
Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis


Abstract
:1. Introduction
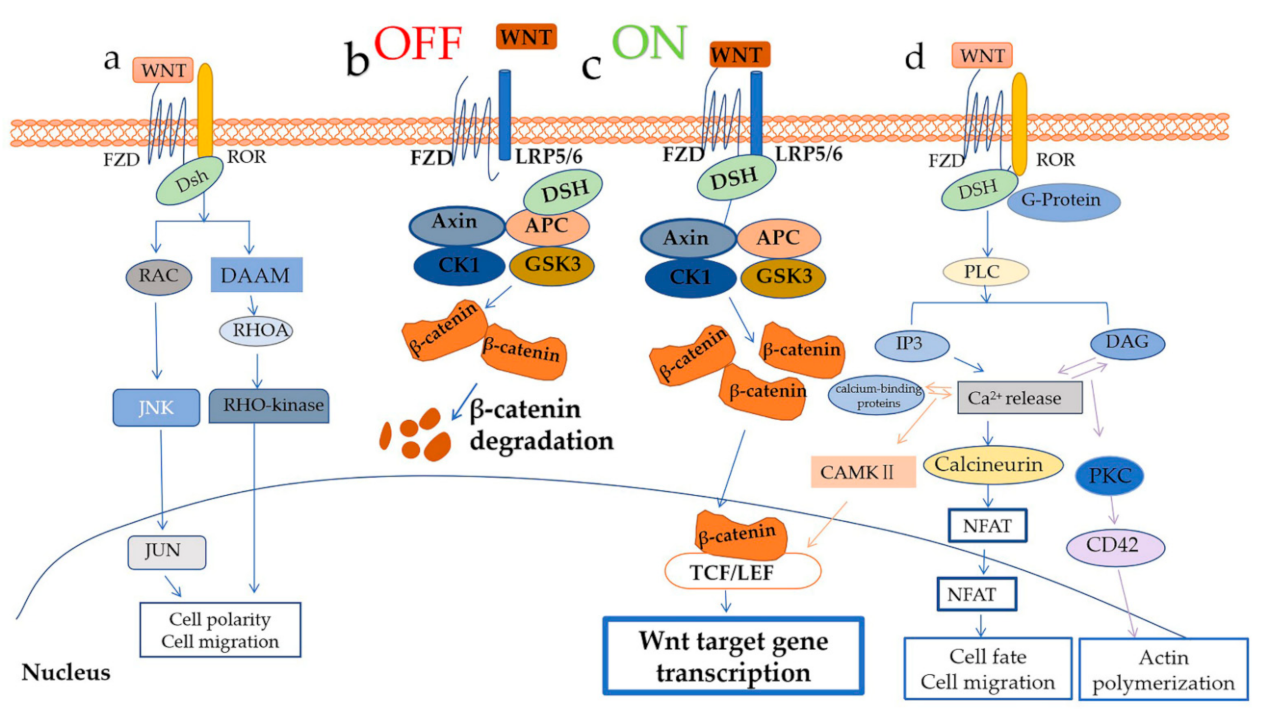
2. An Overview of the WNT Signaling Pathway
2.1. WNT/β–Catenin Signaling
2.2. WNT/PCP Pathway and WNT/Ca2+ Pathway
3. Alteration of WNT Signaling in ALS
3.1. Activated WNT/β–Catenin Signaling in ALS
3.2. Alteration of the WNT/PCP and WNT/Ca2+ Pathways in ALS
4. Dysregulated WNT/β–Catenin Signaling in Neurons and Glial Cells
4.1. Extensive β–Catenin Accumulation in Motor Neurons
4.2. Activation of WNT/β–Catenin Signaling and Astrogliosis
4.3. Activation of WNT/β–Catenin Degenerated Oligodendrocytes
4.4. WNT5A and Activation of WNT/β–Catenin Signaling Induced Proinflammatory Microglia
5. Receptor RYK and Axon Dysregulation
6. WNT Proteins and NMJs in the Progress of ALS
6.1. NMJs in ALS
6.2. WNT Proteins and Receptors Contribute to Neuromuscular Junction Formation
6.3. Frizzled Related Protein (FRZB) and Receptor MUSK in NMJs and Skeletal Muscle: Early ALS Diagnosis
6.4. LRP4 Autoantibody Detection in ALS Cases
7. Special Case in ALS–Spared Extraocular Muscle and WNT Ligands
8. Conclusions and Remarks
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACh | Acetylcholine |
| AChR | acetylcholine receptor |
| ALS | amyotrophic lateral sclerosis |
| APC | adenomatous polyposis coli |
| ATF2 | activating transcription factor 2 |
| CK1 | casein kinase 1 |
| CNS | central nervous system |
| C9orf72 | chromosome 9 open reading frame 72 gene |
| CAMKII | calcium calmodulin mediated kinase II |
| Dsh | Dishevelled |
| DAG | Diacylglycerol |
| EOM | extraocular muscles |
| EAAT 1–5 | excitatory amino acid transporter 1–5 |
| FRZBFZD 1–10 | Frizzled Related ProteinFrizzled1–10 |
| GFAP | glial fibrillary acidic protein |
| GSK3 | glycogen synthase kinase 3 |
| GLT–1 | glutamate transporter 1 |
| Ins (1,4,5)P3 | inositol–1,4,5–trisphosphate |
| JNK | JUN–N–terminal kinase |
| LRP5/6 | low–density lipoprotein receptor–related protein5/6 |
| LRP4 | low–density lipoprotein receptor–related protein 4 |
| mEPSCs | miniature excitatory postsynaptic currents |
| MUSK | muscle skeletal receptor |
| NMJ | neuromuscular junction |
| NPCs | neural progenitor cells |
| NSCs | neural stem cells |
| PCP | planar cell polarity |
| PLC | phospholipase C |
| PTK7 | protein Tyr kinase 7 |
| ROR1/2 | receptor Tyr kinase–like orphan receptor1/2 |
| RYK | receptor Tyr kinase |
| SOD1 | superoxide dismutase–1 |
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef] [Green Version]
- Oskarsson, B.; Gendron, T.F.; Staff, N.P. Amyotrophic Lateral Sclerosis: An Update for 2018. Mayo Clin. Proc. 2018, 93, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Tsitkanou, S.; Lindsay, A.; Della Gatta, P. The role of skeletal muscle in amyotrophic lateral sclerosis: A ‘dying-back’ or ‘dying-forward’ phenomenon? J. Physiol. 2019, 597, 5527–5528. [Google Scholar] [CrossRef]
- Naumann, M.; Pal, A.; Goswami, A.; Lojewski, X.; Japtok, J.; Vehlow, A.; Naujock, M.; Günther, R.; Jin, M.; Stanslowsky, N.; et al. Impaired DNA damage response signaling by FUS-NLS mutations leads to neurodegeneration and FUS aggregate formation. Nat. Commun. 2018, 9, 335. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef]
- Dadon-Nachum, M.; Melamed, E.; Offen, D. The "dying-back" phenomenon of motor neurons in ALS. J. Mol. NeuroSci. 2011, 43, 470–477. [Google Scholar] [CrossRef]
