
Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients


Abstract
:1. Introduction
2. Molecular Basis of Inflammasome Activation
3. Mechanisms of Blood Clotting Induced by Activated Inflammasome
4. Inflammasome Activation in COVID-19 Patients
5. Hypercoagulopathy Associated with SARS-CoV-2 Infection
6. Activation of the NLRP3 Inflammasome Contributed to Cardiovascular Disorders
7. Potential Role and Therapeutic Target of the NLRP3 Inflammasome for Cardiovascular Complications in COVID-19
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Glossary
| ACE2 | Angiotensin-converting enzyme 2 |
| ASC | Apoptosis-associated speck-like protein containing a CARD |
| ATP | Adenosine triphosphate |
| CaMKIIδ | Calcium/calmodulin-dependent protein kinase IIδ |
| CARD | Caspase activation and recruitment domain |
| CCE | Cholecalciferol cholesterol emulsion |
| COVID-19 | Coronavirus disease 2019 |
| DAMPs | Damage-associated molecular patterns |
| EC | Endothelial cell |
| GSDMD | Gasdermin D |
| HIF-1α | Hypoxia-inducible factor 1-alpha |
| ICU | Intensive care unit |
| IFI16 | Interferon gamma inducible protein 16 |
| IFN-γ | Interferon gamma |
| IL-18 | Interleukin-18 |
| IL-1β | Interleukin-1 beta |
| IL-6 | Interleukin-6 |
| IRAK4 | Interleukin-1 receptor-associated kinase 4 |
| LDL | Low density lipoprotein |
| LRR | Leucine rich repeat |
| MI | Myocardial infarction |
| MPs | Microparticles |
| NET | Neutrophil extracellular trap |
| NLR | Nucleotide oligomerization domain-like receptors |
| NLRP3 | NLR family pyrin domain containing 3 |
| NOD | Nucleotide oligomerization domain |
| PAMPs | Pathogen associated molecular patterns |
| PBMCs | Peripheral blood mononuclear cells |
| PRR | Pattern recognition receptors |
| PYD | Pyrin domain |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| TF | Tissue factor |
| TGF-β1 | Transforming growth factor-beta1 |
| Th1 | T helper type 1 |
| TLR | Toll-like receptors |
References
- Bryant, C.; Fitzgerald, K.A. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 2009, 19, 455–464. [Google Scholar] [CrossRef]
- Zhao, C.; Zhao, W. NLRP3 inflammasome-A key player in antiviral responses. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antushevich, H. Interplays between inflammasomes and viruses, bacteria (pathogenic and probiotic), yeasts and parasites. Immunol. Lett. 2020, 228, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, S.L.; Cookson, B.T. Apoptosis, pyroptosis, and necrosis: Mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef] [Green Version]
- Chen, I.Y.; Ichinohe, T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015, 23, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Monack, D.M. Molecular mechanisms of inflammasome activation during microbial infections. Immunol. Rev. 2011, 243, 174–190. [Google Scholar] [CrossRef]
- Li, Y.; Ren, B.; Peng, X.; Hu, T.; Li, J.; Gong, T.; Tang, B.; Xu, X.; Zhou, X. Saliva is a non-negligible factor in the spread of COVID-19. Mol. Oral Microbiol. 2020, 35, 141–145. [Google Scholar] [CrossRef]
- Guo, J.; Xie, H.; Liang, M.; Wu, H. COVID-19: A novel coronavirus and a novel challenge for oral healthcare. Clin. Oral Investig. 2020, 24, 2137–2138. [Google Scholar] [CrossRef]
