
IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. Cell Culture
2.3. Mice
2.4. CS Exposure and CS Extract (CSE) Preparation
2.5. Immunohistochemistry
2.6. Immunofluorescence
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
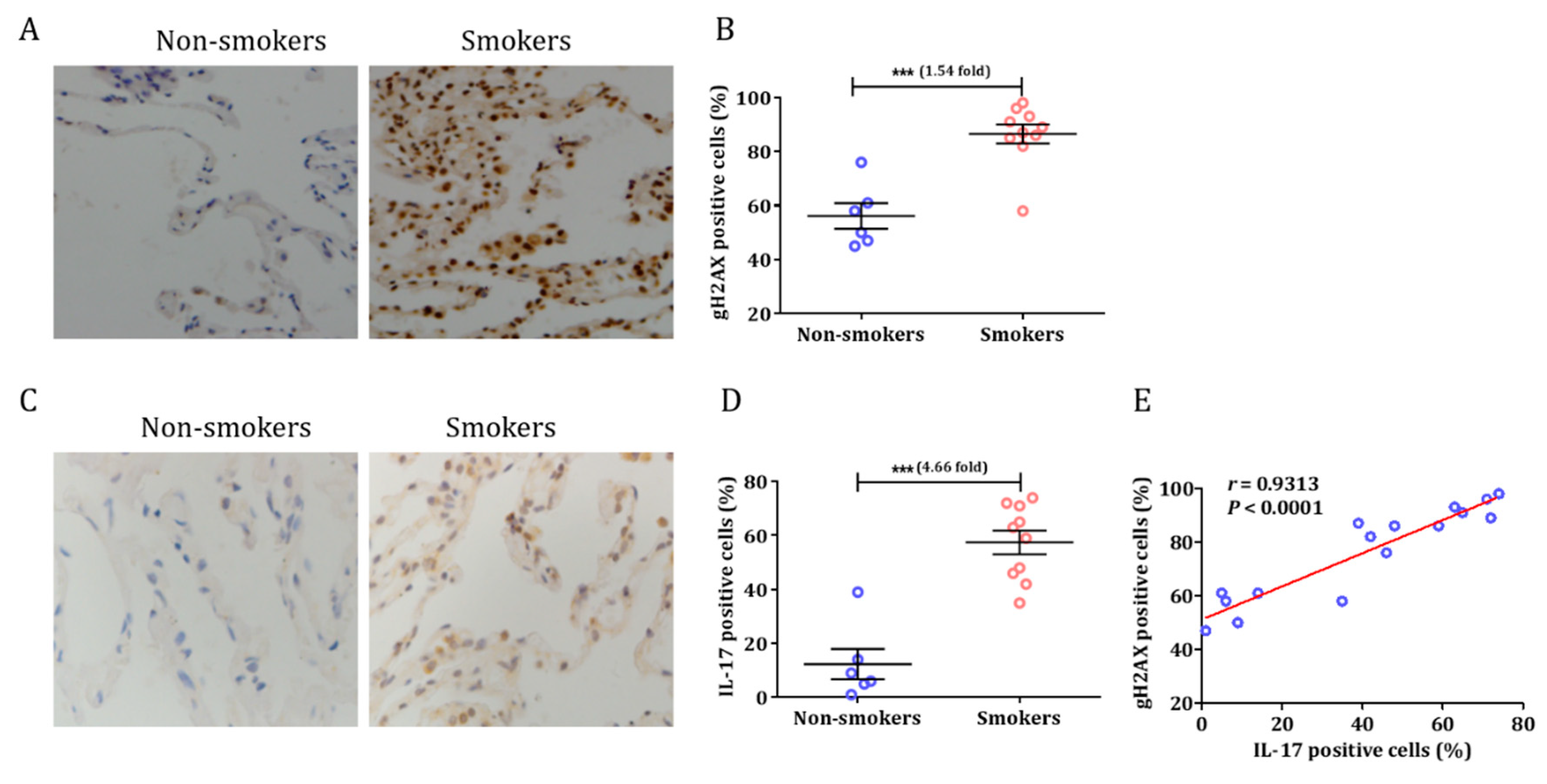
3.1. Smoking Induced a DNA Damage Response and Airway Inflammation in Human Tissue
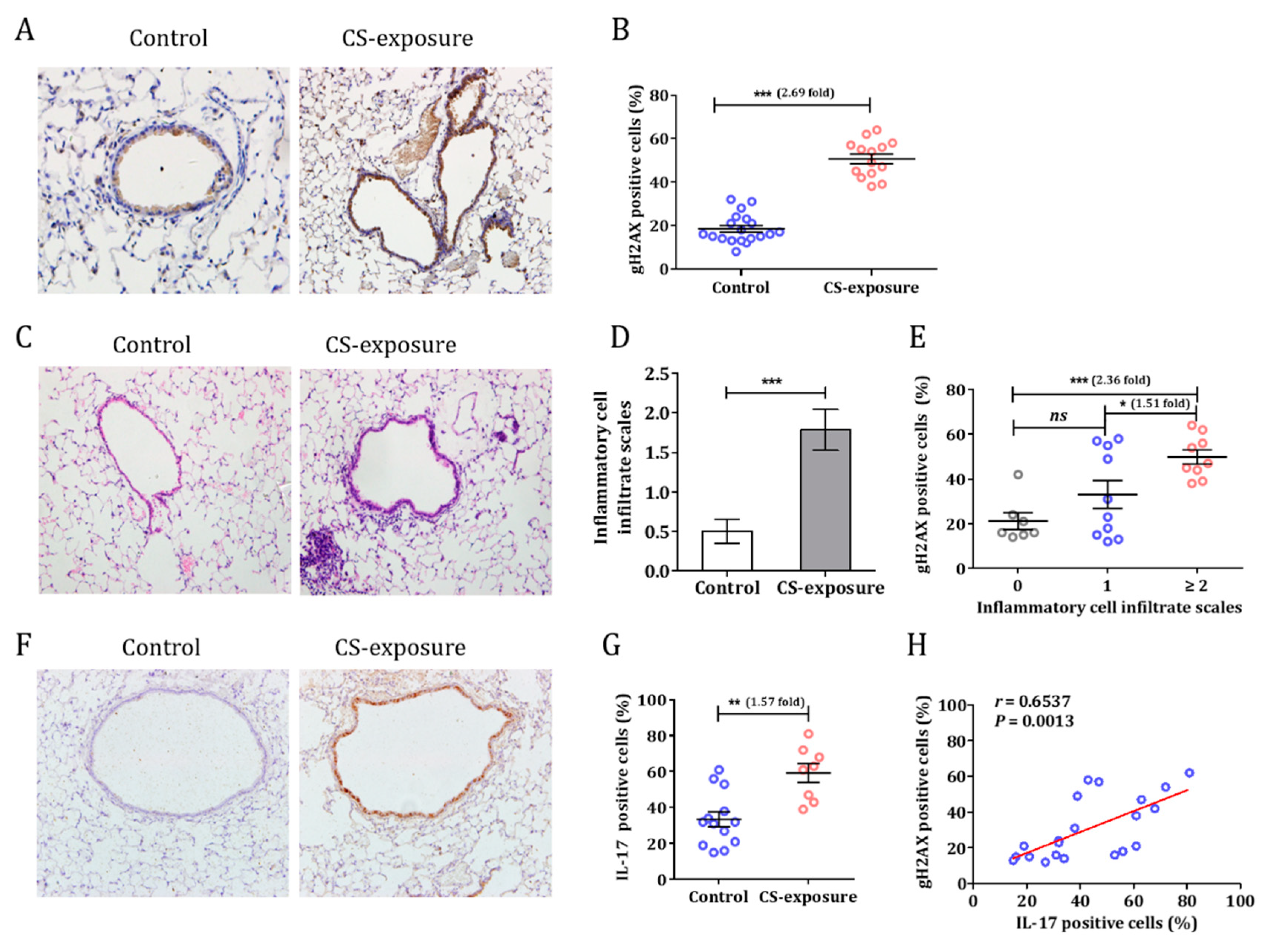
3.2. Cigarette Smoke Induced a DNA Damage Response in C57BL/6 Mice
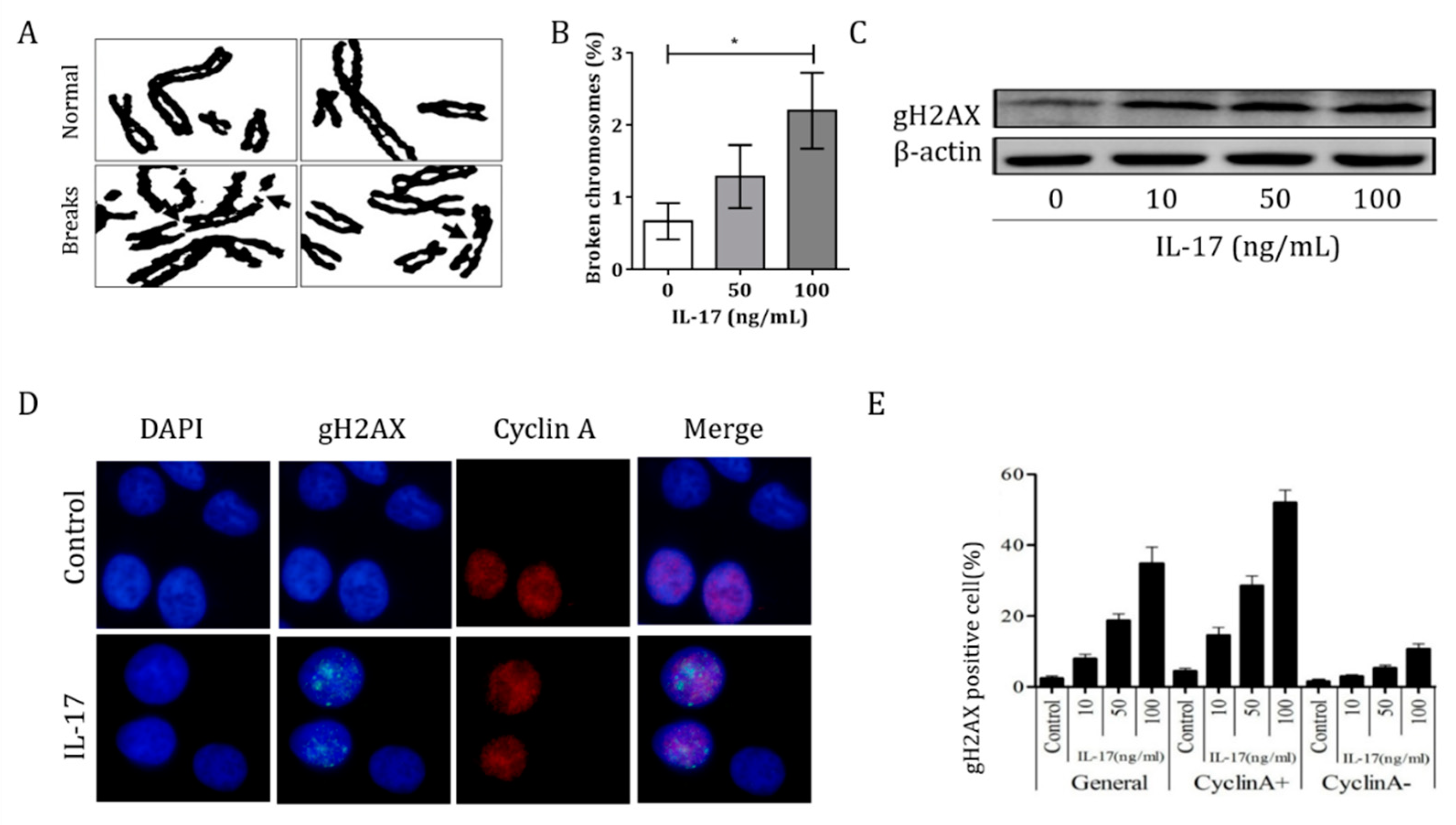
3.3. IL-17 Induced Genomic Instability in Bronchial Epithelial Cells
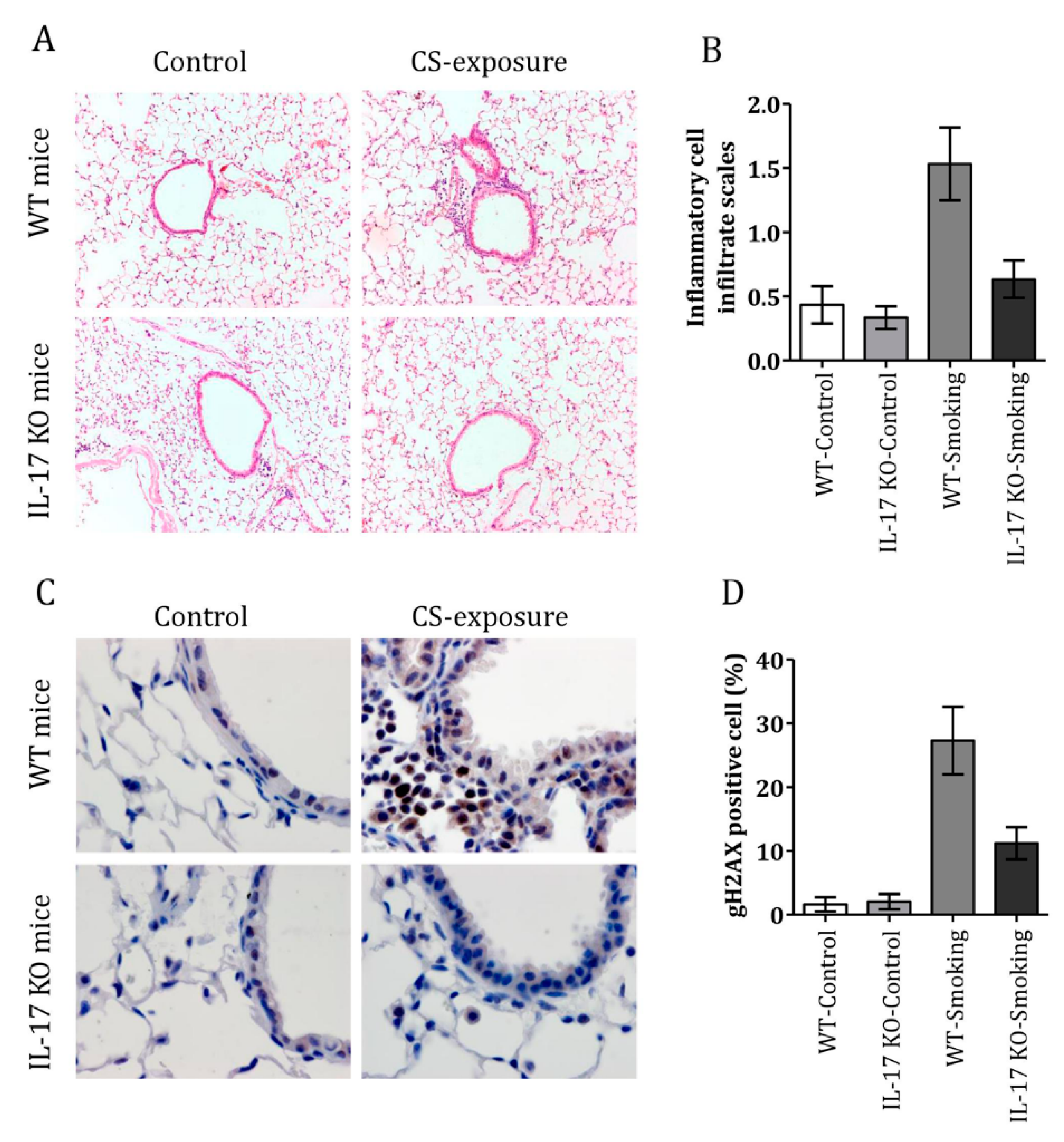
3.4. CS Induced Airway Inflammation and DDR in IL-17 KO Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sekine, Y.; Katsura, H.; Koh, E.; Hiroshima, K.; Fujisawa, T. Early detection of COPD is important for lung cancer surveillance. Eur. Respir. J. 2011, 39, 1230–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durham, A.; Adcock, I. The relationship between COPD and lung cancer. Lung Cancer 2015, 90, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandrov, L.B.; Ju, Y.S.; Haase, K.; Van Loo, P.; Martincorena, I.; Nik-Zainal, S.; Totoki, Y.; Fujimoto, A.; Nakagawa, H.; Shibata, T.; et al. Mutational signatures associated with tobacco smoking in human cancer. Science 2016, 354, 618–622. [Google Scholar] [CrossRef] [Green Version]
