
Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
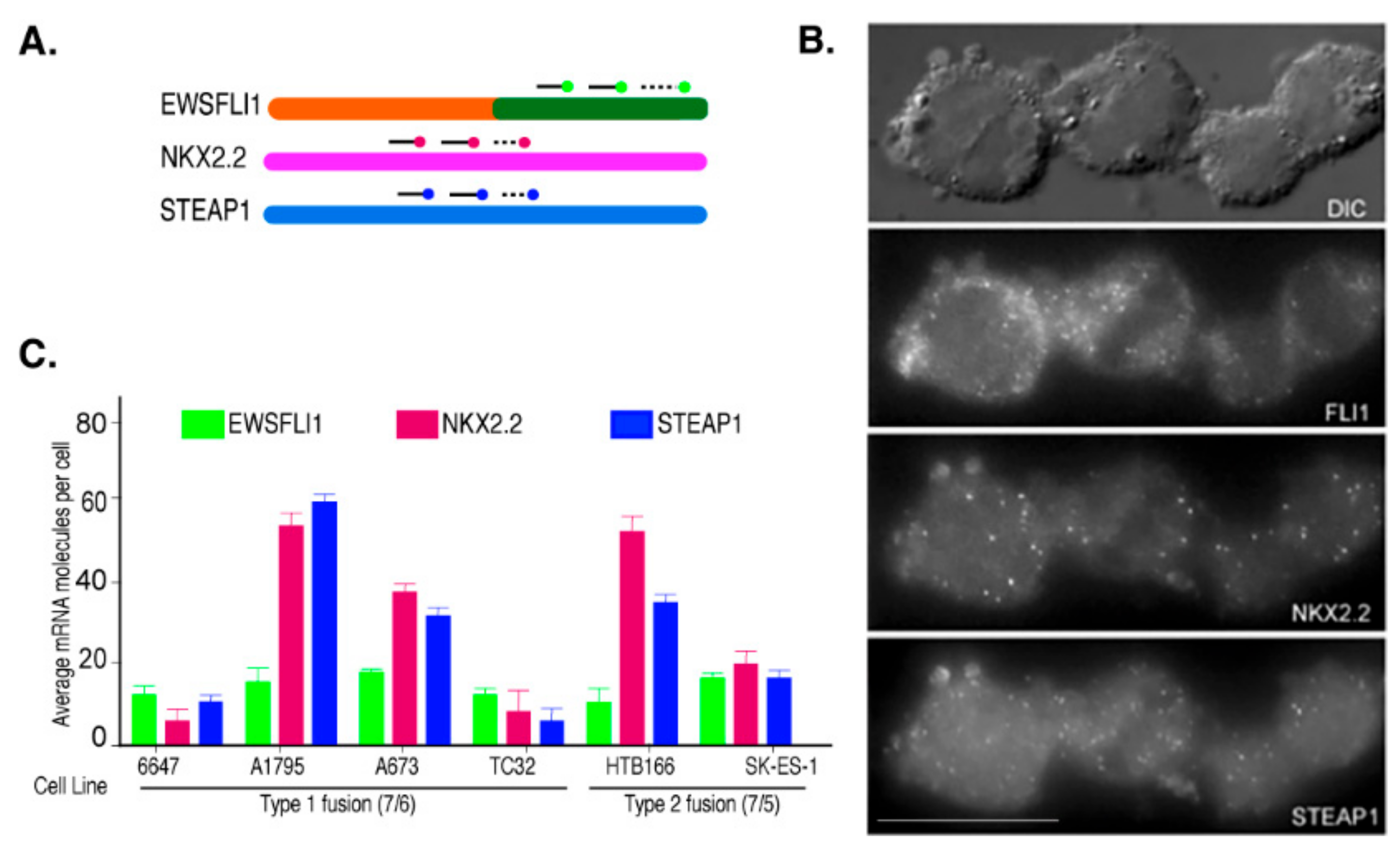
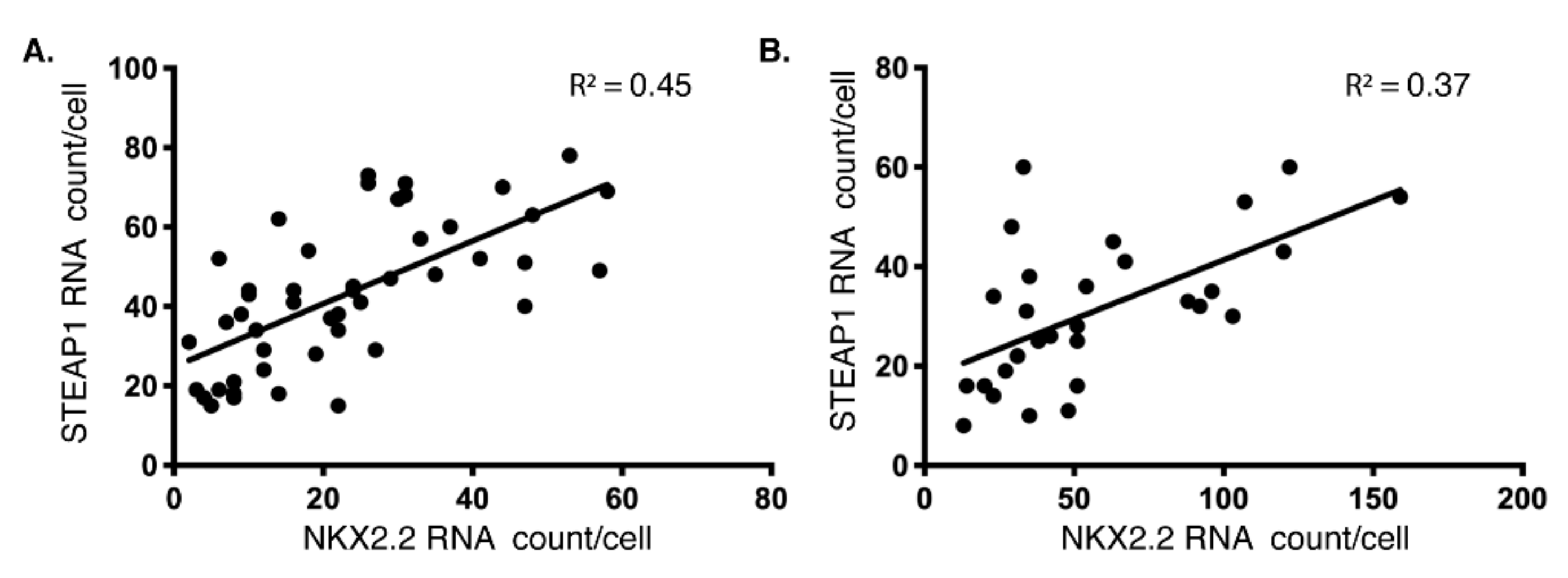
3.1. NKX2.2 and STEAP1 Expression Are Positively Correlated in Ewing’s Sarcoma Cell Lines
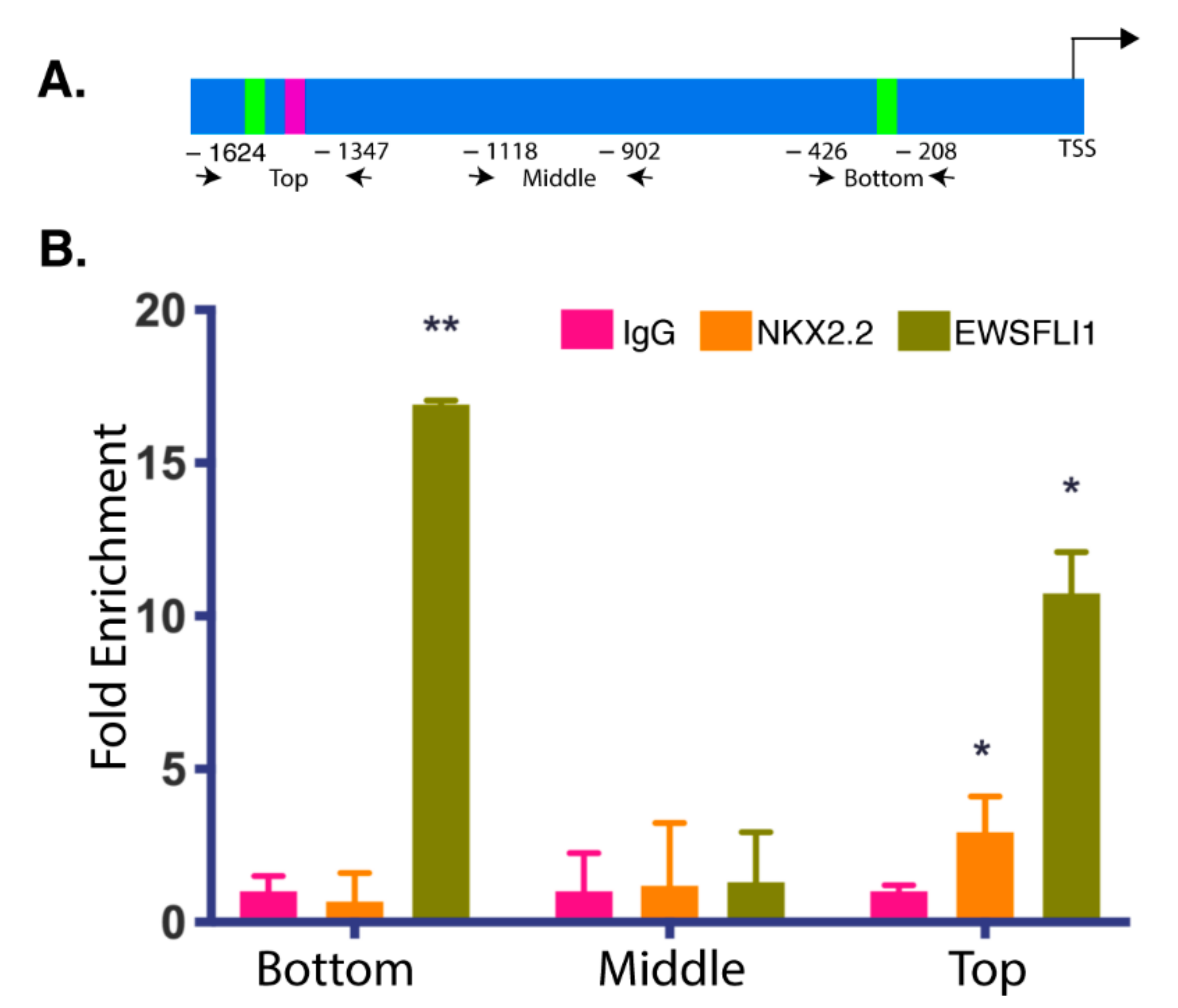
3.2. NKX2.2 and EWSFLI1 Bind within the Same Region of the STEAP1 Promoter
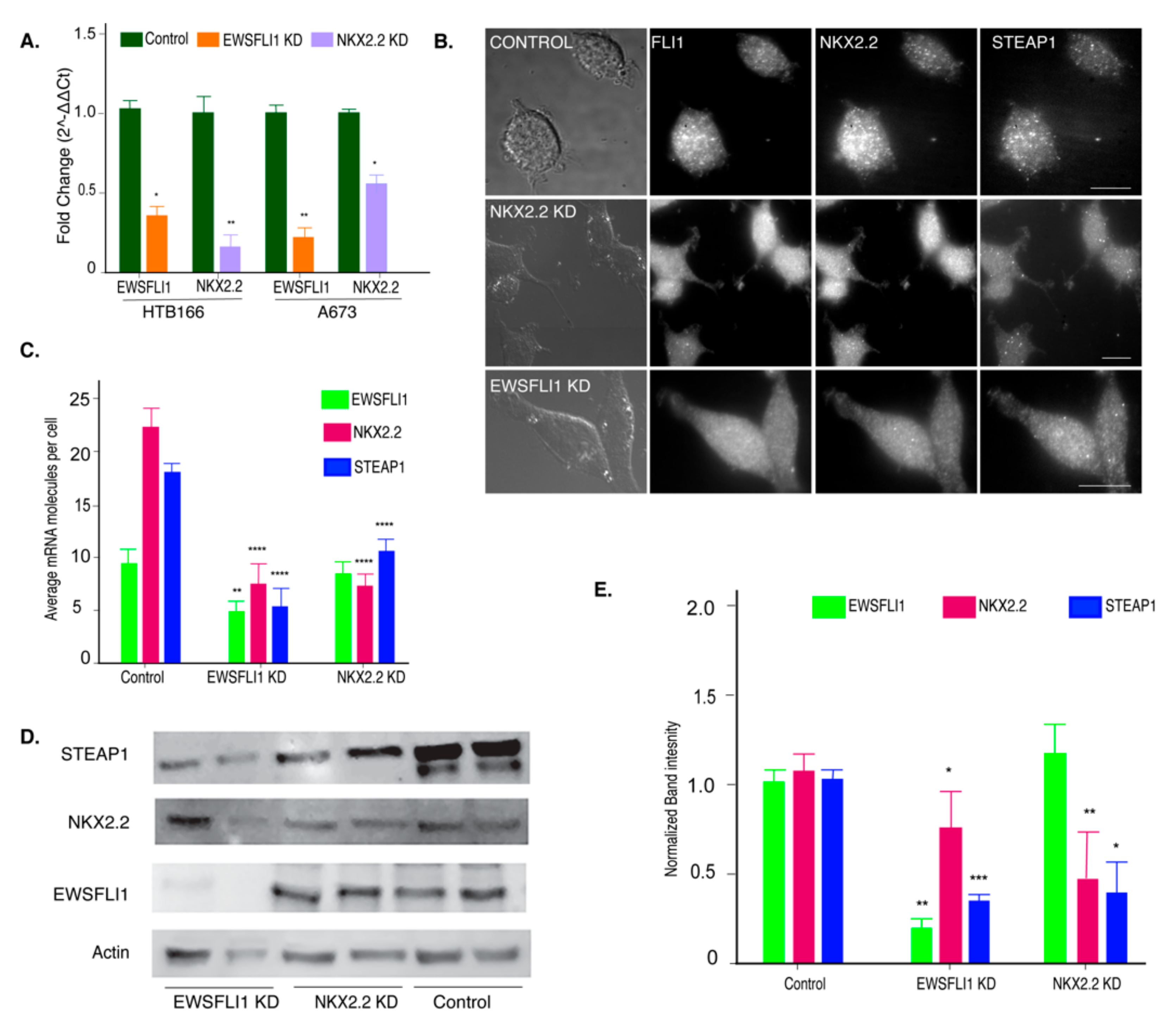
3.3. NKX2.2 Is Regulating STEAP1 Expression in ES
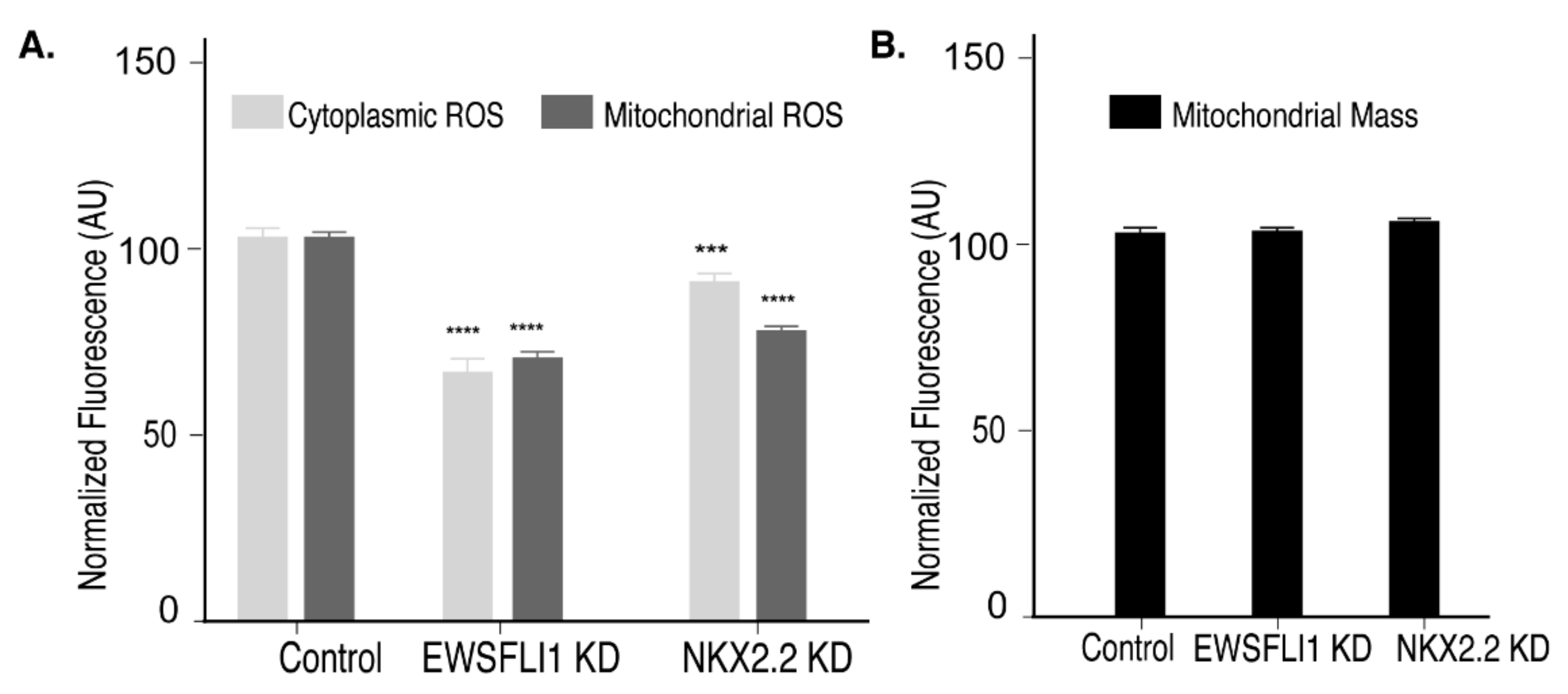
3.4. NKX2.2 Positively Regulates Reactive Oxygen Species in ES
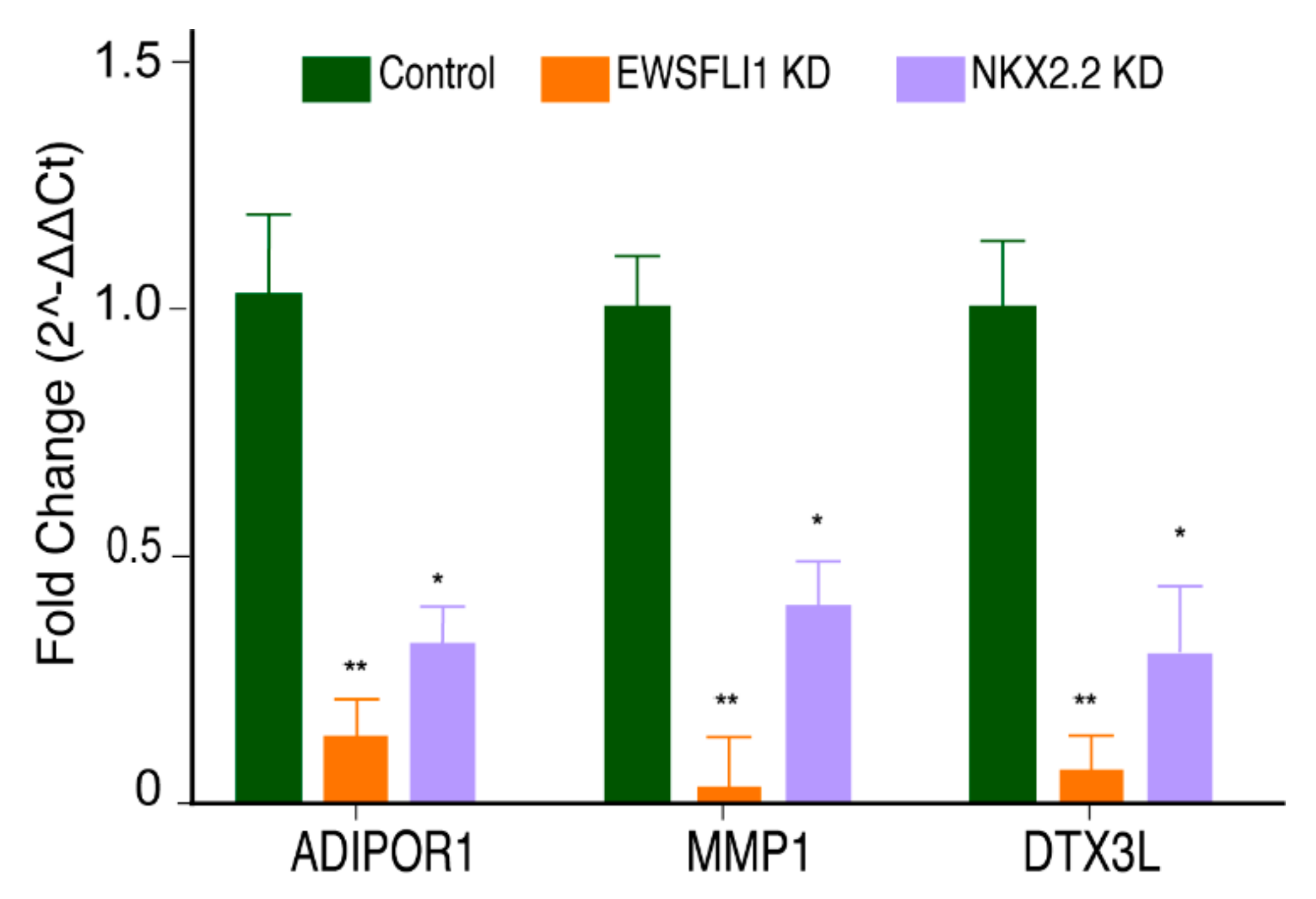
3.5. NKX2.2 Regulates the Function of STEAP1
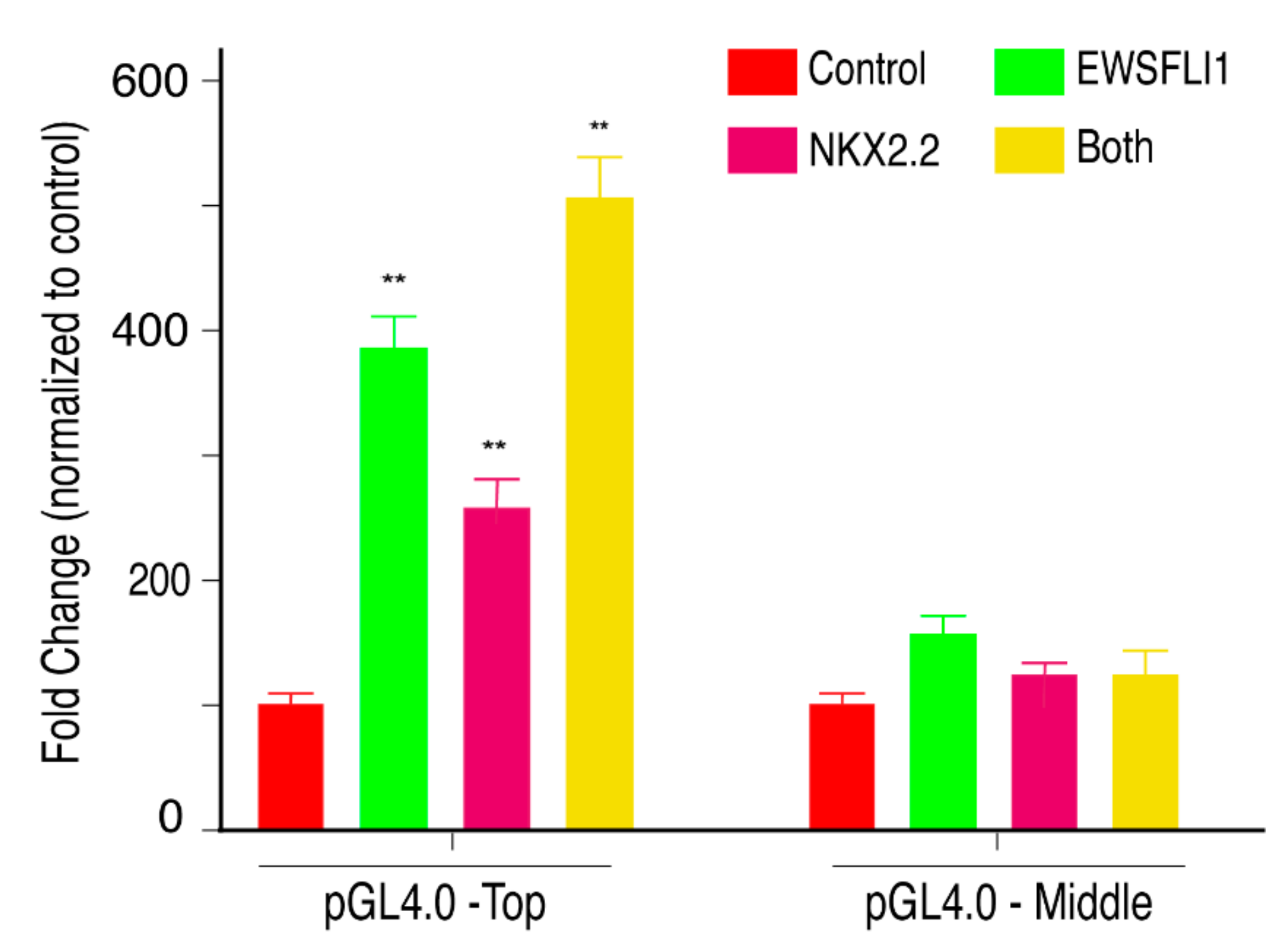
3.6. EWSFLI1 and NKX2.2 Act in a Cooperative Manner to Regulate STEAP1
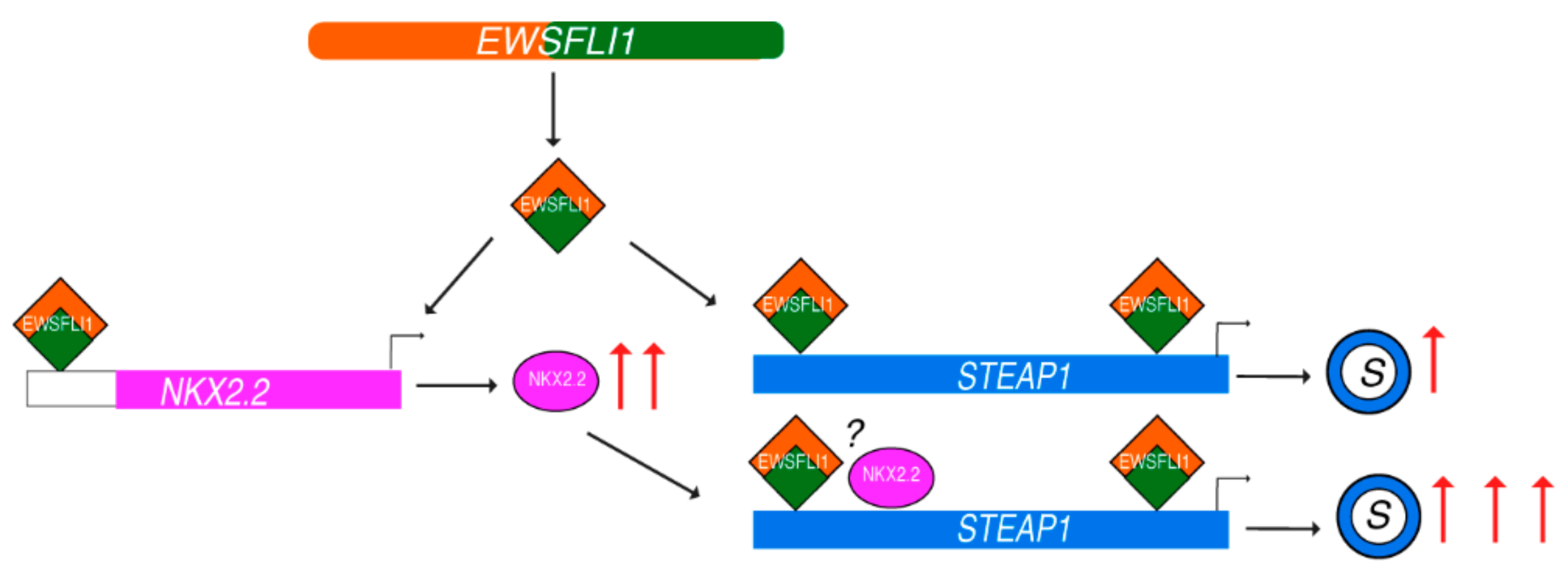
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mackintosh, C.; Madoz-Gurpide, J.; Ordonez, J.L.; Osuna, D.; Herrero-Martin, D. The molecular pathogenesis of Ewing’s sarcoma. Cancer Biol. Ther. 2010, 9, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Lessnick, S.L.; Kovar, H.K.; Houghton, P. The Molecular Basis of Sarcoma. Sarcoma 2011, 2011, 864130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hancock, J.D.; Lessnick, S.L. A transcriptional profiling meta-analysis reveals a core EWS-FLI gene expression signature. Cell Cycle 2008, 7, 250–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uren, A.; Toretsky, J.A. Ewing’s sarcoma oncoprotein EWS-FLI1: The perfect target without a therapeutic agent. Future Oncol. 2005, 1, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Riggi, N.; Knoechel, B.; Gillespie, S.M.; Rheinbay, E.; Boulay, G.; Suva, M.L.; Rossetti, N.E.; Boonseng, W.E.; Oksuz, O.; Cook, E.B.; et al. EWS-FLI1 utilizes divergent chromatin remodeling mechanisms to directly activate or repress enhancer elements in Ewing sarcoma. Cancer Cell 2014, 26, 668–681. [Google Scholar] [CrossRef] [Green Version]
