
Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis

 , ,
, , 
Abstract
:1. Introduction
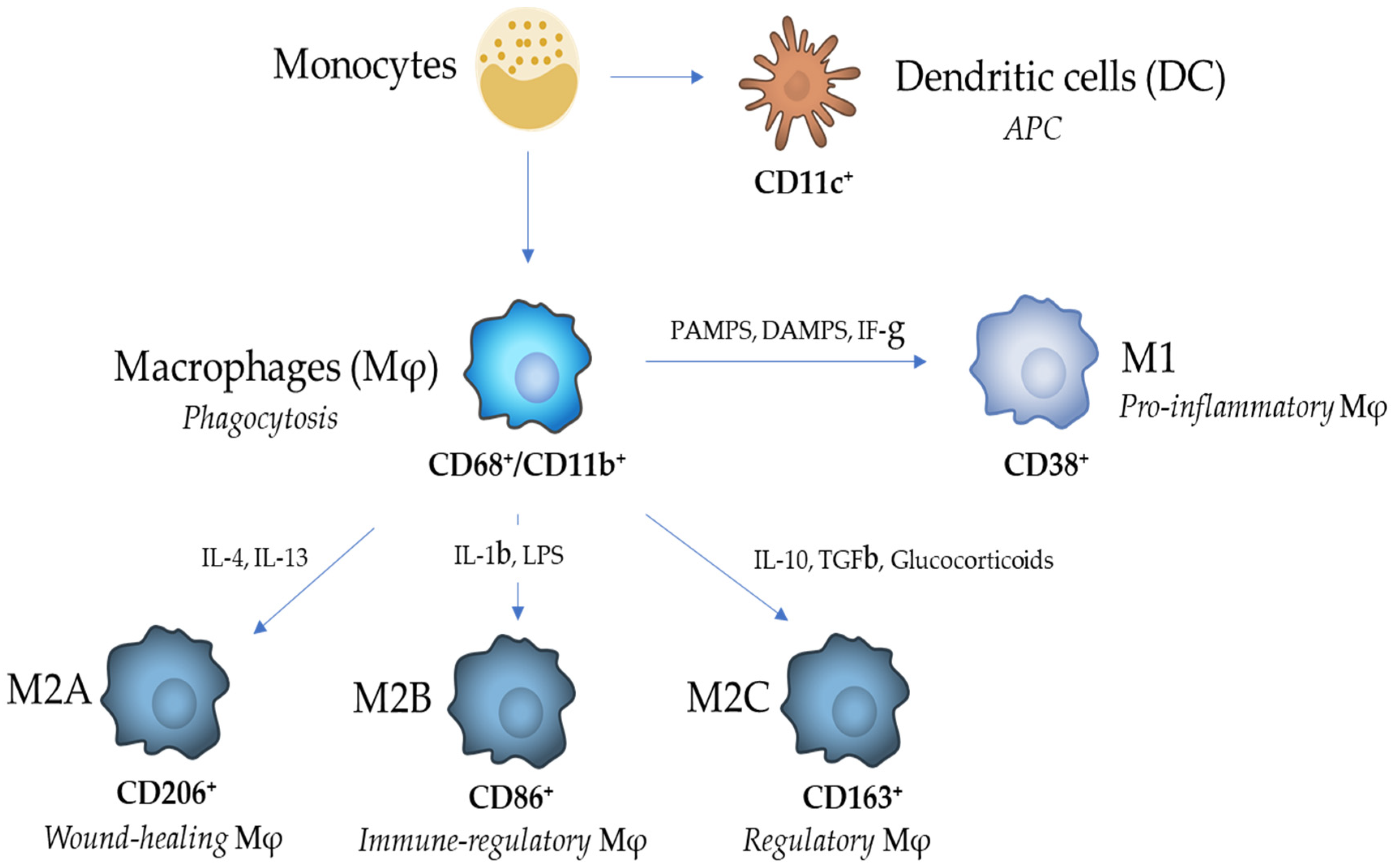
2. Macrophage Heterogeneity and Functions
3. Phenotype Overlap and Renal Compartmentation
4. Glomerular Macrophages in Human Glomerulonephritis
- The number of glomerular macrophages is correlated with the severity of proliferative GN.
- 2.
- Macrophages localize within glomeruli in the most severe lesions.
- 3.
- Macrophages are attracted within glomeruli and glomerular lesions by chemokines produced by intrinsic glomerular cells.
- 4.
- Attracted macrophages are activated in proliferative glomerular lesions.
- 5.
- The difficulty of identifying macrophage subpopulations in proliferative glomerular lesions.
- -
- no differences in numbers of glomerular M2b were observed between M0 and M1, S0 and S1, T0 and T1, C0 and C1. M2b macrophages only had an increased trend in glomeruli of G1 (global glomerulosclerosis), without any significant difference.