- Hays, L.; Zodrow, D.M.; Yates, J.; Deffebach, M.; Jacoby, D.B.; Olson, S.B.; Pankow, J.F.; Bagby, G.C. Cigarette smoke induces genetic instability in airway epithelial cells by suppressing FANCD2 expression. Br. J. Cancer 2008, 98, 1653–1661. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Hua, W.; Jin, Y.; Zhang, C.; Che, L.; Xia, L.; Zhou, J.; Chen, Z.; Li, W.; Shen, H. Tc17 cells are associated with cigarette smoke-induced lung inflammation and emphysema. Respirology 2015, 20, 426–433. [Google Scholar] [CrossRef]
- Zhang, L.; Cheng, Z.; Liu, W.; Wu, K. Expression of Interleukin (IL)-10, IL-17A and IL-22 in Serum and Sputum of Stable Chronic Obstructive Pulmonary Disease Patients. COPD: J. Chronic Obstr. Pulm. Dis. 2013, 10, 459–465. [Google Scholar] [CrossRef]
- Yanagisawa, H.; Hashimoto, M.; Minagawa, S.; Takasaka, N.; Ma, R.; Moermans, C.; Ito, S.; Araya, J.; Budelsky, A.; Goodsell, A.; et al. Role of IL-17A in murine models of COPD airway disease. Am. J. Physiol. Cell. Mol. Physiol. 2017, 312, L122–L130. [Google Scholar] [CrossRef] [PubMed]