- Tomazou, E.M.; Sheffield, N.C.; Schmidl, C.; Schuster, M.; Schonegger, A.; Datlinger, P.; Kubicek, S.; Bock, C.; Kovar, H. Epigenome mapping reveals distinct modes of gene regulation and widespread enhancer reprogramming by the oncogenic fusion protein EWS-FLI1. Cell Rep. 2015, 10, 1082–1095. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Selich-Anderson, J.; Miller, K.R.; Tanner, J.M.; Taslim, C.; Pishas, K.I.; Sharma, S.; Lessnick, S.L. Chromatin profiling reveals relocalization of lysine-specific demethylase 1 by an oncogenic fusion protein. Epigenetics 2020, 16, 1–20. [Google Scholar] [CrossRef]
- Guillon, N.; Tirode, F.; Boeva, V.; Zynovyev, A.; Barillot, E.; Delattre, O. The oncogenic EWS-FLI1 protein binds in vivo GGAA microsatellite sequences with potential transcriptional activation function. PLoS ONE 2009, 4, e4932. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.M.; Taslim, C.; Saund, R.S.; Lessnick, S.L. Identification of two types of GGAA-microsatellites and their roles in EWS/FLI binding and gene regulation in Ewing sarcoma. PLoS ONE 2017, 12, e0186275. [Google Scholar] [CrossRef]
- Lin, L.; Huang, M.; Shi, X.; Mayakonda, A.; Hu, K.; Jiang, Y.Y.; Guo, X.; Chen, L.; Pang, B.; Doan, N.; et al. Super-enhancer-associated MEIS1 promotes transcriptional dysregulation in Ewing sarcoma in co-operation with EWS-FLI1. Nucleic Acids Res. 2019, 47, 1255–1267. [Google Scholar] [CrossRef]
- Selvanathan, S.P.; Graham, G.T.; Grego, A.R.; Baker, T.M.; Hogg, J.R.; Simpson, M.; Batish, M.; Crompton, B.; Stegmaier, K.; Tomazou, E.M.; et al. EWS-FLI1 modulated alternative splicing of ARID1A reveals novel oncogenic function through the BAF complex. Nucleic Acids Res. 2019, 47, 9619–9636. [Google Scholar] [CrossRef]
- Lee, K.A. Ewings family oncoproteins: Drunk, disorderly and in search of partners. Cell Res. 2007, 17, 286–288. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Uversky, V.N.; Toretsky, J.A. Oncogenic partnerships: EWS-FLI1 protein interactions initiate key pathways of Ewing’s sarcoma. Clin. Cancer Res. 2010, 16, 4077–4083. [Google Scholar] [CrossRef] [Green Version]