- Mallineni, S.K.; Bhumireddy, C.J.; Nuvvula, S. Dentistry for children during and post COVID-19 pandemic outbreak. Child. Youth Serv. Rev. 2021, 120, 105734. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020, 71, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Wang, Y.; Shao, C.; Huang, J.; Gan, J.; Huang, X.; Bucci, E.; Piacentini, M.; Ippolito, G.; Melino, G. COVID-19 infection: The perspectives on immune responses. Cell Death Differ. 2020, 27, 1451–1454. [Google Scholar] [CrossRef] [Green Version]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 inflammasome in severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- de Rivero Vaccari, J.C.; Dietrich, W.D.; Keane, R.W.; de Rivero Vaccari, J.P. The inflammasome in times of COVID-19. Front. Immunol. 2020, 11, 583373. [Google Scholar] [CrossRef]
- Conforti-Andreoni, C.; Ricciardi-Castagnoli, P.; Mortellaro, A. The inflammasomes in health and disease: From genetics to molecular mechanisms of autoinflammation and beyond. Cell. Mol. Immunol. 2011, 8, 135–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosso, M.; Prelli Bozzo, C.; Hotter, D.; Volcic, M.; Sturzel, C.M.; Rammelt, A.; Ni, Y.; Urban, S.; Becker, M.; Schelhaas, M.; et al. Nuclear PYHIN proteins target the host transcription factor Sp1 thereby restricting HIV-1 in human macrophages and CD4+ T cells. PLoS Pathog. 2020, 16, e1008752. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Hornung, V. Of inflammasomes and pathogens—Sensing of microbes by the inflammasome. EMBO Mol. Med. 2013, 5, 814–826. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 2009, 182, 3173–3182. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, K. Unique action of interleukin-18 on T Cells and other immune cells. Front. Immunol. 2018, 9, 763. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Nakanishi, K.; Tsutsui, H. Interleukin-18 in health and disease. Int. J. Mol. Sci. 2019, 20, 649. [Google Scholar] [CrossRef] [Green Version]
- Gavrilin, M.A.; McAndrew, C.C.; Prather, E.R.; Tsai, M.; Spitzer, C.R.; Song, M.A.; Mitra, S.; Sarkar, A.; Shields, P.G.; Diaz, P.T.; et al. Inflammasome adaptor ASC is highly elevated in lung over plasma and relates to inflammation and lung diffusion in the absence of speck formation. Front. Immunol. 2020, 11, 461. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2010, 11, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Yuan, J. Caspases in apoptosis and beyond. Oncogene 2008, 27, 6194–6206. [Google Scholar] [CrossRef] [Green Version]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. The interactions between inflammation and coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef] [PubMed]
- Levi, M.; van der Poll, T. Inflammation and coagulation. Crit. Care Med. 2010, 38 (Suppl. 2), S26–S34. [Google Scholar] [CrossRef]
- Levi, M. Infection and inflammation and the coagulation system. Cardiovasc. Res. 2003, 60, 26–39. [Google Scholar] [CrossRef]
- Assinger, A. Platelets and infection—An emerging role of platelets in viral infection. Front. Immunol. 2014, 5, 649. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.G.; Williams, J.C.; Davis, B.K.; Jacobson, K.; Doerschuk, C.M.; Ting, J.P.; Mackman, N. Monocytic microparticles activate endothelial cells in an IL-1beta-dependent manner. Blood 2011, 118, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.; Lu, W.; Zhang, Y.; Zhang, G.; Shi, X.; Hisada, Y.; Grover, S.P.; Zhang, X.; Li, L.; Xiang, B.; et al. Inflammasome activation triggers blood clotting and host death through pyroptosis. Immunity 2019, 50, 1401–1411e4. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Y.; Li, L.; Pandeya, A.; Zhang, G.; Cui, J.; Kirchhofer, D.; Wood, J.P.; Smyth, S.S.; Wei, Y.; et al. Extracellular histones trigger disseminated intravascular coagulation by lytic cell death. bioRxiv 2020. [Google Scholar] [CrossRef]