- Swaidani, S.; Bulek, K.; Kang, Z.; Liu, C.; Lu, Y.; Aronica, M.; Li, X. SY-16 The critical role of epithelial-derived act1 in IL-17- and IL-25-mediated pulmonary inflammation. Cytokine 2008, 43, 279. [Google Scholar] [CrossRef]
- Henderson, W.R.; Lewis, D.B.; Albert, R.K.; Zhang, Y.; Lamm, W.J.; Chiang, G.K.; Jones, F.; Eriksen, P.; Tien, Y.T.; Jonas, M.; et al. The importance of leukotrienes in airway inflammation in a mouse model of asthma. J. Exp. Med. 1996, 184, 1483–1494. [Google Scholar] [CrossRef]
- Laniado-Laborín, R. Smoking and Chronic Obstructive Pulmonary Disease (COPD). Parallel Epidemics of the 21st Century. Int. J. Environ. Res. Public Heal. 2009, 6, 209–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celli, B.; MacNee, W.; Agusti, A.; Anzueto, A.; Berg, B.; Buist, A.; Calverley, P.; Chavannes, N.; Dillard, T.; Fahy, B.; et al. Standards for the diagnosis and treatment of patients with COPD: A summary of the ATS/ERS position paper. Eur. Respir. J. 2004, 23, 932–946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skillrud, D.M.; Offord, K.P.; Miller, R.D. Higher Risk of Lung Cancer in Chronic Obstructive Pulmonary Disease. Ann. Intern. Med. 1986, 105, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Aoshiba, K.; Zhou, F.; Tsuji, T.; Nagai, A. DNA damage as a molecular link in the pathogenesis of COPD in smokers. Eur. Respir. J. 2012, 39, 1368–1376. [Google Scholar] [CrossRef] [Green Version]