- Liu Yang, H.C.; Hickstein, D. EWS-Fli-1 fusion protein interacts with hyperphosphorylated RNA polymerase II and interferes with serine-arginine protein-mediated RNA splicing. J. Biol. Chem. 2000, 275, 37612–37618. [Google Scholar] [CrossRef] [Green Version]
- Jeffrey Toretsky, V.E.; Levenson, A.; Abaan, O.D.; Parvin, J.D.; Cripe, T.P.; Rice, A.M.; Lee, S.B.; Uren, A. Oncoprotein EWS-FLI1 activity is enhanced by RNA helicase A. Cancer Res. 2006, 66, 5574–5581. [Google Scholar] [CrossRef] [Green Version]
- Joo, J.; Christensen, L.; Warner, K.; States, L.; Kang, H.-G.; Vo, K.; Lawlor, E.R.; May, W.A. GLI1 is a central mediator of EWS/FLI1 signaling in Ewing tumors. PLoS ONE 2009, 4, e7608. [Google Scholar] [CrossRef]
- Richter, G.H.; Plehm, S.; Fasan, A.; Rossler, S.; Unland, R.; Bennani-Baiti, I.M.; Hotfilder, M.; Lowel, D.; von Luettichau, I.; Mossbrugger, I.; et al. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc. Natl. Acad. Sci. USA 2009, 106, 5324–5329. [Google Scholar] [CrossRef] [Green Version]
- Xiaodong, L.; McGee-Lawrence, M.E.; Decker, M.; Westendorf, J.J. The Ewing’s Sarcoma Fusion Protein, EWS-FLI, Binds Runx2 and Blocks Osteoblast Differentiation. J. Cell Biochem. 2010, 111, 933–943. [Google Scholar] [CrossRef] [Green Version]
- Bilke, S.; Schwentner, R.; Yang, F.; Kauer, M.; Jug, G.; Walker, R.L.; Davis, S.; Zhu, Y.J.; Pineda, M.; Meltzer, P.S.; et al. Oncogenic ETS fusions deregulate E2F3 target genes in Ewing sarcoma and prostate cancer. Genome Res. 2013, 23, 1797–1809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovar, H. Blocking the road, stopping the engine or killing the driver? Advances in targeting EWS/FLI-1 fusion in Ewing sarcoma as novel therapy. Expert Opin. Ther. Targets 2014, 18, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Ge, Y.; Guo, L.; Huang, L. Potential approaches to the treatment of Ewing’s sarcoma. Oncotarget 2017, 8, 5523–5539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, I.M.; Maia, C.J.; Santos, C.R. STEAP proteins: From structure