- -
- no differences in numbers of glomerular M2c were observed between M1 and M0, S1 and S0, C1 and C0. There were fewer M2c macrophages in glomeruli with T1 and G1.
5. Glomerular Macrophages in Renal Allograft Rejection
- The number of glomerular Mφs is correlated with severe allograft rejection, renal dysfunction and reduced graft survival.
- 2.
- Glomerulitis with predominant Mφ infiltration may represent a histological marker of humoral rejection.
- 3.
- Glomerular CD68+CD163+ are predominant cells in chronic-active antibody-mediated rejection.
- 4.
- Glomerular Mφs may act through complement cascade in worsening AMR.
6. Insights from Animal Models
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Geissmann, F. The origin of dendritic cells. Nat. Immunol. 2007, 8, 558–560. [Google Scholar] [CrossRef]
- Shortman, K.; Liu, Y.J. Mouse and human dendritic cell subtypes. Nat. Rev. Immunol. 2002, 2, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, T.; Omatsu, Y.; Sugiyama, T. Control of hematopoietic stem cells by the bone marrow stromal niche: The role of reticular cells. Trends Immunol. 2011, 32, 315–320. [Google Scholar] [CrossRef]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Mersich, A.T.; Miller, M.R.; Chkourko, H.; Blystone, S.D. The formin frl1 (fmnl1) is an essential component of macrophage podosomes. Cytoskeleton (Hoboken) 2010, 67, 573–585. [Google Scholar] [CrossRef] [Green Version]
- Segerer, S.; Heller, F.; Lindenmeyer, M.T.; Schmid, H.; Cohen, C.D.; Draganovici, D.; Mandelbaum, J.; Nelson, P.J.; Grone, H.J.; Grone, E.F.; et al. Compartment specific expression of dendritic cell markers in human glomerulonephritis. Kidney Int. 2008, 74, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef]
- Liu, K.; Nussenzweig, M.C. Origin and development of dendritic cells. Immunol. Rev. 2010, 234, 45–54. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C., Jr. Innate immune recognition: Mechanisms and pathways. Immunol. Rev. 2000, 173, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Macrophages and immunologic inflammation of the kidney. Semin. Nephrol. 2010, 30, 234–254. [Google Scholar] [CrossRef] [Green Version]
- Rogers, N.M.; Ferenbach, D.A.; Isenberg, J.S.; Thomson, A.W.; Hughes, J. Dendritic cells and macrophages in the kidney: A spectrum of good and evil. Nat. Rev. Nephrol. 2014, 10, 625–643. [Google Scholar] [CrossRef]
- Shortman, K.; Naik, S.H. Steady-state and inflammatory dendritic-cell development. Nat. Rev. Immunol. 2007, 7, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Mellman, I.; Steinman, R.M. Dendritic cells: Specialized and regulated antigen processing machines. Cell 2001, 106, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Serhan, C.N.; Savill, J. Resolution of inflammation: The beginning programs the end. Nat. Immunol. 2005, 6, 1191–1197. [Google Scholar] [CrossRef]
- Sunderkotter, C.; Nikolic, T.; Dillon, M.J.; Van Rooijen, N.; Stehling, M.; Drevets, D.A.; Leenen, P.J. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J. Immunol. 2004, 172, 4410–4417. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, K.A.; Amici, S.A.; Webb, L.M.; Ruiz-Rosado Jde, D.; Popovich, P.G.; Partida-Sanchez, S.; Guerau-de-Arellano, M. Novel markers to delineate murine m1 and m2 macrophages. PLoS ONE 2015, 10, e0145342. [Google Scholar] [CrossRef] [Green Version]
- Fadok, V.A.; Bratton, D.L.; Konowal, A.; Freed, P.W.; Westcott, J.Y.; Henson, P.M. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving tgf-beta, pge2, and paf. J. Clin. Investig. 1998, 101, 890–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, A.; Sica, A.; Sozzani, S.; Allavena, P.; Vecchi, A.; Locati, M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004, 25, 677–686. [Google Scholar] [CrossRef] [PubMed]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Roszer, T. Understanding the mysterious m2 macrophage through activation markers and effector mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Y.; Xu, X.H.; Jin, L. Macrophage polarization in physiological and pathological pregnancy. Front. Immunol. 