albeck, J.G.; Darp, R.; Leon, E.; Wulfkuhle, J.; Petricoin, E.F.; Wittner, B.S.; Ramaswamy, S. Asymmetric cancer cell division regulated by AKT. Proc. Natl. Acad. Sci. USA 2011, 108, 12845–12850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heckmann, B.L.; Zhang, X.; Xie, X.; Liu, J. The G0/G1 switch gene 2 (G0S2): Regulating metabolism and beyond. Biochim. Biophys. Acta 2013, 1831, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; Park, C.S.; Shen, Y.; Rabin, K.R.; Lacorazza, H.D. G0S2 inhibits the proliferation of K562 cells by interacting with nucleolin in the cytosol. Leuk. Res. 2014, 38, 210–217. [Google Scholar] [CrossRef] [Green Version]
- Moore, N.; Houghton, J.; Lyle, S. Slow-cycling therapy-resistant cancer cells. Stem Cells Dev. 2012, 21, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Quayle, L.A.; Ottewell, P.D.; Holen, I. Chemotherapy resistance and stemness in mitotically quiescent human breast cancer cells identified by fluorescent dye retention. Clin. Exp. Metastasis 2018, 35, 831–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kusumbe, A.P.; Bapat, S.A. Cancer stem cells and aneuploid populations within developing tumors are the major determinants of tumor dormancy. Cancer Res. 2009, 69, 9245–9253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Tuo, X.; Zhao, L.; Wang, Q.; Han, L.; Wang, Y.; Ma, S.; Feng, X.; Li, Q.; Sun, C.; Wang, Q.; et al. Validation of molecular typing for endometrial screening test that predicts benign and malignant lesions. Front. Oncol. 2019, 9, 561. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38alpha/beta signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [Green Version]
- Matatall, K.A.; Kadmon, C.S.; King, K.Y. Detecting hematopoietic stem cell proliferation using BrdU incorporation. In Cellular Quiescence. Methods in Molecular Biology; Daniel Lacorazza, H., Ed.; Humana Press: New York, NY, USA, 2018; pp. 91–103. [Google Scholar]
- Yumoto, K.; Berry, J.E.; Taichman, R.S.; Shiozawa, Y. A novel method for monitoring tumor proliferation in vivo using fluorescent dye DiD. Cytom. A 2014, 85, 548–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Silva, L.; Quevedo, L.; Varela, I. Tumor Functional Heterogeneity Unraveled by scRNA-seq Technologies. Trends Cancer 2020, 6, 13–19. [Google Scholar] [CrossRef] [Green Version]
- Andor, N.; Simonds, E.F.; Czerwinski, D.K.; Chen, J.; Grimes, S.M.; Wood-Bouwens, C.; Zheng, G.X.Y.; Kubit, M.A.; Greer, S.; Weiss, W.A.; et al. Single-cell RNA-Seq of follicular lymphoma reveals malignant B-cell types and coexpression of T-cell immune checkpoints. Blood 2019, 133, 1119–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Model | Method | Cell Line | Detection | Molecules/Pathways | Remarks |
|---|---|---|---|---|---|
| Serum deprivation | Serum free [11,12,16,19,22] or low serum medium [20,21,23,24] | Prostate (LNCaP) [12,16,22], ovarian (SKOV3, TOV21G, OVCAR3) [19], lung (H1975) [22], pancreatic (SU86.86, Panc1, AsPc1) [23], colon (HD6) [11], renal (786-0) [24], multiple myeloma (NCI-H929, RPMI8226, U266) [21], osteosarcoma (MNNG-HOS) [25] | Ki-67 [16,22] Hoechst/Pyronin Y [11,12,19,21,22,23], PI [12,20,21,23,24], BrdU [16,20,24], p27 [16,19,24] | Rb [12,19], c-Myc [12], Cyclin D [19], Mirk [11,19], p27 [11,19], cPLA2α [16] | -Some cell lines are not suitable for induction to QCCs, e.g., OVCAR4, OV90, HeLa [19,24]. |
| Nutrient deprivation | Low glucose [19] | Ovarian (SKOV3, TOV21G) [19] | Hoechst/Pyronin Y [19] | AMPK [19], cAMP [19], mTOR [19] | -With limited studies |
| Hypoxia | 1% oxygen [15,26,27,28] or cobalt chloride [15] | Breast (MCF-7, MDA-MB-231) [15], pancreatic (AsPC-1) [27], ovarian (OVCAR-3) [15], liver (HepG2, SNU-449, SNU-387, SNU-398) [28] leukemia (KCL22, K562) [8], MEF [26] | HIF-α [8,15,27], Pimonidazole [27], Hoechst/Pyronin Y [8], Hoechst [28], Ki-67 [15,28], BrdU [27], PI [15,27], DAPI [15], p38 [27], ERK [27], acridine orange [26] | HIF [8,15], AKT [27], ERK [27] p21 [15], Myc [15], VHL [15], catenin [8] | -Tumor cell types have dissimilar resistance towards acute and chronic hypoxia [27]. -Suitable for cancer intervention that causes hypoxia [28]. |