- Nusse, R.; Varmus, H.E. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell 1982, 31, 99–109. [Google Scholar] [CrossRef]
- Niehrs, C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 767–779. [Google Scholar] [CrossRef]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkawara, B.; Kobayakawa, A.; Kanbara, S.; Hattori, T.; Kubota, S.; Ito, M.; Masuda, A.; Takigawa, M.; Lyons, K.M.; Ishiguro, N.; et al. CTGF/CCN2 facilitates LRP4-mediated formation of the embryonic neuromuscular junction. EMBO Rep. 2020, 21, e48462. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, A.; Yamamoto, H.; Sato, A.; Matsumoto, S. New insights into the mechanism of Wnt signaling pathway activation. Int. Rev. Cell Mol. Biol. 2011, 291, 21–71. [Google Scholar] [CrossRef]
- Niessen, C.M.; Gottardi, C.J. Molecular components of the adherens junction. Biochim. Biophys. Acta 2008, 1778, 562–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanneberger, K.; Pfister, A.S.; Brauburger, K.; Schneikert, J.; Hadjihannas, M.V.; Kriz, V.; Schulte, G.; Bryja, V.; Behrens, J. Amer1/WTX couples Wnt-induced formation of PtdIns(4,5)P2 to LRP6 phosphorylation. Embo J. 2011, 30, 1433–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, C.; Chen, Y.G. Dishevelled: The hub of Wnt signaling. Cell Signal. 2010, 22, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. beta-catenin is a target for the ubiquitin-proteasome pathway. Embo J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Cadigan, K.M.; Waterman, M.L. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- Vlad, A.; Röhrs, S.; Klein-Hitpass, L.; Müller, O. The first five years of the Wnt targetome. Cell Signal. 2008, 20, 795–802. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Mattes, B.; Dang, Y.; Greicius, G.; Kaufmann, L.T.; Prunsche, B.; Rosenbauer, J.; Stegmaier, J.; Mikut, R.; Özbek, S.; Nienhaus, G.U.; et al. Wnt/PCP controls spreading of Wnt/β-catenin signals by cytonemes in vertebrates. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- De, A. Wnt/Ca2+ signaling pathway: A brief overview. Acta Biochim. Biophys. Sin. 2011, 43, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 1999, 9, 695–698. [Google Scholar] [CrossRef] [Green Version]
- Braun, A.P.; Schulman, H. The multifunctional calcium/calmodulin-dependent protein kinase: From form to function. Annu. Rev. Physiol. 1995, 57, 417–445. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef] [Green Version]
- Meffre, D.; Grenier, J.; Bernard, S.; Courtin, F.; Dudev, T.; Shackleford, G.; Jafarian-Tehrani, M.; Massaad, C. Wnt and lithium: A common destiny in the therapy of nervous system pathologies? Cell Mol. Life Sci. 2014, 71, 1123–1148. [Google Scholar] [CrossRef]
- Yu, L.; Guan, Y.; Wu, X.; Chen, Y.; Liu, Z.; Du, H.; Wang, X. Wnt Signaling is altered by spinal cord neuronal dysfunction in amyotrophic lateral sclerosis transgenic mice. Neurochem. Res. 2013, 38, 1904–1913. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Guan, Y.; Chen, Y.; Zhang, C.; Shi, C.; Zhou, F.; Yu, L.; Juan, J.; Wang, X. Expression of Wnt5a and its receptor Fzd2 is changed in the spinal cord of adult amyotrophic lateral sclerosis transgenic mice. Int. J. Clin. Exp. Pathol. 2013, 6, 1245–1260. [Google Scholar]