2019, 10, 792. [Google Scholar] [CrossRef]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates il-6 expression in macrophages and promotes the generation of m2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an m2-like phenotype is independent of interleukin-4 receptor alpha (il-4ralpha) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [Green Version]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, mechanisms, and significance of macrophage plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.; Harris, D.C.; Wang, Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology 2015, 30, 183–194. [Google Scholar] [CrossRef]
- Anders, H.J.; Ryu, M. Renal microenvironments and macrophage phenotypes determine progression or resolution of renal inflammation and fibrosis. Kidney Int. 2011, 80, 915–925. [Google Scholar] [CrossRef] [Green Version]
- Ferenbach, D.; Hughes, J. Macrophages and dendritic cells: What is the difference? Kidney Int. 2008, 74, 5–7. [Google Scholar] [CrossRef] [Green Version]
- Cao, Q.; Wang, Y.; Wang, X.M.; Lu, J.; Lee, V.W.; Ye, Q.; Nguyen, H.; Zheng, G.; Zhao, Y.; Alexander, S.I.; et al. Renal f4/80+ cd11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J. Am. Soc. Nephrol. 2015, 26, 349–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huen, S.C.; Cantley, L.G. Macrophages in renal injury and repair. Annu. Rev. Physiol. 2017, 79, 449–469. [Google Scholar] [CrossRef]
- Woltman, A.M.; de Fijter, J.W.; Zuidwijk, K.; Vlug, A.G.; Bajema, I.M.; van der Kooij, S.W.; van Ham, V.; van Kooten, C. Quantification of dendritic cell subsets in human renal tissue under normal and pathological conditions. Kidney Int. 2007, 71, 1001–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geijtenbeek, T.B.; Torensma, R.; van Vliet, S.J.; van Duijnhoven, G.C.; Adema, G.J.; van Kooyk, Y.; Figdor, C.G. Identification of dc-sign, a novel dendritic cell-specific icam-3 receptor that supports primary immune responses. Cell 2000, 100, 575–585. [Google Scholar] [CrossRef] [Green Version]
- Steinman, R.M. Dc-sign: A guide to some mysteries of dendritic cells. Cell 2000, 100, 491–494. [Google Scholar] [CrossRef] [Green Version]
- De Saint-Vis, B.; Vincent, J.; Vandenabeele, S.; Vanbervliet, B.; Pin, J.J.; Ait-Yahia, S.; Patel, S.; Mattei, M.G.; Banchereau, J.; Zurawski, S.; et al. A novel lysosome-associated membrane glycoprotein, dc-lamp, induced upon dc maturation, is transiently expressed in mhc class ii compartment. Immunity 1998, 9, 325–336. [Google Scholar] [CrossRef] [Green Version]
- Lindenmeyer, M.; Noessner, E.; Nelson, P.J.; Segerer, S. Dendritic cells in experimental renal inflammation—Part i. Nephron Exp. Nephrol. 2011, 119, e83–e90. [Google Scholar] [CrossRef] [Green Version]
- Noessner, E.; Lindenmeyer, M.; Nelson, P.J.; Segerer, S. Dendritic cells in human renal inflammation—Part ii. Nephron Exp. Nephrol. 2011, 119, e91–e98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klessens, C.Q.F.; Zandbergen, M.; Wolterbeek, R.; Bruijn, J.A.; Rabelink, T.J.; Bajema, I.M.; Jpelaar, D.H.T.I. Macrophages in diabetic nephropathy in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2017, 32, 1322–1329. [Google Scholar] [CrossRef]
- Ferrario, F.; Castiglione, A.; Colasanti, G.; Barbiano di Belgioioso, G.; Bertoli, S.; D’Amico, G. The detection of monocytes in human glomerulonephritis. Kidney Int. 1985, 28, 513–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolasco, F.E.; Cameron, J.S.; Hartley, B.; Coelho, A.; Hildreth, G.; Reuben, R. Intraglomerular t cells and monocytes in nephritis: Study with monoclonal antibodies. Kidney Int. 1987, 31, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Bolton, W.K.; Innes, D.J.; Sturgill, B.C., Jr.; Kaiser, D.L. T-cells and macrophages in rapidly progressive glomerulonephritis: Clinicopathologic correlations. Kidney Int. 1987, 32, 869–876. [Google Scholar] [CrossRef] [Green Version]