| Contact inhibition | Confluent culture cells [20,29] | Prostate (PC-3) [12,22], glioma (T98G, NAC6) [30], renal (786-0) [24], lung (A549) [22] | Ki-67 [22], Hoechst/Pyronin Y [12,22], PI [12,24,30], BrdU [24], mVenus-p27K [22], p27 [24,30] | C-Myc [12,22], VHL [29], pRb [12,22], p27 [22,30], FACT [22], Wnt [29], Cyclin D [29] | -Unsuitable for cell lines that continue to proliferate at confluent state [24,30] |
| 3D culture | (a) scaffold method [31]–BME [32], agarose [33], poly (2-hydroxyethyl methacrylate matrix) [26,34] (b) non-scaffold method-agitation, force-floating or hanging-drop method [35] | Colon (DLD-1) [33], breast (MCF-7 and MDA-MB-231) [32]; tongue (SAS) [31], osteosarcoma (MHM, MNNG-HOS, SJSA-I) [34], mouse mammary cancer cell (D2.0R and D2A1) [32], MEF [26] | Ki-67 [32,33,34], FUCCI [31], HIF-α [31], Pimonidazole [31], Hoechst [31], EdU [31], DAPI [32], p27 [33], acridine orange [26] | Low oxygen and nutrient supply, accumulation of metabolite [10], HIF-1α [36], ATG7 [32], p27 [33], Wnt-β catenin [34] | -Irregular size of spheroids [10,35]. -Irregular proportion of QCCs in spheroids [35]. |
| Cancer treatment | Chemotherapy or radiotherapy | Breast (MCF7, MDA-MB-231) [37], colon (HCT 116 [37,38], LoVo [38], HT29 [39]), lung (PC9) [37], melanoma (A375) [37], pancreas (SU86.86) [23], stomach (MKN45) [17] | Ki-67 [38,39], Hoechst/Pyronin Y [23], PI [23,38], DAPI [37], CFSE [38], FUCCI [17] | Autophagy [40], G6PD [41], Yes tyrosine kinase [39], Mirk [23] | -Resemble QCCs in tumors induced by treatments |
| Coculture | (a) organotypic coculture [42] (b) simple coculture [43,44,45,46,47] | Coculture endothelial cells (HUVECs) with breast cancer cells (T4-2, MCF-7, MDA-MB-231) [42]; coculture prostate PC-3 cancer cells with murine bone marrow stromal cell line ST2 cells [43]; coculture breast cancer cells (MDA-MB-231, T47D) with bone marrow stroma [44]; coculture prostate cancer cells (PC-3, DU145) with pre-osteoblastic cells (MC3T3-E1) [46]; coculture murine myeloma cancer cells (5TGM1) with pre-osteoblastic cells (MC3T3) [47]; coculture bone marrow-metastatic breast cancer cell (BM2) with bone marrow-mesenchymal stem cells (BM-MSCs) from human donor [45] | Ki-67 [42,43,46], Hoechst/Pyronin Y [44,46], DAPI [46], DiD [47], Hoechst [45], PKH26 [45] | TSP-1 [42], TBK1 [43], IKKE [43], miRNAs passage through GIJC [44], Axl [46,47], miR-23b [45] | -Mimic microenvironment in actual tumor, which allow for examining effect of microvascular niche [42] and tumor microenvironment [43] on QCCs |
| In vivo | Xenograft on BALB/c nude mice [48,49], BKAL mice [47], NOD/SCID mice [42,50], SCID mice [46] | Breast (MCF-7 [48], MDA-MB-231 [42,48,50], T4-2 [42]), mouse mammary cancer cell (D2.0R, D2A1) [48], prostate (PC-3, DU145) [46], murine myeloma (5TGM1) [47], lymphoma (BCL1) cell [49] | Ki-67 [42,46], DAPI [42,46,48], Hoechst [42], DiD [47], CFSE [49] | ATG7 [32], myosin light chain kinase (MLCK) [48], integrin β1 signaling [48], FAK [51], ERK [51], p38 [50], TGF-β [46,50], Axl [46,47] | -With limited studies |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nik Nabil, W.N.; Xi, Z.; Song, Z.; Jin, L.; Zhang, X.D.; Zhou, H.; De Souza, P.; Dong, Q.; Xu, H. Towards a Framework for Better Understanding of Quiescent Cancer Cells. Cells 2021, 10, 562. https://doi.org/10.3390/cells10030562
Nik Nabil WN, Xi Z, Song Z, Jin L, Zhang XD, Zhou H, De Souza P, Dong Q, Xu H. Towards a Framework for Better Understanding of Quiescent Cancer Cells. Cells. 2021; 10(3):562. https://doi.org/10.3390/cells10030562
Chicago/Turabian StyleNik Nabil, Wan Najbah, Zhichao Xi, Zejia Song, Lei Jin, Xu Dong Zhang, Hua Zhou, Paul De Souza, Qihan Dong, and Hongxi Xu. 2021. "Towards a Framework for Better Understanding of Quiescent Cancer Cells" Cells 10, no. 3: 562. https://doi.org/10.3390/cells10030562
APA StyleNik Nabil, W. N., Xi, Z., Song, Z., Jin, L., Zhang, X. D., Zhou, H., De Souza, P., Dong, Q., & Xu, H. (2021). Towards a Framework for Better Understanding of Quiescent Cancer Cells. Cells, 10(3), 562. https://doi.org/10.3390/cells10030562