- Oit-Wiscombe, I.; Virag, L.; Soomets, U.; Altraja, A. Increased DNA Damage in Progression Of COPD: A Response By Poly(ADP-Ribose) Polymerase-1. PLoS ONE 2013, 8, e70333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorbanihaghjo, A.; Safa, J.; Alizadeh, S.; Argani, H.; Rashtchizadeh, N.; Taghinia, M.V.; Abbasi, M.M. Protective Effect of Fish Oil Supplementation on DNA Damage Induced by Cigarette Smoking. J. Heal. Popul. Nutr. 2013, 31, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Hattotuwa, K.L.; Gizycki, M.J.; Ansari, T.W.; Jeffery, P.K.; Barnes, N.C. The effects of inhaled fluticasone on airway inflammation in chronic obstructive pulmonary disease: A double-blind, placebo-controlled biopsy study. Am. J. Respir. Crit. Care Med. 2002, 165, 1592–1596. [Google Scholar] [CrossRef]
- Parimon, T.; Chien, J.W.; Bryson, C.L.; McDonell, M.B.; Udris, E.M.; Au, D.H. Inhaled Corticosteroids and Risk of Lung Cancer among Patients with Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2007, 175, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Xiang, T.; Long, H.; He, L.; Han, X.; Lin, K.; Liang, Z.; Zhuo, W.; Xie, R.; Zhu, B. Interleukin-17 produced by tumor microenvironment promotes self-renewal of CD133+ cancer stem-like cells in ovarian cancer. Oncogene 2013, 34, 165–176. [Google Scholar] [CrossRef]
