
Myostatin Deficiency Protects C2C12 Cells from Oxidative Stress by Inhibiting Intrinsic Activation of Apoptosis

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Myoblast Culturing
2.2. Generation of a C2C12-Mstn−/− Cell Line
2.3. IR/HR In Vitro Model
2.4. Scratch Assay
2.5. Fluorescein Diacetate/Propidium Iodide (FDA/PI) Stain
2.6. CellRox Green Assay
2.7. Immunofluorescence
2.8. Statistics
2.9. Study Approval
3. Results
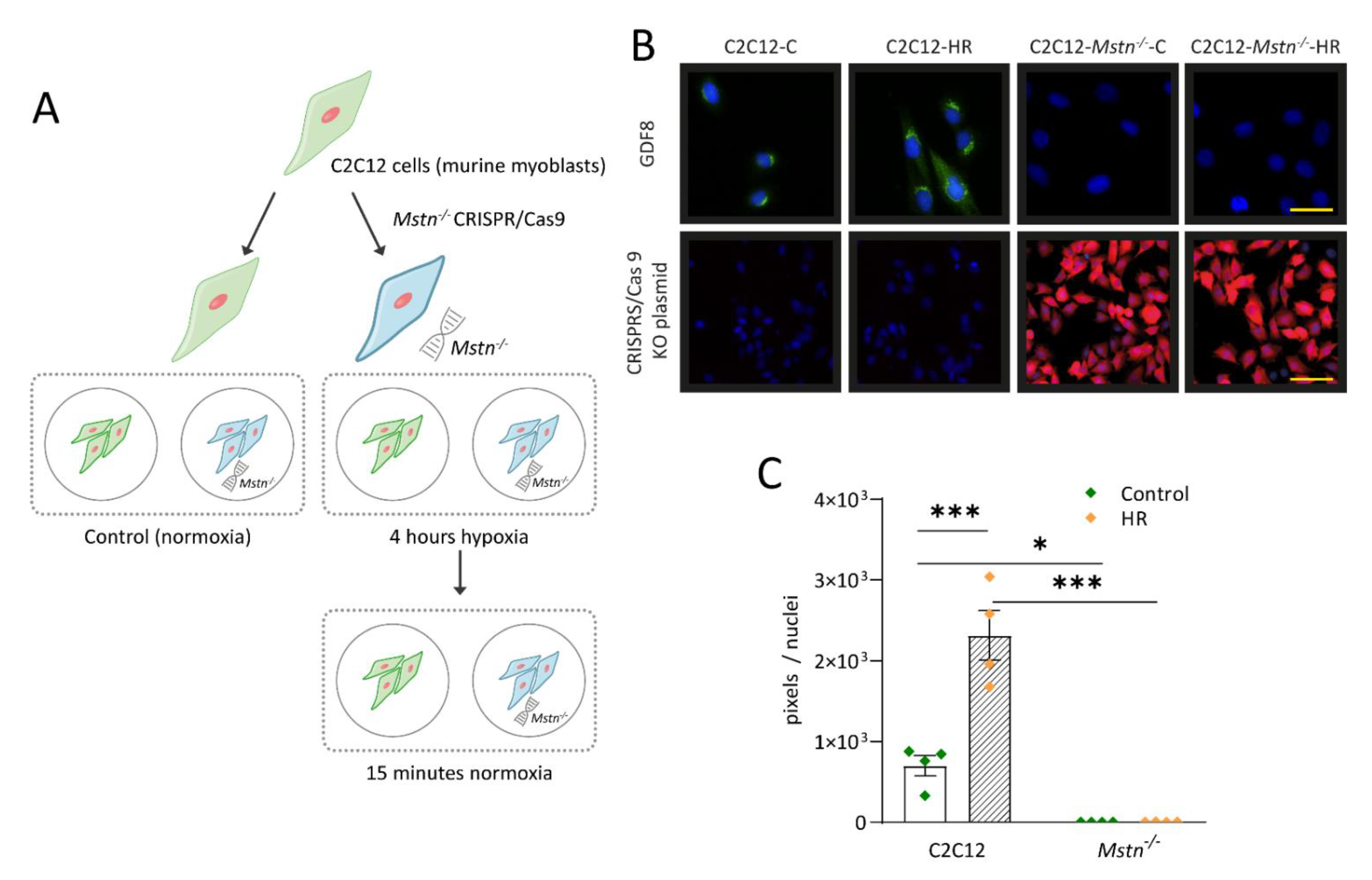
3.1. Generation of a C2C12-Mstn−/− Cell Line