to applications in cancer therapy. Mol. Cancer Res. 2012, 10, 573–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, T.G.; Bach, H.; Cossarizza, A.; Matsumoto, I. The STEAP protein family: Versatile oxidoreductases and targets for cancer immunotherapy with overlapping and distinct cellular functions. Biol. Cell 2012, 104, 641–657. [Google Scholar] [CrossRef] [PubMed]
- Moreaux, J.; Kassambara, A.; Hose, D.; Klein, B. STEAP1 is overexpressed in cancers: A promising therapeutic target. Biochem. Biophys. Res. Commun. 2012, 429, 148–155. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Diebold, I.; Esposito, I.; Plehm, S.; Hauer, K.; Thiel, U.; da Silva-Buttkus, P.; Neff, F.; Unland, R.; Muller-Tidow, C.; et al. STEAP1 is associated with the invasive and oxidative stress phenotype of Ewing tumors. Mol. Cancer Res. 2012, 10, 52–65. [Google Scholar] [CrossRef] [Green Version]
- Grunewald, T.G.; Ranft, A.; Esposito, I.; da Silva-Buttkus, P.; Aichler, M.; Baumhoer, D.; Schaefer, K.L.; Ottaviano, L.; Poremba, C.; Jundt, G.; et al. High STEAP1 expression is associated with improved outcome of Ewing’s sarcoma patients. Ann. Oncol. 2012, 23, 2185–2190. [Google Scholar] [CrossRef]
- Huo, S.F.; Shang, W.L.; Yu, M.; Ren, X.P.; Wen, H.X.; Chai, C.Y.; Sun, L.; Hui, K.; Liu, L.H.; Wei, S.H.; et al. STEAP1 facilitates metastasis and epithelial-mesenchymal transition of lung adenocarcinoma via the JAK2/STAT3 signaling pathway. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Wu, H.T.; Chen, W.J.; Xu, Y.; Shen, J.X.; Chen, W.T.; Liu, J. The Tumor Suppressive Roles and Prognostic Values of STEAP Family Members in Breast Cancer. BioMed Res. Int. 2020, 2020, 9578484. [Google Scholar] [CrossRef]
- Zhang, Z.; Hou, W.B.; Zhang, C.; Tan, Y.E.; Zhang, D.D.; An, W.; Pan, S.W.; Wu, W.D.; Chen, Q.C.; Xu, H.M. A research of STEAP1 regulated gastric cancer cell proliferation, migration and invasion in vitro and in vivos. J. Cell Mol. Med. 2020, 24, 14217–14230. [Google Scholar] [CrossRef]