- Qiao, J.; Wu, X.; Luo, Q.; Wei, G.; Xu, M.; Wu, Y.; Liu, Y.; Li, X.; Zi, J.; Ju, W.; et al. NLRP3 regulates platelet integrin alphaIIbbeta3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica 2018, 103, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Chanchal, S.; Mishra, A.; Singh, M.K.; Ashraf, M.Z. Understanding inflammatory responses in the manifestation of prothrombotic phenotypes. Front. Cell Dev. Biol. 2020, 8, 73. [Google Scholar] [CrossRef] [Green Version]
- Boone, B.A.; Murthy, P.; Miller-Ocuin, J.L.; Liang, X.; Russell, K.L.; Loughran, P.; Gawaz, M.; Lotze, M.T.; Zeh, H.J., 3rd; Vogel, S. The platelet NLRP3 inflammasome is upregulated in a murine model of pancreatic cancer and promotes platelet aggregation and tumor growth. Ann. Hematol. 2019, 98, 1603–1610. [Google Scholar] [CrossRef]
- Gupta, N.; Sahu, A.; Prabhakar, A.; Chatterjee, T.; Tyagi, T.; Kumari, B.; Khan, N.; Nair, V.; Bajaj, N.; Sharma, M.; et al. Activation of NLRP3 inflammasome complex potentiates venous thrombosis in response to hypoxia. Proc. Natl. Acad. Sci. USA 2017, 114, 4763–4768. [Google Scholar] [CrossRef] [Green Version]
- Hottz, E.D.; Monteiro, A.P.; Bozza, F.A.; Bozza, P.T. Inflammasome in platelets: Allying coagulation and inflammation in infectious and sterile diseases? Mediat. Inflamm. 2015, 2015, 435783. [Google Scholar] [CrossRef]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Hottz, E.D.; Lopes, J.F.; Freitas, C.; Valls-de-Souza, R.; Oliveira, M.F.; Bozza, M.T.; Da Poian, A.T.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; et al. Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 2013, 122, 3405–3414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, T.S.; Sa, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasome activation in COVID-19 patients. medRxiv 2020. [Google Scholar] [CrossRef]
- Toldo, S.; Bussani, R.; Nuzzi, V.; Bonaventura, A.; Mauro, A.G.; Cannata, A.; Pillappa, R.; Sinagra, G.; Nana-Sinkam, P.; Sime, P.; et al. Inflammasome formation in the lungs of patients with fatal COVID-19. Inflamm. Res. 2020, 70, 7–10. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Moriyama, M.; Iwasaki, A. Inflammasomes and pyroptosis as therapeutic targets for COVID-19. J. Immunol. 2020, 205, 307–312. [Google Scholar] [CrossRef]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of severe acute respiratory syndrome coronavirus viroporins E, 3a, and 8a in replication and pathogenesis. mBio 2018, 9, e02325-17. [Google Scholar] [CrossRef] [Green Version]
- Theobald, S.J.; Alexander, S.; Kreer, C.; Zehner, M.; Fischer, J.; Albert, M.-C.; Malin, J.J.; Gräb, J.; Winter, S.; Silva, U.S.d.; et al. The SARS-CoV-2 spike protein primes inflammasome mediated interleukin 1β secretionin COVID-19 drived macrophages. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-coronavirus open reading frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Chitre, S.A.; Akinyemi, I.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin triggers the NLRP3 inflammatory pathway. bioRxiv 2020. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal. Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Goncalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Zou, Y.; Guo, H.; Zhang, Y.; Zhang, Z.; Liu, Y.; Wang, J.; Lu, H.; Qian, Z. Analysis of coagulation parameters in patients with COVID-19 in Shanghai, China. Biosci. Trends 2020, 14, 285–289. [Google Scholar] [CrossRef]