- Chen, Y.; Guan, Y.; Liu, H.; Wu, X.; Yu, L.; Wang, S.; Zhao, C.; Du, H.; Wang, X. Activation of the Wnt/β-catenin signaling pathway is associated with glial proliferation in the adult spinal cord of ALS transgenic mice. Biochem. Biophys. Res. Commun. 2012, 420, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Guan, Y.; Zhang, Z.; Liu, H.; Wang, S.; Yu, L.; Wu, X.; Wang, X. Wnt signaling pathway is involved in the pathogenesis of amyotrophic lateral sclerosis in adult transgenic mice. Neurol. Res. 2012, 34, 390–399. [Google Scholar] [CrossRef]
- Wang, S.; Guan, Y.; Chen, Y.; Li, X.; Zhang, C.; Yu, L.; Zhou, F.; Wang, X. Role of Wnt1 and Fzd1 in the spinal cord pathogenesis of amyotrophic lateral sclerosis-transgenic mice. Biotechnol. Lett. 2013, 35, 1199–1207. [Google Scholar] [CrossRef]
- Ripps, M.E.; Huntley, G.W.; Hof, P.R.; Morrison, J.H.; Gordon, J.W. Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 1995, 92, 689–693. [Google Scholar] [CrossRef] [Green Version]
- Dal Canto, M.C.; Gurney, M.E. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar] [PubMed]
- Pinto, C.; Medinas, D.B.; Fuentes-Villalobos, F.; Maripillán, J.; Castro, A.F.; Martínez, A.D.; Osses, N.; Hetz, C.; Henríquez, J.P. β-catenin aggregation in models of ALS motor neurons: GSK3β inhibition effect and neuronal differentiation. Neuro Biol. Dis 2019, 130, 104497. [Google Scholar] [CrossRef]
- Veeman, M.T.; Axelrod, J.D.; Moon, R.T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell 2003, 5, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.; Schaer, D.J.; Bachli, E.B.; Kurrer, M.O.; Schoedon, G. Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the antiinflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 504–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Fernández, C.; Gonzalez, P.; Andres-Benito, P.; Ferrer, I.; Rodríguez, F.J. Wnt Signaling Alterations in the Human Spinal Cord of Amyotrophic Lateral Sclerosis Cases: Spotlight on Fz2 and Wnt5a. Mol. NeuroBiol. 2019, 56, 6777–6791. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, Y.; Smallwood, P.M.; Nathans, J. An essential role for Frizzled5 in neuronal survival in the parafascicular nucleus of the thalamus. J. NeuroSci. 2008, 28, 5641–5653. [Google Scholar] [CrossRef]
- Sahores, M.; Gibb, A.; Salinas, P.C. Frizzled-5, a receptor for the synaptic organizer Wnt7a, regulates activity-mediated synaptogenesis. Development 2010, 137, 2215–2225. [Google Scholar] [CrossRef] [Green Version]
- González-Fernández, C.; Mancuso, R.; Del Valle, J.; Navarro, X.; Rodríguez, F.J. Wnt Signaling Alteration in the Spinal Cord of Amyotrophic Lateral Sclerosis Transgenic Mice: Special Focus on Frizzled-5 Cellular Expression Pattern. PLoS ONE 2016, 11, e0155867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinonen, K.M.; Vanegas, J.R.; Lew, D.; Krosl, J.; Perreault, C. Wnt4 enhances murine hematopoietic progenitor cell expansion through a planar cell polarity-like pathway. PLoS ONE 2011, 6, e19279. [Google Scholar] [CrossRef] [PubMed]
- Messéant, J.; Ezan, J.; Delers, P.; Glebov, K.; Marchiol, C.; Lager, F.; Renault, G.; Tissir, F.; Montcouquiol, M.; Sans, N.; et al. Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development 2017, 144, 1712–1724. [Google Scholar] [CrossRef] [Green Version]