- Briscoe, J.L.; Sussel, L.; Serup, P.; Hartigan-O’Connor, D.; Jessell, T.M.; Rubenstein, J.L.; Ericson, J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 1999, 398, 622–627. [Google Scholar] [CrossRef]
- Altaba, A.R.; Sánchez, P.; Dahmane, N. Gli and hedgehog in cancer: Tumours, embryos and stem cells. Nature Rev. 2002, 2, 361–372. [Google Scholar] [CrossRef]
- Papizan, J.B.; Singer, R.A.; Tschen, S.-I.; Dhawan, S.; Friel, J.M.; Hipkens, S.B.; Magnuson, M.A.; Bhushan, A.; Sussel, L. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha reprogramming. Genes Dev. 2011, 25, 2291–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.; Torres, C.A.; Solomon, K.; Becker, T.C.; Newgard, C.B.; Wright, C.W.; Hagman, J.; Sussel, L. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J. Biol Chem. 2009, 284, 31236–31248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.; Bulut, G.; Abaan, O.; Chen, K.; Merchant, A.; Matsui, W.; Endo, Y.; Rubin, J.S.; Toretsky, J.; Uren, A. GLI1 is a direct transcriptional target of EWS-FLI1 oncoprotein. J. Biol. Chem. 2009, 284, 9074–9082. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.; Owen, L.A.; Trem, D.J.; Wong, J.S.; Whangbo, J.S.; Golub, T.R.; Lessnick, S.L. Expression profiling of EWS/FLI identifies NKX2.2 as a critical target gene in Ewing’s sarcoma. Cancer Cell 2006, 9, 405–416. [Google Scholar] [CrossRef] [Green Version]
- Owen, L.A.; Kowalewski, A.A.; Lessnick, S.L. EWS/FLI mediates transcriptional repression via NKX2.2 during oncogenic transformation in Ewing’s sarcoma. PLoS ONE 2008, 3, e1965. [Google Scholar] [CrossRef] [Green Version]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E. DNA microarrays reveal relationship of Ewing family tumors to both endothelial and fetal neural crest-derived cells and define novel targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.; Meltzer, S.P. Ewing’s sarcoma: General insights from a rare model. Cancer Cell 2006, 331–332. [Google Scholar] [CrossRef] [Green Version]