- Zhang, Y.; He, L.; Chen, H.; Lu, S.; Xiong, Y.; Liu, J.; Zheng, Y.; Wang, S.; Liu, L. Manifestations of blood coagulation and its relation to clinical outcomes in severe COVID-19 patients: Retrospective analysis. Int. J. Lab. Hematol. 2020, 42, 766–772. [Google Scholar] [CrossRef]
- Panigada, M.; Bottino, N.; Tagliabue, P.; Grasselli, G.; Novembrino, C.; Chantarangkul, V.; Pesenti, A.; Peyvandi, F.; Tripodi, A. Hypercoagulability of COVID-19 patients in intensive care unit: A report of thromboelastography findings and other parameters of hemostasis. J. Thromb. Haemost. 2020, 18, 1738–1742. [Google Scholar] [CrossRef] [PubMed]
- Debuc, B.; Smadja, D.M. Is COVID-19 a New Hematologic Disease? Stem Cell Rev. Rep. 2021, 17, 4–8. [Google Scholar] [CrossRef]
- Tang, Y.W.; Schmitz, J.E.; Persing, D.H.; Stratton, C.W. The laboratory diagnosis of COVID-19 infection: Current issues and challenges. J. Clin. Microbiol. 2020, 58, e00512–e00520. [Google Scholar] [CrossRef] [Green Version]
- Klok, F.A.; Kruip, M.; van der Meer, N.J.M.; Arbous, M.S.; Gommers, D.; Kant, K.M.; Kaptein, F.H.J.; van Paassen, J.; Stals, M.A.M.; Huisman, M.V.; et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb. Res. 2020, 191, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Joly, S.B.; Siguret, V.; Veyradier, A. Understanding pathophysiology of hemostasis disorders in critically ill patients with COVID-19. Intensive Care Med. 2020, 46, 1603–1606. [Google Scholar] [CrossRef]
- Stefan, N.; Birkenfeld, A.L.; Schulze, M.B.; Ludwig, D.S. Obesity and impaired metabolic health in patients with COVID-19. Nat. Rev. Endocrinol. 2020, 16, 341–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef]
- Mehta, J.L.; Calcaterra, G.; Bassareo, P.P. COVID-19, thromboembolic risk, and Virchow’s triad: Lesson from the past. Clin. Cardiol. 2020, 43, 1362–1367. [Google Scholar] [CrossRef]
- Ghoshal, K.; Bhattacharyya, M. Overview of platelet physiology: Its hemostatic and nonhemostatic role in disease pathogenesis. Sci. World J. 2014, 2014, 781857. [Google Scholar] [CrossRef] [Green Version]
- Fan, B.E.; Chong, V.C.L.; Chan, S.S.W.; Lim, G.H.; Lim, K.G.E.; Tan, G.B.; Mucheli, S.S.; Kuperan, P.; Ong, K.H. Hematologic parameters in patients with COVID-19 infection. Am. J. Hematol. 2020, 95, E131–E134. [Google Scholar] [CrossRef] [Green Version]
- Lippi, G.; Plebani, M.; Henry, B.M. Thrombocytopenia is associated with severe coronavirus disease 2019 (COVID-19) infections: A meta-analysis. Clin. Chim. Acta 2020, 506, 145–148. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in COVID-19 Patients. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Nishiga, M.; Wang, D.W.; Han, Y.; Lewis, D.B.; Wu, J.C. COVID-19 and cardiovascular disease: From basic mechanisms to clinical perspectives. Nat. Rev. Cardiol. 2020, 17, 543–558. [Google Scholar] [CrossRef]
- Li, X.; Deroide, N.; Mallat, Z. The role of the inflammasome in cardiovascular diseases. J. Mol. Med. 2014, 92, 307–319. [Google Scholar] [CrossRef]
- WTO. 2016. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 29 December 2020).
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, X.; Shi, H.; Yu, Y.; Yu, Y.; Li, M.; Chen, R. NLRP3 inflammasome, an immune-inflammatory target in pathogenesis and treatment of cardiovascular diseases. Clin. Transl. Med. 2020, 10, 91–106. [Google Scholar] [CrossRef]