- Alexopoulos, E.; Seron, D.; Hartley, R.B.; Nolasco, F.; Cameron, J.S. The role of interstitial infiltrates in iga nephropathy: A study with monoclonal antibodies. Nephrol. Dial. Transplant. 1989, 4, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Young, B.A.; Burdmann, E.A.; Johnson, R.J.; Andoh, T.; Bennett, W.M.; Couser, W.G.; Alpers, C.E. Cyclosporine a induced arteriolopathy in a rat model of chronic cyclosporine nephropathy. Kidney Int. 1995, 48, 431–438. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Doe, N.; Tan, L.C. Monocyte chemoattractant protein-1 levels in patients with glomerular disease. Am. J. Kidney Dis. 1996, 27, 640–646. [Google Scholar] [CrossRef]
- Wilde, B.; van Paassen, P.; Damoiseaux, J.; Heerings-Rewinkel, P.; van Rie, H.; Witzke, O.; Tervaert, J.W. Dendritic cells in renal biopsies of patients with anca-associated vasculitis. Nephrol. Dial. Transplant. 2009, 24, 2151–2156. [Google Scholar] [CrossRef] [Green Version]
- Tucci, M.; Quatraro, C.; Lombardi, L.; Pellegrino, C.; Dammacco, F.; Silvestris, F. Glomerular accumulation of plasmacytoid dendritic cells in active lupus nephritis: Role of interleukin-18. Arthritis Rheum. 2008, 58, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tanaka, H.; Hirukawa, T.; Endoh, M.; Fukagawa, M. Characterization and quantification of proliferating cell patterns in endocapillary proliferation. Nephrol. Dial. Transplant. 2012, 27, 3234–3241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rastaldi, M.P.; Ferrario, F.; Crippa, A.; Dell’Antonio, G.; Casartelli, D.; Grillo, C.; D’Amico, G. Glomerular monocyte-macrophage features in anca-positive renal vasculitis and cryoglobulinemic nephritis. J. Am. Soc. Nephrol. 2000, 11, 2036–2043. [Google Scholar]
- Li, J.; Liu, C.H.; Gao, B.; Xu, D.L. Clinical-pathologic significance of cd163 positive macrophage in iga nephropathy patients with crescents. Int. J. Clin. Exp. Med. 2015, 8, 9299–9305. [Google Scholar]
- Olmes, G.; Buttner-Herold, M.; Ferrazzi, F.; Distel, L.; Amann, K.; Daniel, C. Cd163+ m2c-like macrophages predominate in renal biopsies from patients with lupus nephritis. Arthritis Res. Ther. 2016, 18, 90. [Google Scholar] [CrossRef] [Green Version]
- Baggiolini, M. Chemokines and leukocyte traffic. Nature 1998, 392, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Mackay, C.R. Chemokines: Immunology’s high impact factors. Nat. Immunol. 2001, 2, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Cockwell, P.; Howie, A.J.; Adu, D.; Savage, C.O. In situ analysis of c-c chemokine mrna in human glomerulonephritis. Kidney Int. 1998, 54, 827–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovin, B.H.; Rumancik, M.; Tan, L.; Dickerson, J. Glomerular expression of monocyte chemoattractant protein-1 in experimental and human glomerulonephritis. Lab. Investig. 1994, 71, 536–542. [Google Scholar]
- Segerer, S.; Cui, Y.; Hudkins, K.L.; Goodpaster, T.; Eitner, F.; Mack, M.; Schlondorff, D.; Alpers, C.E. Expression of the chemokine monocyte chemoattractant protein-1 and its receptor chemokine receptor 2 in human crescentic glomerulonephritis. J. Am. Soc. Nephrol. 2000, 11, 2231–2242. [Google Scholar]
- Lan, H.Y.; Yang, N.; Nikolic-Paterson, D.J.; Yu, X.Q.; Mu, W.; Isbel, N.M.; Metz, C.N.; Bucala, R.; Atkins, R.C. Expression of macrophage migration inhibitory factor in human glomerulonephritis. Kidney Int. 2000, 57, 499–509. [Google Scholar] [CrossRef]
- Brown, F.G.; Nikolic-Paterson, D.J.; Hill, P.A.; Isbel, N.M.; Dowling, J.; Metz, C.M.; Atkins, R.C. Urine macrophage migration inhibitory factor reflects the severity of renal injury in human glomerulonephritis. J. Am. Soc. Nephrol. 2002, 13 (Suppl. 1), S7–S13. [Google Scholar]
- Matsumoto, K.; Kanmatsuse, K. Urinary levels of macrophage migration inhibitory factor in patients with iga nephropathy. Nephron 2002, 92, 309–315. [Google Scholar] [CrossRef]
- Bhardwaj, R.S.; Zotz, C.; Zwadlo-Klarwasser, G.; Roth, J.; Goebeler, M.; Mahnke, K.; Falk, M.; Meinardus-Hager, G.; Sorg, C. The calcium-binding proteins mrp8 and mrp14 form a membrane-associated heterodimer in a subset of monocytes/macrophages present in acute but absent in chronic inflammatory lesions. Eur. J. Immunol. 1992, 22, 1891–1897. [Google Scholar] [CrossRef]