- Liu, S.; Zhang, Q.; Chen, C.; Ge, D.; Qu, Y.; Chen, R.; Fan, Y.-M.; Li, N.; Tang, W.W.; Zhang, W.; et al. Hyperinsulinemia enhances interleukin-17-induced inflammation to promote prostate cancer development in obese mice through inhibiting glycogen synthase kinase 3-mediated phosphorylation and degradation of interleukin-17 receptor. Oncotarget 2016, 7, 13651–13666. [Google Scholar] [CrossRef]
- Wu, H.-H.; Hwang-Verslues, W.W.; Lee, W.-H.; Huang, C.-K.; Wei, P.-C.; Chen, C.-L.; Shew, J.-Y.; Lee, E.Y.-H.; Jeng, Y.-M.; Tien, Y.-W.; et al. Targeting IL-17B–IL-17RB signaling with an anti–IL-17RB antibody blocks pancreatic cancer metastasis by silencing multiple chemokines. J. Exp. Med. 2015, 212, 333–349. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Togo, S.; Al-Mugotir, M.; Kim, H.; Fang, Q.; Kobayashi, T.; Wang, X.; Mao, L.; Bitterman, P.; Rennard, S. NF-kappaB mediates the survival of human bronchial epithelial cells exposed to cigarette smoke extract. Respir. Res. 2008, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Conner, H.; Kobayashi, T.; Kim, H.; Wen, F.; Abe, S.; Fang, Q.; Wang, X.; Hashimoto, M.; Bitterman, P.; et