- Yin, Y.; Yan, Y.; Jiang, X.; Mai, J.; Chen, N.C.; Wang, H.; Yang, F.G. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 2009, 22, 311–322. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, H.; Qi, W.; Zhang, Y.; Li, J.; Li, Z.; Lin, Y.; Bai, X.; Liu, X.; Chen, X.; et al. Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death. Dis. 2018, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Lu, Y.; Cao, Z.; Ma, Q.; Pi, H.; Fang, Y.; Yu, Z.; Hu, H.; Zhou, Z. Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol. Lett. 2016, 246, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Jiang, L.; Li, Q.; Liu, X.; Zhang, T.; Dong, L.; Liu, T.; Liu, L.; Hu, G.; Sun, X.; et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology 2018, 410, 26–40. [Google Scholar] [CrossRef]
- Varghese, J.F.; Patel, R.; Yadav, U.C.S. Sterol regulatory element binding protein (SREBP)-1 mediates oxidized low-density lipoprotein (oxLDL) induced macrophage foam cell formation through NLRP3 inflammasome activation. Cell. Signal. 2019, 53, 316–326. [Google Scholar] [CrossRef]
- An, N.; Gao, Y.; Si, Z.; Zhang, H.; Wang, L.; Tian, C.; Yuan, M.; Yang, X.; Li, X.; Shang, H.; et al. Regulatory mechanisms of the NLRP3 Inflammasome, a novel immune-inflammatory marker in cardiovascular diseases. Front. Immunol. 2019, 10, 1592. [Google Scholar] [CrossRef]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Tong, Y.; Wang, Z.; Cai, L.; Lin, L.; Liu, J.; Cheng, J. NLRP3 inflammasome and its central role in the cardiovascular diseases. Oxid. Med. Cell. Longev. 2020, 2020, 4293206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Rev. Cardiol. 2014, 11, 255–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M. NLRP3 inflammasome as a novel player in myocardial infarction. Int. Heart J. 2014, 55, 101–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toldo, S.; Mezzaroma, E.; Mauro, A.G.; Salloum, F.; Van Tassell, B.W.; Abbate, A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid. Redox. Signal. 2015, 22, 1146–1161. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef] [Green Version]
- Moriyama, M.; Chen, I.Y.; Kawaguchi, A.; Koshiba, T.; Nagata, K.; Takeyama, H.; Hasegawa, H.; Ichinohe, T. The RNA- and TRIM25-binding domains of influenza virus NS1 protein are essential for suppression of NLRP3 inflammasome-mediated interleukin-1beta secretion. J. Virol. 2016, 90, 4105–4114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cero, F.T.; Hillestad, V.; Sjaastad, I.; Yndestad, A.; Aukrust, P.; Ranheim, T.; Lunde, I.G.; Olsen, M.B.; Lien, E.; Zhang, L.; et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 309, L378–L387. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lian, K.; Zhang, L.; Wang, R.; Yi, F.; Gao, C.; Xin, C.; Zhu, D.; Li, Y.; Yan, W.; et al. TXNIP mediates NLRP3 inflammasome activation in cardiac microvascular endothelial cells as a novel mechanism in myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2014, 109, 415. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, C.; Chojnacki, J.; Toldo, S.; Mezzaroma, E.; Tranchida, N.; Rose, S.W.; Federici, M.; Van Tassell, B.W.; Zhang, S.; Abbate, A. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J. Cardiovasc. Pharmacol. 2014, 63, 316–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhang, X.; Zhao, M.; Zhang, X.; Chi, J.; Liu, Y.; Lin, F.; Fu, Y.; Ma, D.; Yin, X. Activation in M1 but not M2 macrophages contributes to cardiac remodeling after myocardial infarction in rats: A critical role of the calcium sensing receptor/NRLP3 inflammasome. Cell. Physiol. Biochem. 2015, 35, 2483–2500. [Google Scholar] [CrossRef]