- Ferrario, F.; Napodano, P.; Rastaldi, M.P.; D’Amico, G. Capillaritis in iga nephropathy. Contrib. Nephrol. 1995, 111, 8–12. [Google Scholar]
- D’Amico, G.; Napodano, P.; Ferrario, F.; Rastaldi, M.P.; Arrigo, G. Idiopathic iga nephropathy with segmental necrotizing lesions of the capillary wall. Kidney. Int. 2001, 59, 682–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, C.H.; Xu, D.L.; Gao, B. Significance of cd163-positive macrophages in proliferative glomerulonephritis. Am. J. Med. Sci. 2015, 350, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; David, M.Z.; Hyjek, E.; Chang, A.; Meehan, S.M. M2 macrophage infiltrates in the early stages of anca-associated pauci-immune necrotizing gn. Clin. J. Am. Soc. Nephrol. 2015, 10, 54–62. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Jinde, K.; Endoh, M.; Sakai, H. Costimulatory molecules cd80 and cd86 in human crescentic glomerulonephritis. Am. J. Kidney Dis. 2003, 41, 950–961. [Google Scholar] [CrossRef]
- Hu, W.; Lin, J.; Lian, X.; Yu, F.; Liu, W.; Wu, Y.; Fang, X.; Liang, X.; Hao, W. M2a and m2b macrophages predominate in kidney tissues and m2 subpopulations were associated with the severity of disease of igan patients. Clin. Immunol. 2019, 205, 8–15. [Google Scholar] [CrossRef]
- Tang, Z.; Niven-Fairchild, T.; Tadesse, S.; Norwitz, E.R.; Buhimschi, C.S.; Buhimschi, I.A.; Guller, S. Glucocorticoids enhance cd163 expression in placental hofbauer cells. Endocrinology 2013, 154, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekerkova, A.; Krepsova, E.; Brabcova, E.; Slatinska, J.; Viklicky, O.; Lanska, V.; Striz, I. Cd14+cd16+ and cd14+cd163+ monocyte subpopulations in kidney allograft transplantation. BMC Immunol. 2014, 15, 4. [Google Scholar] [CrossRef] [Green Version]
- Zizzo, G.; Guerrieri, J.; Dittman, L.M.; Merrill, J.T.; Cohen, P.L. Circulating levels of soluble mer in lupus reflect m2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res. Ther. 2013, 15, R212. [Google Scholar] [CrossRef] [Green Version]
- Trimarchi, H.; Barratt, J.; Cattran, D.C.; Cook, H.T.; Coppo, R.; Haas, M.; Liu, Z.H.; Roberts, I.S.; Yuzawa, Y.; Zhang, H.; et al. Oxford classification of iga nephropathy 2016: An update from the iga nephropathy classification working group. Kidney Int. 2017, 91, 1014–1021. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.M.; Rao, V.M.; Franklin, W.A.; Schiffer, M.S.; Aronson, A.J.; Spargo, B.H.; Katz, A.I. Iga nephropathy: Morphologic predictors of progressive renal disease. Hum. Pathol. 1982, 13, 314–322. [Google Scholar] [CrossRef]
- Feucht, H.E.; Felber, E.; Gokel, M.J.; Hillebrand, G.; Nattermann, U.; Brockmeyer, C.; Held, E.; Riethmuller, G.; Land, W.; Albert, E. Vascular deposition of complement-split products in kidney allografts with cell-mediated rejection. Clin. Exp. Immunol. 1991, 86, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Loupy, A.; Haas, M.; Roufosse, C.; Naesens, M.; Adam, B.; Afrouzian, M.; Akalin, E.; Alachkar, N.; Bagnasco, S.; Becker, J.U.; et al. The banff 2019 kidney meeting report (i): Updates on and clarification of criteria for t cell- and antibody-mediated rejection. Am. J. Transplant. 2020, 20, 2318–2331. [Google Scholar] [CrossRef]
- Voora, S.; Adey, D.B. Management of kidney transplant recipients by general nephrologists: Core curriculum 2019. Am. J. Kidney Dis. 2019, 73, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Raza, A.; Firasat, S.; Khaliq, S.; Khan, A.R.; Mahmood, S.; Aziz, T.; Mubarak, M.; Naqvi, S.A.; Rizvi, S.A.; Abid, A. Monocyte chemoattractant protein-1 (mcp-1/ccl2) levels and its association with renal allograft rejection. Immunol. Investig. 2017, 46, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Harry, T.R.; Coles, G.A.; Davies, M.; Bryant, D.; Williams, G.T.; Griffin, P.J. The significance of monocytes in glomeruli of human renal transplants. Transplantation 1984, 37, 70–73. [Google Scholar] [CrossRef]