- de Oliveira, G.P.; Maximino, J.R.; Maschietto, M.; Zanoteli, E.; Puga, R.D.; Lima, L.; Carraro, D.M.; Chadi, G. Early gene expression changes in skeletal muscle from SOD1(G93A) amyotrophic lateral sclerosis animal model. Cell Mol. NeuroBiol. 2014, 34, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant iPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.E.; Hong, Y.H.; Kim, J.Y.; Jeon, G.S.; Jung, J.H.; Yoon, B.N.; Son, S.Y.; Lee, K.W.; Kim, J.I.; Sung, J.J. Altered nucleocytoplasmic proteome and transcriptome distributions in an in vitro model of amyotrophic lateral sclerosis. PLoS ONE 2017, 12, e0176462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, T.; Kazamel, M.; Thoenes, K.; Si, Y.; Jiang, N.; King, P.H. Wnt antagonist FRZB is a muscle biomarker of denervation atrophy in amyotrophic lateral sclerosis. Sci. Rep. 2020, 10, 16679. [Google Scholar] [CrossRef]
- McLoon, L.K.; Harandi, V.M.; Brännström, T.; Andersen, P.M.; Liu, J.X. Wnt and extraocular muscle sparing in amyotrophic lateral sclerosis. Investig. Ophthalmol. Vis. Sci. 2014, 55, 5482–5496. [Google Scholar] [CrossRef]
- Tzartos, J.S.; Zisimopoulou, P.; Rentzos, M.; Karandreas, N.; Zouvelou, V.; Evangelakou, P.; Tsonis, A.; Thomaidis, T.; Lauria, G.; Andreetta, F.; et al. LRP4 antibodies in serum and CSF from amyotrophic lateral sclerosis patients. Ann. Clin. Transl. Neurol. 2014, 1, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Shen, X.M.; Wang, S.Y.; Lu, Y.; Wang, S.B.; Chen, H.; Liu, Z.; Ouyang, Y.S.; Duo, J.Y.; Da, Y.W.; et al. Presence of antibodies against low-density lipoprotein receptor-related protein 4 and impairment of neuromuscular junction in a Chinese cohort of amyotrophic lateral sclerosis. Chin. Med. J. 2019, 132, 1487–1489. [Google Scholar] [CrossRef]
- Pinto, C.; Cárdenas, P.; Osses, N.; Henríquez, J.P. Characterization of Wnt/β-catenin and BMP/Smad signaling pathways in an in vitro model of amyotrophic lateral sclerosis. Front. Cell NeuroSci. 2013, 7, 239. [Google Scholar] [CrossRef] [Green Version]
- Godin, J.D.; Poizat, G.; Hickey, M.A.; Maschat, F.; Humbert, S. Mutant huntingtin-impaired degradation of beta-catenin causes neurotoxicity in Huntington’s disease. Embo J. 2010, 29, 2433–2445. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Piña-Crespo, J.; Li, Y. Restoring Wnt/β-catenin signaling is a promising therapeutic strategy for Alzheimer’s disease. Mol. Brain 2019, 12, 104. [Google Scholar] [CrossRef]
- Blackburn, D.; Sargsyan, S.; Monk, P.N.; Shaw, P.J. Astrocyte function and role in motor neuron disease: A future therapeutic target? Glia 2009, 57, 1251–1264. [Google Scholar] [CrossRef]
- Vargas, M.R.; Johnson, D.A.; Sirkis, D.W.; Messing, A.; Johnson, J.A. Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J. NeuroSci. 2008, 28, 13574–13581. [Google Scholar] [CrossRef]
- Li, W.; Fotinos, A.; Wu, Q.; Chen, Y.; Zhu, Y.; Baranov, S.; Tu, Y.; Zhou, E.W.; Sinha, B.; Kristal, B.S.; et al. N-acetyl-L-tryptophan delays disease onset and extends survival in an amyotrophic lateral sclerosis transgenic mouse model. NeuroBiol. Dis. 2015, 80, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Cook, A.; Kim, J.; Baranov, S.V.; Jiang, J.; Smith, K.; Cormier, K.; Bennett, E.; Browser, R.P.; Day, A.L.; et al. Melatonin inhibits the caspase-1/cytochrome c/caspase-3 cell death pathway, inhibits MT1 receptor loss and delays disease progression in a mouse model of amyotrophic lateral sclerosis. NeuroBiol. Dis. 2013, 55, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sofroniew, M.V. Astrogliosis. Cold Spring Harb. Perspect. Biol. 2014, 7, a020420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, M.; Re, D.B.; Nagata, T.; Chalazonitis, A.; Jessell, T.M.; Wichterle, H.; Przedborski, S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat. NeuroSci. 2007, 10, 615–622. [Google Scholar] [CrossRef] [Green Version]