- Li, F.; Zhang, H.; Yang, L.; Yong, H.; Qin, Q.; Tan, M.; Xu, L.; Liang, K.; Zong, J.; Qian, W. NLRP3 deficiency accelerates pressure overload-induced cardiac remodeling via increased TLR4 expression. J. Mol. Med. 2018, 96, 1189–1202. [Google Scholar] [CrossRef]
- Li, R.; Lu, K.; Wang, Y.; Chen, M.; Zhang, F.; Shen, H.; Yao, D.; Gong, K.; Zhang, Z. Triptolide attenuates pressure overload-induced myocardial remodeling in mice via the inhibition of NLRP3 inflammasome expression. Biochem. Biophys. Res. Commun. 2017, 485, 69–75. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Y.; Chen, J.; Zhao, S.; Li, H. Pirfenidone attenuates cardiac fibrosis in a mouse model of TAC-induced left ventricular remodeling by suppressing NLRP3 inflammasome formation. Cardiology 2013, 126, 1–11. [Google Scholar] [CrossRef]
- Butts, B.; Butler, J.; Dunbar, S.B.; Corwin, E.; Gary, R.A. Effects of exercise on ASC methylation and IL-1 cytokines in heart failure. Med. Sci. Sports. Exerc. 2018, 50, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Sossalla, S.; Fluschnik, N.; Schotola, H.; Ort, K.R.; Neef, S.; Schulte, T.; Wittkopper, K.; Renner, A.; Schmitto, J.D.; Gummert, J.; et al. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ. Res. 2010, 107, 1150–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willeford, A.; Suetomi, T.; Nickle, A.; Hoffman, H.M.; Miyamoto, S.; Brown, J.H. CaMKIIdelta-mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight 2018, 3, e97054. [Google Scholar] [CrossRef] [Green Version]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/Calmodulin-dependent protein kinase II delta signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [Green Version]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-mediated clonal hematopoiesis accelerates heart failure through a mechanism involving the IL-1beta/NLRP3 inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA. Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kochi, A.N.; Tagliari, A.P.; Forleo, G.B.; Fassini, G.M.; Tondo, C. Cardiac and arrhythmic complications in patients with COVID-19. J. Cardiovasc. Electrophysiol. 2020, 31, 1003–1008. [Google Scholar] [CrossRef] [Green Version]
- Nasoohi, S.; Ismael, S.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) in cerebrovascular and neurodegenerative diseases: Regulation and implication. Mol. Neurobiol. 2018, 55, 7900–7920. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Shen, T.; Hu, J.; Zhang, L.; Zhang, Y.; Bao, L.; Cui, C.; Jin, G.; Zan, K.; Zhang, Z.; et al. Purinergic 2X7 receptor/NLRP3 pathway triggers neuronal apoptosis after ischemic stroke in the mouse. Exp. Neurol. 2017, 292, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Fann, D.Y.; Lim, Y.A.; Cheng, Y.L.; Lok, K.Z.; Chunduri, P.; Baik, S.H.; Drummond, G.R.; Dheen, S.T.; Sobey, C.G.; Jo, D.G.; et al. Evidence that NF-kappaB and MAPK signaling promotes NLRP inflammasome activation in neurons following ischemic stroke. Mol. Neurobiol. 2018, 55, 1082–1096. [Google Scholar] [CrossRef]
- Faried, A.; Dian, S.; Halim, D.; Hermanto, Y.; Pratama, D.M.A.; Arifin, M.Z. The neurological significance of COVID-19: Lesson learn from the pandemic. Interdiscip. Neurosurg. 2020, 22, 100809. [Google Scholar] [CrossRef] [PubMed]
- Gerc, V.; Masic, I.; Salihefendic, N.; Zildzic, M. Cardiovascular diseases (CVDs) in COVID-19 pandemic era. Mater. Sociomed. 2020, 32, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Thankam, F.G.; Agrawal, D.K. Molecular chronicles of cytokine burst in patients with coronavirus disease 2019 (COVID-19) with cardiovascular diseases. J. Thorac. Cardiovasc. Surg. 2021, 161, e217–e226. [Google Scholar] [CrossRef] [PubMed]