al. Cigarette Smoke Extract Induces DNA Damage but Not Apoptosis in Human Bronchial Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2005, 33, 121–129. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, C.; Tian, B.; Geng, X.; Zhou, H.; Xu, Z.; Lai, T.; Wu, Y.; Bao, Z.; Chen, Z.; Li, W.; et al. IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability. Cells 2021, 10, 1173. https://doi.org/10.3390/cells10051173
Cao C, Tian B, Geng X, Zhou H, Xu Z, Lai T, Wu Y, Bao Z, Chen Z, Li W, et al. IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability. Cells. 2021; 10(5):1173. https://doi.org/10.3390/cells10051173
Chicago/Turabian StyleCao, Chao, Baoping Tian, Xinwei Geng, Hongbin Zhou, Zhiwei Xu, Tianwen Lai, Yanping Wu, Zhengqiang Bao, Zhihua Chen, Wen Li, and et al. 2021. "IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability" Cells 10, no. 5: 1173. https://doi.org/10.3390/cells10051173
APA StyleCao, C., Tian, B., Geng, X., Zhou, H., Xu, Z., Lai, T., Wu, Y., Bao, Z., Chen, Z., Li, W., Shen, H., & Ying, S. (2021). IL-17-Mediated Inflammation Promotes Cigarette Smoke-Induced Genomic Instability. Cells, 10(5), 1173. https://doi.org/10.3390/cells10051173