- Ozdemir, B.H.; Demirhan, B.; Gungen, Y. The presence and prognostic importance of glomerular macrophage infiltration in renal allografts. Nephron 2002, 90, 442–446. [Google Scholar] [CrossRef]
- Sund, S.; Reisaeter, A.V.; Scott, H.; Mollnes, T.E.; Hovig, T. Glomerular monocyte/macrophage influx correlates strongly with complement activation in 1-week protocol kidney allograft biopsies. Clin. Nephrol. 2004, 62, 121–130. [Google Scholar] [CrossRef]
- Toki, D.; Zhang, W.; Hor, K.L.; Liuwantara, D.; Alexander, S.I.; Yi, Z.; Sharma, R.; Chapman, J.R.; Nankivell, B.J.; Murphy, B.; et al. The role of macrophages in the development of human renal allograft fibrosis in the first year after transplantation. Am. J. Transplant. 2014, 14, 2126–2136. [Google Scholar] [CrossRef]
- Magil, A.B. Monocytes/macrophages in renal allograft rejection. Transplant. Rev. 2009, 23, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Reitamo, S.; Konttinen, Y.T.; Ranki, A.; Hayry, P. The relation of different inflammatory cell types to the various parenchymal components of rejecting kidney allografts. Histopathology 1980, 4, 517–532. [Google Scholar] [CrossRef]
- Hancock, W.W.; Thomson, N.M.; Atkins, R.C. Composition of interstitial cellular infiltrate identified by monoclonal antibodies in renal biopsies of rejecting human renal allografts. Transplantation 1983, 35, 458–463. [Google Scholar] [CrossRef] [PubMed]
- Girlanda, R.; Kleiner, D.E.; Duan, Z.; Ford, E.A.; Wright, E.C.; Mannon, R.B.; Kirk, A.D. Monocyte infiltration and kidney allograft dysfunction during acute rejection. Am. J. Transplant. 2008, 8, 600–607. [Google Scholar] [CrossRef] [Green Version]
- Om, A.; Baquero, A.; Raja, R.; Kim, P.; Bannett, A.D. The prognostic significance of the presence of monocytes in glomeruli of renal transplant allografts. Transplant. Proc. 1987, 19 Pt 2, 1618–1622. [Google Scholar]
- Croker, B.P.; Clapp, W.L.; Abu Shamat, A.R.; Kone, B.C.; Peterson, J.C. Macrophages and chronic renal allograft nephropathy. Kidney Int. Suppl. 1996, 57, S42–S49. [Google Scholar] [PubMed]
- Grimm, P.C.; McKenna, R.; Nickerson, P.; Russell, M.E.; Gough, J.; Gospodarek, E.; Liu, B.; Jeffery, J.; Rush, D.N. Clinical rejection is distinguished from subclinical rejection by increased infiltration by a population of activated macrophages. J. Am. Soc. Nephrol. 1999, 10, 1582–1589. [Google Scholar]
- Tinckam, K.J.; Djurdjev, O.; Magil, A.B. Glomerular monocytes predict worse outcomes after acute renal allograft rejection independent of c4d status. Kidney Int. 2005, 68, 1866–1874. [Google Scholar] [CrossRef] [Green Version]
- Magil, A.B.; Tinckam, K. Monocytes and peritubular capillary c4d deposition in acute renal allograft rejection. Kidney Int. 2003, 63, 1888–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magil, A.B. Infiltrating cell types in transplant glomerulitis: Relationship to peritubular capillary c4d deposition. Am. J. Kidney Dis. 2005, 45, 1084–1089. [Google Scholar] [CrossRef]
- Bishop, G.A.; Hall, B.M.; Duggin, G.G.; Horvath, J.S.; Sheil, A.G.; Tiller, D.J. Immunopathology of renal allograft rejection analyzed with monoclonal antibodies to mononuclear cell markers. Kidney Int. 1986, 29, 708–717. [Google Scholar] [CrossRef] [Green Version]
- Hiki, Y.; Leong, A.S.; Mathew, T.H.; Seymour, A.E.; Pascoe, V.; Woodroffe, A.J. Typing of intraglomerular mononuclear cells associated with transplant glomerular rejection. Clin. Nephrol. 1986, 26, 244–249. [Google Scholar]
- Nickeleit, V.; Zeiler, M.; Gudat, F.; Thiel, G.; Mihatsch, M.J. Detection of the complement degradation product c4d in renal allografts: Diagnostic and therapeutic implications. J. Am. Soc. Nephrol. 2002, 13, 242–251. [Google Scholar]
- Fahim, T.; Bohmig, G.A.; Exner, M.; Huttary, N.; Kerschner, H.; Kandutsch, S.; Kerjaschki, D.; Brambock, A.; Nagy-Bojarszky, K.; Regele, H. The cellular lesion of humoral rejection: Predominant recruitment of monocytes to peritubular and glomerular capillaries. Am. J. Transplant. 2007, 7, 385–393. [Google Scholar] [CrossRef]