- Kadosh, B.S.; Garshick, M.S.; Gaztanaga, J.; Moore, K.J.; Newman, J.D.; Pillinger, M.; Ramasamy, R.; Reynolds, H.R.; Shah, B.; Hochman, J.; et al. COVID-19 and the heart and vasculature: Novel approaches to reduce virus-induced inflammation in patients with cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2045–2053. [Google Scholar] [CrossRef]
- Zheng, Y.Y.; Ma, Y.T.; Zhang, J.Y.; Xie, X. COVID-19 and the cardiovascular system. Nat. Rev. Cardiol. 2020, 17, 259–260. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.L.N.; Wang, X.; Sun, Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J. Thromb. Haemost. 2020, 18, 844–847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikdeli, B.; Madhavan, M.V.; Jimenez, D.; Chuich, T.; Dreyfus, I.; Driggin, E.; Nigoghossian, C.; Ageno, W.; Madjid, M.; Guo, Y.; et al. COVID-19 and thrombotic or thromboembolic disease: Implications for prevention, antithrombotic therapy, and follow-up: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 2950–2973. [Google Scholar] [CrossRef]
- Boukhris, M.; Hillani, A.; Moroni, F.; Annabi, M.S.; Addad, F.; Ribeiro, M.H.; Mansour, S.; Zhao, X.; Ybarra, L.F.; Abbate, A.; et al. Cardiovascular implications of the COVID-19 pandemic: A global perspective. Can. J. Cardiol. 2020, 36, 1068–1080. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini, P.E.; Boutjdir, M.; Capecchi, P.L. COVID-19, arrhythmic risk, and inflammation: Mind the gap! Circulation 2020, 142, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Li, B.; Wang, W.; Liu, X.; Liu, X.; Xia, Y.; Zhang, C.; Zhang, Y.; Zhang, M.; An, F. Rosuvastatin alleviates diabetic cardiomyopathy by inhibiting NLRP3 inflammasome and MAPK pathways in a type 2 diabetes rat model. Cardiovasc. Drugs Ther. 2014, 28, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xie, X.; Lei, T.; Zhang, K.; Lai, B.; Zhang, Z.; Guan, Y.; Mao, G.; Xiao, L.; Wang, N. Statins attenuate activation of the NLRP3 inflammasome by oxidized LDL or TNF alpha in vascular endothelial cells through a PXR-dependent mechanism. Mol. Pharmacol. 2017, 92, 256–264. [Google Scholar] [CrossRef] [Green Version]
- Kang, L.L.; Zhang, D.M.; Ma, C.H.; Zhang, J.H.; Jia, K.K.; Liu, J.H.; Wang, R.; Kong, L.D. Cinnamaldehyde and allopurinol reduce fructose-induced cardiac inflammation and fibrosis by attenuating CD36-mediated TLR4/6-IRAK4/1 signaling to suppress NLRP3 inflammasome activation. Sci. Rep. 2016, 6, 27460. [Google Scholar] [CrossRef] [PubMed]
- Fujisue, K.; Sugamura, K.; Kurokawa, H.; Matsubara, J.; Ishii, M.; Izumiya, Y.; Kaikita, K.; Sugiyama, S. Colchicine improves survival, left ventricular remodeling, and chronic cardiac function after acute myocardial infarction. Circ. J. 2017, 81, 1174–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Su, H.; Zhang, Y.; Liu, Z.; Kong, J. Cholecalciterol cholesterol emulsion attenuates experimental autoimmune myocarditis in mice via inhibition of the pyroptosis signaling pathway. Biochem. Biophys. Res. Commun. 2017, 493, 422–428. [Google Scholar] [CrossRef]