- Nagy, D.; Kato, T.; Kushner, P.D. Reactive astrocytes are widespread in the cortical gray matter of amyotrophic lateral sclerosis. J. NeuroSci. Res. 1994, 38, 336–347. [Google Scholar] [CrossRef]
- Valori, C.F.; Guidotti, G.; Brambilla, L.; Rossi, D. Astrocytes in Motor Neuron Diseases. Adv. Exp. Med. Biol. 2019, 1175, 227–272. [Google Scholar] [CrossRef]
- Pehar, M.; Harlan, B.A.; Killoy, K.M.; Vargas, M.R. Role and Therapeutic Potential of Astrocytes in Amyotrophic Lateral Sclerosis. Curr. Pharm. Des. 2017, 23, 5010–5021. [Google Scholar] [CrossRef] [Green Version]
- Sun, S.; Zhu, X.J.; Huang, H.; Guo, W.; Tang, T.; Xie, B.; Xu, X.; Zhang, Z.; Shen, Y.; Dai, Z.M.; et al. WNT signaling represses astrogliogenesis via Ngn2-dependent direct suppression of astrocyte gene expression. Glia 2019, 67, 1333–1343. [Google Scholar] [CrossRef]
- Miller, S.J.; Glatzer, J.C.; Hsieh, Y.C.; Rothstein, J.D. Cortical astroglia undergo transcriptomic dysregulation in the G93A SOD1 ALS mouse model. J. Neurogenet. 2018, 32, 322–335. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Tsai, G.; Kuncl, R.W.; Clawson, L.; Cornblath, D.R.; Drachman, D.B.; Pestronk, A.; Stauch, B.L.; Coyle, J.T. Abnormal excitatory amino acid metabolism in amyotrophic lateral sclerosis. Ann. Neurol. 1990, 28, 18–25. [Google Scholar] [CrossRef]
- Parkin, G.M.; Udawela, M.; Gibbons, A.; Dean, B. Glutamate transporters, EAAT1 and EAAT2, are potentially important in the pathophysiology and treatment of schizophrenia and affective disorders. World J. Psychiatry 2018, 8, 51–63. [Google Scholar] [CrossRef]
- Wei, L.; Chen, C.; Ding, L.; Mo, M.; Zou, J.; Lu, Z.; Li, H.; Wu, H.; Dai, Y.; Xu, P.; et al. Wnt1 Promotes EAAT2 Expression and Mediates the Protective Effects of Astrocytes on Dopaminergic Cells in Parkinson’s Disease. Neural. Plast. 2019, 2019, 1247276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.G.; Su, Z.Z.; Emdad, L.; Gupta, P.; Sarkar, D.; Borjabad, A.; Volsky, D.J.; Fisher, P.B. Mechanism of ceftriaxone induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J. Biol. Chem. 2008, 283, 13116–13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, J.D.; Shefner, J.M.; Conwit, R.; Schoenfeld, D.; Keroack, M.; Felsenstein, D.; Krivickas, L.; David, W.S.; Vriesendorp, F.; Pestronk, A.; et al. Design and initial results of a multi-phase randomized trial of ceftriaxone in amyotrophic lateral sclerosis. PLoS ONE 2013, 8, e61177. [Google Scholar] [CrossRef]
- Sun, X.D.; Li, L.; Liu, F.; Huang, Z.H.; Bean, J.C.; Jiao, H.F.; Barik, A.; Kim, S.M.; Wu, H.; Shen, C.; et al. Lrp4 in astrocytes modulates glutamatergic transmission. Nat. NeuroSci. 2016, 19, 1010–1018. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain 2013, 136, 471–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. NeuroSci. 2013, 16, 571–579. [Google Scholar] [CrossRef] [Green Version]
- Nave, K.A. Myelination and support of axonal integrity by glia. Nature 2010, 468, 244–252. [Google Scholar] [CrossRef]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef]