- Fadul, J.; Bell, R.; Hoffman, L.M.; Beckerle, M.C.; Engel, M.E.; Lessnick, S.L. EWS/FLI utilizes NKX2-2 to repress mesenchymal features of Ewing sarcoma. Genes Cancer 2015, 6, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Batish, M.; Tyagi, S. Fluorescence In Situ Imaging of Dendritic RNAs at Single-Molecule Resolution. Curr. Protoc. Neurosci. 2019, 89, e79. [Google Scholar] [CrossRef]
- Batish, M.; Raj, A.; Tyagi, S. Single molecule imaging of RNA in situ. Methods Mol. Biol. 2011, 714, 3–13. [Google Scholar] [CrossRef]
- Lazar, A. Molecular diagnosis of sarcomas: Chromosomal translocations in sarcomas. Arch. Pathol. Lab. Med. 2006, 130, 1199–1207. [Google Scholar] [CrossRef]
- Raj, A.; Peskin, C.S.; Tranchina, D.; Vargas, D.Y.; Tyagi, S. Stochastic mRNA synthesis in mammalian cells. PLoS Biol. 2006, 4, e309. [Google Scholar] [CrossRef]
- Markey, F.B.; Ruezinsky, W.; Tyagi, S.; Batish, M. Fusion FISH imaging: Single-molecule detection of gene fusion transcripts in situ. PLoS ONE 2014, 9, e93488. [Google Scholar] [CrossRef] [Green Version]
- Watada, H.; Mirmira, R.G.; Kalamaras, J.; German, M.S. Intramolecular control of transcriptional activity by the NK2-specific domain in NK-2 homeodomain proteins. Proc. Natl. Acad. Sci. USA 2000, 97, 9443–9448. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.T.; Anderson, K.R.; Mastracci, T.L.; Kaestner, K.H.; Sussel, L. Novel computational analysis of protein binding array data identifies direct targets of Nkx2.2 in the pancreas. BMC Bioinform. 2011, 12, 62. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.Z.; Li, Y.; Guo, L.R.; Zhao, Y.Y.; Zhao, X.J. Influence of expression of six transmembrane epithelial antigen of the prostate-1 on intracellular reactive oxygen species level and cell growth: An in vitro experiment. Zhonghua Yi Xue Za Zhi 2008, 88, 641–644. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Grek, C.L.; Tew, K.D. Redox metabolism and malignancy. Curr. Opin. Pharmacol. 2010, 10, 362–368. [Google Scholar] [CrossRef] [Green Version]