- Yang, F.; Qin, Y.; Wang, Y.; Meng, S.; Xian, H.; Che, H.; Lv, J.; Li, Y.; Yu, Y.; Bai, Y.; et al. Metformin inhibits the NLRP3 inflammasome via AMPK/mTOR-dependent effects in diabetic cardiomyopathy. Int. J. Biol. Sci. 2019, 15, 1010–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Che, H.; Wang, Y.; Li, H.; Li, Y.; Sahil, A.; Lv, J.; Liu, Y.; Yang, Z.; Dong, R.; Xue, H.; et al. Melatonin alleviates cardiac fibrosis via inhibiting lncRNA MALAT1/miR-141-mediated NLRP3 inflammasome and TGF-beta1/Smads signaling in diabetic cardiomyopathy. FASEB J. 2020, 34, 5282–5298. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Li, T.; Wang, Y.; Chang, Y.; Cheng, Y.; Lu, Y.; Liu, X.; Xu, L.; Li, X.; Yu, X.; et al. Empagliflozin prevents cardiomyopathy via sGC-cGMP-PKG pathway in type 2 diabetes mice. Clin. Sci. 2019, 133, 1705–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Heijden, T.; Kritikou, E.; Venema, W.; Van Duijn, J.; Van Santbrink, P.J.; Slutter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E-deficient Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, M.; Ma, H.; Fan, X.; Chen, X.; Miao, M.; Wu, H. Dexamethasone alleviate allergic airway inflammation in mice by inhibiting the activation of NLRP3 inflammasome. Int. Immunopharmacol. 2020, 78, 106017. [Google Scholar] [CrossRef] [PubMed]
- Cauchoisa, R.; Koubia, M.; Delarbreb, D.; Manetc, C.; Carvellid, J.; Blascoe, V.B.; Jeana, R.; Fouchef, L.; Bornetg, C.; Paulyh, V.; et al. Early IL-1 receptor blockade in severe inflammatory respiratory failure complicating COVID-19. Proc. Natl. Acad. Sci. USA 2020, 117, 22604. [Google Scholar]
- Reyes, A.Z.; Hu, K.A.; Teperman, J.; Muskardin, T.L.W.; Tardif, J.C.; Shah, B.; Pillinger, M.H. Anti-inflammatory therapy for COVID-19 infection: The case for colchicine. Ann. Rheum. Dis. 2020, 1–8. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
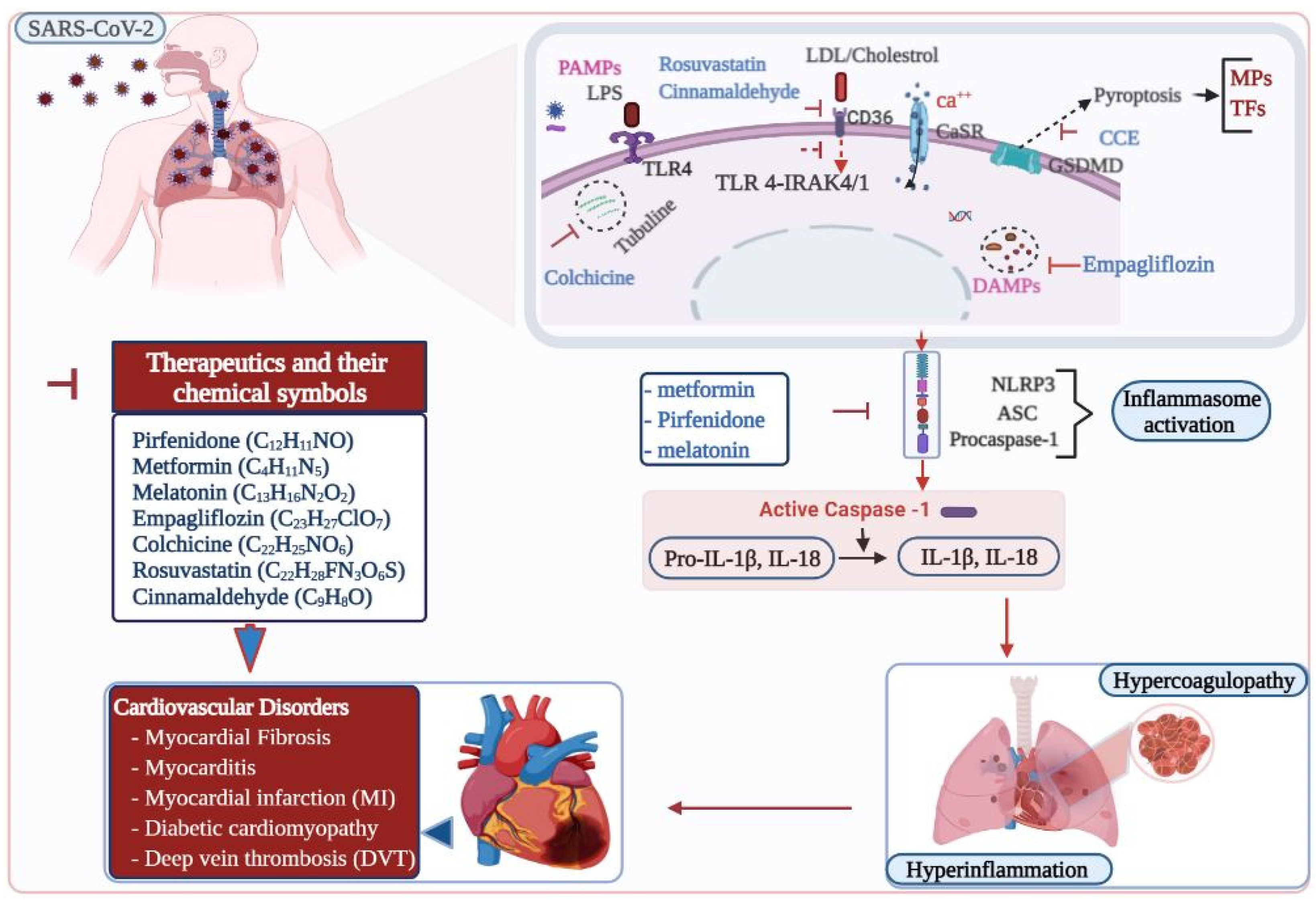{kind=link}
| Cardiac Disorders | Therapeutic Drugs | Mechanisms of NLRP3 Inflammasome Regulation |
|---|---|---|
| Myocardial fibrosis | Pirfenidone | Inhibit NLRP3-induced inflammatory and profibrotic responses [90] |
| Cardiomyopathy | Statins, Rosuvastatin | Inhibit oxidized low-density lipoprotein or tumor necrosis factor-α [110,111] |
| Cardiac inflammation and fibrosis | Cinnamaldehyde | Blockage of CD36-induced TLR4/6-IRAK4/1 signaling pathway [112] |
| Acute myocardial infarction (AMI) myocarditis | Colchicine | Inhibition of excessive tubulin polymerization [113] |
| CCE 1 | Downregulates pyroptosis pathway [114] | |
| Diabetic cardiomyopathy | Metformin | Activate AMPK 2, enhanced autophagy via inhibition of the mTOR pathway [115] |
| Melatonin | Inhibiting lncRMALAT1/miR-141-mediated NLRP3 inflammasome activation and TGF-β1/Smads signaling [116] | |
| Cardiac hypertrophy and fibrosis | Empagliflozin | Inhibition of oxidative stress-induced injury via sGC-cGMP-PKG pathway [117] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gedefaw, L.; Ullah, S.; Leung, P.H.M.; Cai, Y.; Yip, S.-P.; Huang, C.-L. Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells 2021, 10, 916. https://doi.org/10.3390/cells10040916
Gedefaw L, Ullah S, Leung PHM, Cai Y, Yip S-P, Huang C-L. Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells. 2021; 10(4):916. https://doi.org/10.3390/cells10040916
Chicago/Turabian StyleGedefaw, Lealem, Sami Ullah, Polly H. M. Leung, Yin Cai, Shea-Ping Yip, and Chien-Ling Huang. 2021. "Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients" Cells 10, no. 4: 916. https://doi.org/10.3390/cells10040916
APA StyleGedefaw, L., Ullah, S., Leung, P. H. M., Cai, Y., Yip, S. -P., & Huang, C. -L. (2021). Inflammasome Activation-Induced Hypercoagulopathy: Impact on Cardiovascular Dysfunction Triggered in COVID-19 Patients. Cells, 10(4), 916. https://doi.org/10.3390/cells10040916