- Nonneman, A.; Robberecht, W.; Van Den Bosch, L. The role of oligodendroglial dysfunction in amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2014, 4, 223–239. [Google Scholar] [CrossRef] [Green Version]
- Ortega, F.; Gascón, S.; Masserdotti, G.; Deshpande, A.; Simon, C.; Fischer, J.; Dimou, L.; Chichung Lie, D.; Schroeder, T.; Berninger, B. Oligodendrogliogenic and neurogenic adult subependymal zone neural stem cells constitute distinct lineages and exhibit differential responsiveness to Wnt signalling. Nat. Cell Biol. 2013, 15, 602–613. [Google Scholar] [CrossRef] [Green Version]
- Azim, K.; Butt, A.M. GSK3β negatively regulates oligodendrocyte differentiation and myelination in vivo. Glia 2011, 59, 540–553. [Google Scholar] [CrossRef]
- Makoukji, J.; Belle, M.; Meffre, D.; Stassart, R.; Grenier, J.; Shackleford, G.; Fledrich, R.; Fonte, C.; Branchu, J.; Goulard, M.; et al. Lithium enhances remyelination of peripheral nerves. Proc. Natl. Acad. Sci. USA 2012, 109, 3973–3978. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, T.; Kagawa, T.; Wada, T.; Muroyama, Y.; Takada, S.; Ikenaka, K. Wnt signaling controls the timing of oligodendrocyte development in the spinal cord. Dev. Biol. 2005, 282, 397–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feigenson, K.; Reid, M.; See, J.; Crenshaw, E.B., 3rd; Grinspan, J.B. Wnt signaling is sufficient to perturb oligodendrocyte maturation. Mol. Cell NeuroSci. 2009, 42, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Chen, Y.; Hoang, T.; Montgomery, R.L.; Zhao, X.H.; Bu, H.; Hu, T.; Taketo, M.M.; van Es, J.H.; Clevers, H.; et al. HDAC1 and HDAC2 regulate oligodendrocyte differentiation by disrupting the beta-catenin-TCF interaction. Nat. NeuroSci. 2009, 12, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Kawamata, T.; Akiyama, H.; Yamada, T.; McGeer, P.L. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. Am. J. Pathol. 1992, 140, 691–707. [Google Scholar]
- Turner, M.R.; Cagnin, A.; Turkheimer, F.E.; Miller, C.C.; Shaw, C.E.; Brooks, D.J.; Leigh, P.N.; Banati, R.B. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: An [11C](R)-PK11195 positron emission tomography study. NeuroBiol. Dis. 2004, 15, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. NeuroSci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef] [PubMed]
- Frakes, A.E.; Ferraiuolo, L.; Haidet-Phillips, A.M.; Schmelzer, L.; Braun, L.; Miranda, C.J.; Ladner, K.J.; Bevan, A.K.; Foust, K.D.; Godbout, J.P.; et al. Microglia induce motor neuron death via the classical NF-κB pathway in amyotrophic lateral sclerosis. Neuron 2014, 81, 1009–1023. [Google Scholar] [CrossRef] [Green Version]
- Hall, E.D.; Oostveen, J.A.; Gurney, M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 1998, 23, 249–256. [Google Scholar] [CrossRef]
- Ouali Alami, N.; Schurr, C.; Olde Heuvel, F.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. Embo J. 2018, 37. [Google Scholar] [CrossRef] [PubMed]
- Halleskog, C.; Dijksterhuis, J.P.; Kilander, M.B.; Becerril-Ortega, J.; Villaescusa, J.C.; Lindgren, E.; Arenas, E.; Schulte, G. Heterotrimeric G protein-dependent WNT-5A signaling to ERK1/2 mediates distinct aspects of microglia proinflammatory transformation. J. Neuroinflamm. 2012, 9, 111. [Google Scholar] [CrossRef] [Green Version]