- Iwabu, M.; Yamauchi, T.; Okada-Iwabu, M.; Sato, K.; Nakagawa, T.; Funata, M.; Yamaguchi, M.; Namiki, S.; Nakayama, R.; Tabata, M.; et al. Adiponectin and AdipoR1 regulate PGC-1alpha and mitochondria by Ca(2+) and AMPK/SIRT1. Nature 2010, 464, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- DiMascio, L.; Voermans, C.; Uqoezwa, M.; Duncan, A.; Lu, D.; Wu, J.; Sankar, U.; Reya, T. Identification of adiponectin as a novel hemopoietic stem cell growth factor. J. Immunol. 2007, 178, 3511–3520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, N.; Ahonen, M.; Kahari, V.M. Matrix metalloproteinases in tumor invasion. Cell Mol. Life Sci 2000, 57, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; Dutt, S.; Xu, R.; Graves, K.; Juszczynski, P.; Manis, J.P.; Shipp, M.A. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 2009, 36, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Lawlor, E.R.; Sorensen, P.H. Twenty Years on: What Do We Really Know about Ewing Sarcoma and What Is the Path Forward? Crit. Rev. Oncog. 2015, 20, 155–171. [Google Scholar] [CrossRef] [Green Version]
- Minas, T.Z.; Han, J.; Javaheri, T.; Hong, S.H.; Schlederer, M.; Saygideger-Kont, Y.; Celik, H.; Mueller, K.M.; Temel, I.; Ozdemirli, M.; et al. YK-4-279 effectively antagonizes EWS-FLI1 induced leukemia in a transgenic mouse model. Oncotarget 2015, 6, 37678–37694. [Google Scholar] [CrossRef] [Green Version]
- Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Ramamoorthy, V.; Aiyer, R.A.; Maywald, R.L.; Buford, A.S.; Doolittle, D.K.; Culotta, K.S.; O’Dorisio, J.E.; Ludwig, J.A. An Oral Formulation of YK-4-279: Preclinical Efficacy and Acquired Resistance Patterns in Ewing Sarcoma. Mol. Cancer Ther. 2015, 14, 1591–1604. [Google Scholar] [CrossRef] [Green Version]
- Erkizan, H.V.; Kong, Y.; Merchant, M.; Schlottmann, S.; Barber-Rotenberg, J.S.; Yuan, L.; Abaan, O.D.; Chou, T.H.; Dakshanamurthy, S.; Brown, M.L.; et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat. Med. 2009, 15, 750–756. [Google Scholar] [CrossRef] [Green Version]