- Bergler, T.; Jung, B.; Bourier, F.; Kuhne, L.; Banas, M.C.; Rummele, P.; Wurm, S.; Banas, B. Infiltration of macrophages correlates with severity of allograft rejection and outcome in human kidney transplantation. PLoS ONE 2016, 11, e0156900. [Google Scholar] [CrossRef]
- Haas, M.; Loupy, A.; Lefaucheur, C.; Roufosse, C.; Glotz, D.; Seron, D.; Nankivell, B.J.; Halloran, P.F.; Colvin, R.B.; Akalin, E.; et al. The banff 2017 kidney meeting report: Revised diagnostic criteria for chronic active t cell-mediated rejection, antibody-mediated rejection, and prospects for integrative endpoints for next-generation clinical trials. Am. J. Transplant. 2018, 18, 293–307. [Google Scholar] [CrossRef] [Green Version]
- Sablik, K.A.; Jordanova, E.S.; Pocorni, N.; Clahsen-van Groningen, M.C.; Betjes, M.G.H. Immune cell infiltrate in chronic-active antibody-mediated rejection. Front. Immunol. 2019, 10, 3106. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Choi, S.E.; Lim, B.J.; Kim, Y.S.; Huh, K.H.; Lee, J.; Kim, S.I.; Kim, M.S.; Jeong, H.J. Clinical significance of macrophage polarization in antibody-mediated rejection of renal allograft. Transplant. Proc. 2018, 50, 1005–1008. [Google Scholar] [CrossRef]
- Lefaucheur, C.; Viglietti, D.; Hidalgo, L.G.; Ratner, L.E.; Bagnasco, S.M.; Batal, I.; Aubert, O.; Orandi, B.J.; Oppenheimer, F.; Bestard, O.; et al. Complement-activating anti-hla antibodies in kidney transplantation: Allograft gene expression profiling and response to treatment. J. Am. Soc. Nephrol. 2018, 29, 620–635. [Google Scholar] [CrossRef] [Green Version]
- Desai, H.S.; Parasuraman, R.K.; Samarpungavan, D.; Rooney, M.T.; Cohn, S.R.; Reddy, G.H.; Rocher, L.L.; Dumler, F.; Zhang, P.L. Glomerulitis during acute cellular rejection may be a surrogate marker of vasculitis in renal allografts—Better index for diagnosis of vasculitis. Transplant. Proc. 2011, 43, 1629–1633. [Google Scholar] [CrossRef]
- Van den Bosch, T.P.P.; Hilbrands, L.B.; Kraaijeveld, R.; Litjens, N.H.R.; Rezaee, F.; Nieboer, D.; Steyerberg, E.W.; van Gestel, J.A.; Roelen, D.L.; Clahsen-van Groningen, M.C.; et al. Pretransplant numbers of cd16(+) monocytes as a novel biomarker to predict acute rejection after kidney transplantation: A pilot study. Am. J. Transplant. 2017, 17, 2659–2667. [Google Scholar] [CrossRef] [Green Version]
- Cailhier, J.F.; Partolina, M.; Vuthoori, S.; Wu, S.; Ko, K.; Watson, S.; Savill, J.; Hughes, J.; Lang, R.A. Conditional macrophage ablation demonstrates that resident macrophages initiate acute peritoneal inflammation. J. Immunol. 2005, 174, 2336–2342. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.; Unutmaz, D.; Wong, P.; Sano, G.; De los Santos, K.; Sparwasser, T.; Wu, S.; Vuthoori, S.; Ko, K.; Zavala, F.; et al. In vivo depletion of cd11c+ dendritic cells abrogates priming of cd8+ t cells by exogenous cell-associated antigens. Immunity 2002, 17, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Alexander, J.J.; Chaves, L.D.; Chang, A.; Jacob, A.; Ritchie, M.; Quigg, R.J. Cd11b is protective in complement-mediated immune complex glomerulonephritis. Kidney Int. 2015, 87, 930–939. [Google Scholar] [CrossRef] [Green Version]
- Ikezumi, Y.; Hurst, L.A.; Masaki, T.; Atkins, R.C.; Nikolic-Paterson, D.J. Adoptive transfer studies demonstrate that macrophages can induce proteinuria and mesangial cell proliferation. Kidney Int. 2003, 63, 83–95. [Google Scholar] [CrossRef] [Green Version]
- Ikezumi, Y.; Atkins, R.C.; Nikolic-Paterson, D.J. Interferon-gamma augments acute macrophage-mediated renal injury via a glucocorticoid-sensitive mechanism. J. Am. Soc. Nephrol. 2003, 14, 888–898. [Google Scholar] [CrossRef] [Green Version]
- Duffield, J.S.; Tipping, P.G.; Kipari, T.; Cailhier, J.F.; Clay, S.; Lang, R.; Bonventre, J.V.; Hughes, J. Conditional ablation of macrophages halts progression of crescentic glomerulonephritis. Am. J. Pathol. 2005, 167, 1207–1219. [Google Scholar] [CrossRef] [Green Version]