- Halleskog, C.; Mulder, J.; Dahlström, J.; Mackie, K.; Hortobágyi, T.; Tanila, H.; Kumar Puli, L.; Färber, K.; Harkany, T.; Schulte, G. WNT signaling in activated microglia is proinflammatory. Glia 2011, 59, 119–131. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Zhang, X.; Chen, J.; Liu, X.; Xue, J.; Zhang, L.; Lan, X. Wnt canonical pathway activator TWS119 drives microglial anti-inflammatory activation and facilitates neurological recovery following experimental stroke. J. Neuroinflamm. 2019, 16, 256. [Google Scholar] [CrossRef]
- Akiyama, T.; Suzuki, N.; Ishikawa, M.; Fujimori, K.; Sone, T.; Kawada, J.; Funayama, R.; Fujishima, F.; Mitsuzawa, S.; Ikeda, K.; et al. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine 2019, 45, 362–378. [Google Scholar] [CrossRef] [Green Version]
- Onishi, K.; Hollis, E.; Zou, Y. Axon guidance and injury-lessons from Wnts and Wnt signaling. Curr. Opin NeuroBiol. 2014, 27, 232–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tury, A.; Tolentino, K.; Zou, Y. Altered expression of atypical PKC and Ryk in the spinal cord of a mouse model of amyotrophic lateral sclerosis. Dev. NeuroBiol. 2014, 74, 839–850. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wang, X.; Lu, C.C.; Kerman, R.; Steward, O.; Xu, X.M.; Zou, Y. Repulsive Wnt signaling inhibits axon regeneration after CNS injury. J. NeuroSci. 2008, 28, 8376–8382. [Google Scholar] [CrossRef] [PubMed]
- Picchiarelli, G.; Demestre, M.; Zuko, A.; Been, M.; Higelin, J.; Dieterlé, S.; Goy, M.A.; Mallik, M.; Sellier, C.; Scekic-Zahirovic, J.; et al. FUS-mediated regulation of acetylcholine receptor transcription at neuromuscular junctions is compromised in amyotrophic lateral sclerosis. Nat. NeuroSci. 2019, 22, 1793–1805. [Google Scholar] [CrossRef]
- Li, J.; Ito, M.; Ohkawara, B.; Masuda, A.; Ohno, K. Differential effects of spinal motor neuron-derived and skeletal muscle-derived Rspo2 on acetylcholine receptor clustering at the neuromuscular junction. Sci. Rep. 2018, 8, 13577. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef] [Green Version]
- Burden, S.J. SnapShot: Neuromuscular Junction. Cell 2011, 144, 826.e821. [Google Scholar] [CrossRef] [Green Version]
- Gordon, L.R.; Gribble, K.D.; Syrett, C.M.; Granato, M. Initiation of synapse formation by Wnt-induced MuSK endocytosis. Development 2012, 139, 1023–1033. [Google Scholar] [CrossRef] [Green Version]
- Henriquez, J.P.; Webb, A.; Bence, M.; Bildsoe, H.; Sahores, M.; Hughes, S.M.; Salinas, P.C. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc. Natl. Acad. Sci. USA 2008, 105, 18812–18817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Ruan, N.J.; Qian, L.; Lei, W.L.; Chen, F.; Luo, Z.G. Wnt/beta-catenin signaling suppresses Rapsyn expression and inhibits acetylcholine receptor clustering at the neuromuscular junction. J. Biol. Chem. 2008, 283, 21668–21675. [Google Scholar] [CrossRef] [Green Version]
- Pérez-García, M.J.; Burden, S.J. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Rep. 2012, 2, 497–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cantor, S.; Zhang, W.; Delestrée, N.; Remédio, L.; Mentis, G.Z.; Burden, S.J. Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta-Ghosh, A.; Dominguez, S.L.; Xie, L.; Barck, K.H.; Jiang, Z.; Earr, T.; Imperio, J.; Phu, L.; Budayeva, H.G.; Kirkpatrick, D.S.; et al. Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1(G93A) mouse model of ALS. NeuroBiol. Dis. 2019, 124, 340–352. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Methods | Research Subject | Inhibitor and Antagonist | WNT Ligands | Receptors | Downstream Molecules | |