- Wagner, M.J.; Gopalakrishnan, V.; Ravi, V.; Livingston, J.A.; Conley, A.P.; Araujo, D.; Somaiah, N.; Zarzour, M.A.; Ratan, R.; Wang, W.L.; et al. Vincristine, Ifosfamide, and Doxorubicin for Initial Treatment of Ewing Sarcoma in Adults. Oncologist 2017, 22, 1271–1277. [Google Scholar] [CrossRef] [Green Version]
- Theisen, E.R.; Pishas, K.I.; Saund, R.S.; Lessnick, S.L. Therapeutic opportunities in Ewing sarcoma: EWS-FLI inhibition via LSD1 targeting. Oncotarget 2016. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Huang, H.; Chen, Z.; Zhang, Z.; Lu, W.; Qiu, M. The transcription factor NKX2-2 regulates oligodendrocyte differentiation through domain-specific interactions with transcriptional corepressors. J. Biol. Chem. 2020, 295, 1879–1888. [Google Scholar] [CrossRef]
- Shi, X.; Zheng, Y.; Jiang, L.; Zhou, B.; Yang, W.; Li, L.; Ding, L.; Huang, M.; Gery, S.; Lin, D.C.; et al. EWS-FLI1 regulates and cooperates with core regulatory circuitry in Ewing sarcoma. Nucleic Acids Res. 2020, 48, 11434–11451. [Google Scholar] [CrossRef]
- McCuiston, A.; Bishop, J.A. Usefulness of NKX2.2 Immunohistochemistry for Distinguishing Ewing Sarcoma from Other Sinonasal Small Round Blue Cell Tumors. Head Neck Pathol. 2018, 12, 89–94. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Markey, F.B.; Romero, B.; Parashar, V.; Batish, M. Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells 2021, 10, 1300. https://doi.org/10.3390/cells10061300
Markey FB, Romero B, Parashar V, Batish M. Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells. 2021; 10(6):1300. https://doi.org/10.3390/cells10061300
Chicago/Turabian StyleMarkey, Fatu Badiane, Brigette Romero, Vijay Parashar, and Mona Batish. 2021. "Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma" Cells 10, no. 6: 1300. https://doi.org/10.3390/cells10061300
APA StyleMarkey, F. B., Romero, B., Parashar, V., & Batish, M. (2021). Identification of a New Transcriptional Co-Regulator of STEAP1 in Ewing’s Sarcoma. Cells, 10(6), 1300. https://doi.org/10.3390/cells10061300