- Wada, T.; Yokoyama, H.; Furuichi, K.; Kobayashi, K.I.; Harada, K.; Naruto, M.; Su, S.B.; Akiyama, M.; Mukaida, N.; Matsushima, K. Intervention of crescentic glomerulonephritis by antibodies to monocyte chemotactic and activating factor (mcaf/mcp-1). FASEB J. 1996, 10, 1418–1425. [Google Scholar] [CrossRef] [Green Version]
- Garcia, G.E.; Truong, L.D.; Li, P.; Zhang, P.; Johnson, R.J.; Wilson, C.B.; Feng, L. Inhibition of cxcl16 attenuates inflammatory and progressive phases of anti-glomerular basement membrane antibody-associated glomerulonephritis. Am. J. Pathol. 2007, 170, 1485–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kluth, D.C.; Ainslie, C.V.; Pearce, W.P.; Finlay, S.; Clarke, D.; Anegon, I.; Rees, A.J. Macrophages transfected with adenovirus to express il-4 reduce inflammation in experimental glomerulonephritis. J. Immunol. 2001, 166, 4728–4736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, H.M.; Stewart, K.N.; Brown, P.A.; Anegon, I.; Chettibi, S.; Rees, A.J.; Kluth, D.C. Bone-marrow-derived macrophages genetically modified to produce il-10 reduce injury in experimental glomerulonephritis. Mol. Ther. 2002, 6, 710–717. [Google Scholar] [CrossRef]
- Lee, V.W.; Harris, D.C. Adriamycin nephropathy: A model of focal segmental glomerulosclerosis. Nephrology 2011, 16, 30–38. [Google Scholar] [CrossRef]
- Wang, Y.; Mahajan, D.; Tay, Y.C.; Bao, S.; Spicer, T.; Kairaitis, L.; Rangan, G.K.; Harris, D.C. Partial depletion of macrophages by ed7 reduces renal injury in adriamycin nephropathy. Nephrology 2005, 10, 470–477. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.P.; Zheng, G.; Lee, V.W.; Ouyang, L.; Chang, D.H.; Mahajan, D.; Coombs, J.; Wang, Y.M.; Alexander, S.I.; et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007, 72, 290–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, X.; Valenzuela, N.M.; Rossetti, M.; Sosa, R.A.; Nevarez-Mejia, J.; Fishbein, G.A.; Mulder, A.; Dhar, J.; Keslar, K.S.; Baldwin, W.M.; et al. Antibody-induced vascular inflammation skews infiltrating macrophages to a novel remodeling phenotype in a model of transplant rejection. Am. J. Transplant. 2020, 20, 2686–2702. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Glomerular Pathology | Glomerular Macrophages Phenotypic Marker CD68 | Referefnce | Glomerular Macrophages Phenotypic Marker LeuM3 1, FM32 2, Esterase 3, EDI 4 | Reference |
|---|---|---|---|---|
| Post-infectious Gn | - | 27 (intense 3) | [26] | |
| IgA Gn | 2 (positive) | [31] | 35 (negative 2) | [29] |
| 5 (intense 3) | [26] | |||
| 8 (positive 3) | [27] | |||
| Membranous Gn | 6 (positive) | [31] | - | |
| 8 (intense) | [31] | |||
| RPGN | 20 (intense) | [28] | 20 (positive 1) | [28] |
| 20 (intense 3) | [26] | |||
| S Henoch GN | - | 8 (intense 1) | [27] | |
| Lupus Nephritis | 3 (positive) | [31] | 61 (intense 3) | [27] |
| 17 (positive) | [33] | 7 (intense 1) | [27] | |
| 8 (intense) | [33] | |||
| ANCA vasculitis | 1 (positive) | [31] | 6 (intense 1) | [27] |
| 25 (intense) | [32] | |||
| 8 (intense) | [32] | |||
| Cryoglobulinemic Gn | - | 29 (intense 3) | [26] | |
| Anti-GBM Gn | - | 3 (intense 1) | [27] | |
| Cyclosporin Toxicity | - | 6 (positive) | [30] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moll, S.; Angeletti, A.; Scapozza, L.; Cavalli, A.; Ghiggeri, G.M.; Prunotto, M. Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis. Cells 2021, 10, 603. https://doi.org/10.3390/cells10030603
Moll S, Angeletti A, Scapozza L, Cavalli A, Ghiggeri GM, Prunotto M. Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis. Cells. 2021; 10(3):603. https://doi.org/10.3390/cells10030603
Chicago/Turabian StyleMoll, Solange, Andrea Angeletti, Leonardo Scapozza, Andrea Cavalli, Gian Marco Ghiggeri, and Marco Prunotto. 2021. "Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis" Cells 10, no. 3: 603. https://doi.org/10.3390/cells10030603
APA StyleMoll, S., Angeletti, A., Scapozza, L., Cavalli, A., Ghiggeri, G. M., & Prunotto, M. (2021). Glomerular Macrophages in Human Auto- and Allo-Immune Nephritis. Cells, 10(3), 603. https://doi.org/10.3390/cells10030603