|---|---|---|---|---|---|---|
| Gene Expression Profiling PCR Array | SOD1–G93A transgenic mouse spinal cords [27] | 95 d | ↑ Wnt1, Wnt3a, Wnt7b, Wnt8a | ↑ Fzd3 | ↑ Fosl1, Frat1 ↓ Myc, T, Tcf7 | |
| 108 d | ↑ Wif1 and sFrp4 | ↑ Wnt4 ↓ Wnt16, Wnt8b | ↑ Fgf4, Fosl1, Sfrp4, ↓ Csnk1a1, Frat1, FrzbLef1, Nkd1, Pitx2, Sfrp1, T, Tle2 | |||
| 122 d | ↑ Wnt10a, Wnt11, Wnt16, Wnt2, Wnt3, Wnt4, Wnt5a, Wnt5b, Wnt7a, Wnt7b, Wnt9a | ↑ Fzd1, Fzd2, Fzd3, Fzd4, Fzd5, Fzd6, Fzd7, Fzd8, Lrp5 | ↑ Btrc, Ccnd1/2/3, Csnk1a–1, Ctnnb1, Dvl1, Ep300, Fbxw4/11, Fosl1, Frzb, Jun, Myc, Nlk, Pitx2, Ppp2r5d, Rhou, Senp2, Sfrp4, Slc9a3r1, Sox17, T, Tcf3, Wif1 ↓ Tcf7 | |||
| Gene Expression Profiling PCR Array | ALS human spinal cords [37] | ↑ sFRP3 | ↑ WNT3, WNT4, WNT2B, WNT5A | ↑ FZD2, and FZD8, FZD3, LRP5, | ||
| Gene Expression Profiling PCR Array | SOD1–G93A transgenic mouse Skeletal Muscle [43] | 40 d | ↑ Prkx, Dner | |||
| 80 d | ↓ Fzd2 | ↓ Cd44 | ||||
| RNA sequencing | SOD1 mutant motor neurons(iPSC) [44] | ↑ Fzd2 | ↑ Lef, Tcf7l2 β–catenin | |||
| RNA sequencing | hSOD1–G93A mutant motor neurons (NSC34 cell) [45] | ↑ Cltc, Plcb3, Plec, Psmd3, Ruvbl1 | ||||
| Immunofluorescence, Western blot, RT–PCR | astrocytes in SOD1–G93A transgenic mouse spinal cords | ↑ WNT2A, WNT7A [30] WNT3A [29] WNT5A [28] | ↑ FZD2 [28] | ↑ nuclear β–catenin [29] | ||
| Immunofluorescence | astrocytes in ALS human spinal cords [37] | ↑ WNT5A | ↑ FZD2 | |||
| Immunofluorescence, Western blot, RT–PCR | neurons in SOD1–G93A transgenic mouse spinal cords | ↓ WNT3A [29] WNT5A [28] | ↑ FZD5 [40] | ↑ nuclear β–catenin, Phospho–GSK–3β (Ser 9) [29] | ||
| Immunofluorescence | hSOD1–G93A mutant motor neurons (NSC34 cell) [34] | ↑ cytoplasm β–catenin | ||||
| RNA sequencing, Western blot | Skeletal muscle and the neuromuscular junction in ALS human and SOD1–G93A mouse [46] | ↑ FRZB | ↑ β–catenin | |||
| Immunofluorescence [47] | limb muscles in ALS human | ↓ WNT1, WNT3A, WNT7A | ↑ β–catenin | |||
| myofiber in ALS human | ↑ WNT7A ↓ WNT1, WNT3A, | |||||
| neuromuscular junctions in SOD1–G93A mouse | ↓ WNT1, WNT3A, WNT5A WNT7A | |||||
| extraocular muscles in ALS human and SOD1–G93A mouse | ↑ β–catenin | |||||
| cell–based assay and radio– immunoassay | sera from sporadic ALS patients [48,49] | ↑ LRP4 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, X.; Guan, Y.; Zhao, Z.; Meng, F.; Wang, X.; Gao, X.; Liu, J.; Chen, Y.; Zhou, F.; Zhou, S.; et al. Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis. Cells 2021, 10, 839. https://doi.org/10.3390/cells10040839
Jiang X, Guan Y, Zhao Z, Meng F, Wang X, Gao X, Liu J, Chen Y, Zhou F, Zhou S, et al. Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis. Cells. 2021; 10(4):839. https://doi.org/10.3390/cells10040839
Chicago/Turabian StyleJiang, Xin, Yingjun Guan, Zhenhan Zhao, Fandi Meng, Xuemei Wang, Xueshuai Gao, Jinmeng Liu, Yanchun Chen, Fenghua Zhou, Shuanhu Zhou, and et al. 2021. "Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis" Cells 10, no. 4: 839. https://doi.org/10.3390/cells10040839
APA StyleJiang, X., Guan, Y., Zhao, Z., Meng, F., Wang, X., Gao, X., Liu, J., Chen, Y., Zhou, F., Zhou, S., & Wang, X. (2021). Potential Roles of the WNT Signaling Pathway in Amyotrophic Lateral Sclerosis. Cells, 10(4), 839. https://doi.org/10.3390/cells10040839