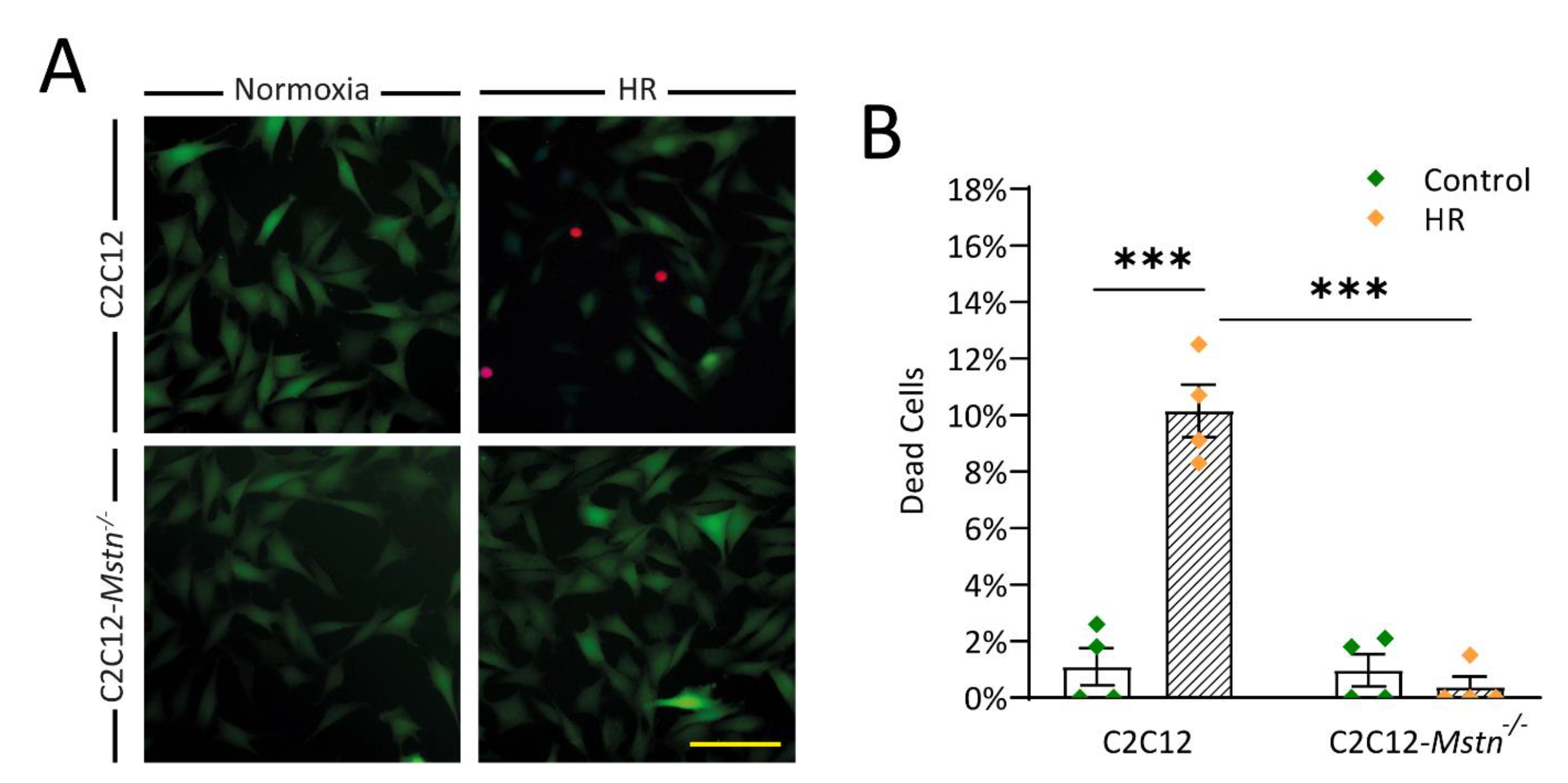
3.2. C2C12-Mstn−/− Cells Are Less Susceptible to HR Injury In Vitro
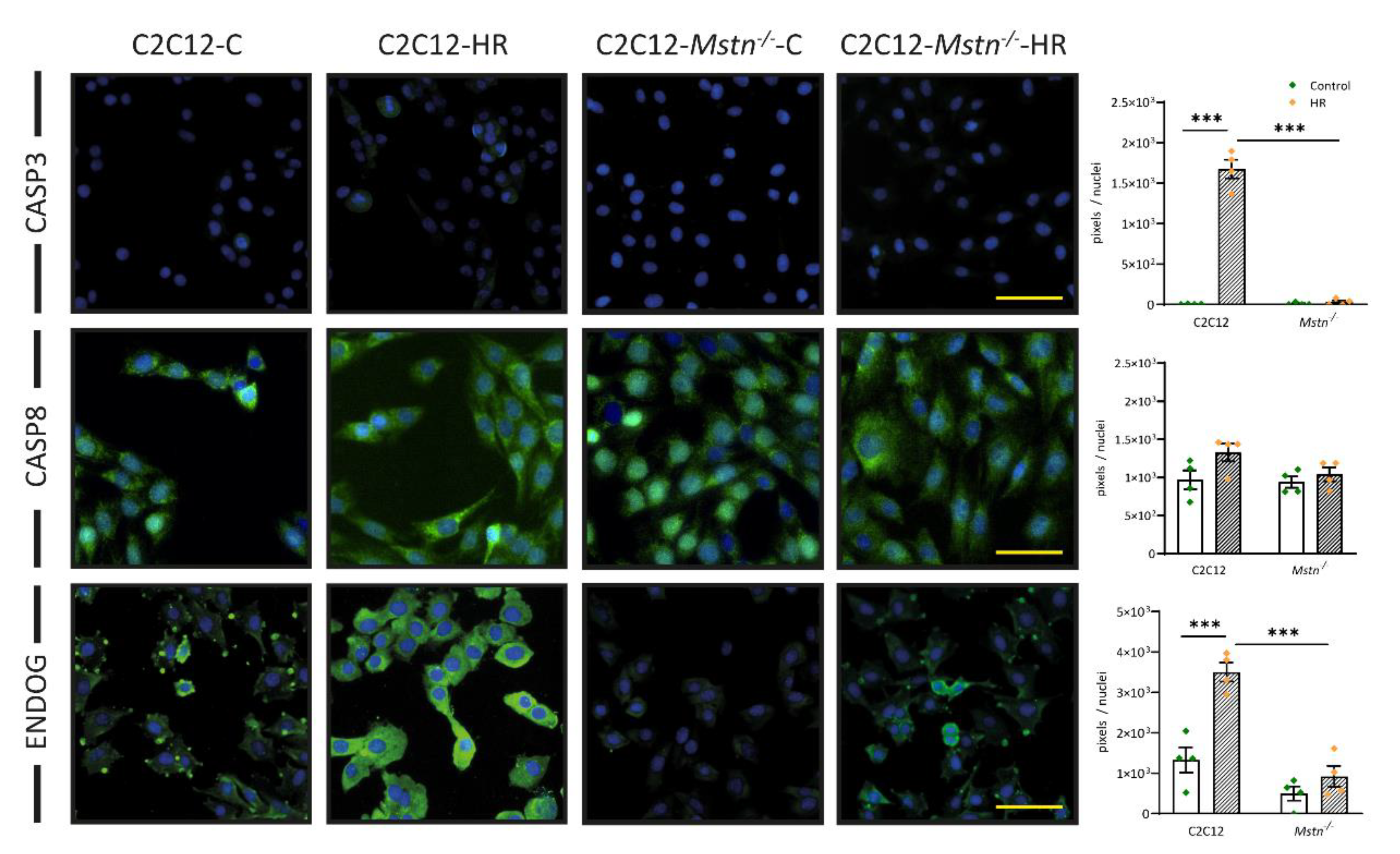
3.3. Mstn Inhibition Mitigates HR-Induced Activation of the Intrisic Apoptotic Pathway
3.4. HR Induces Signaling in the Myostatin–pp38 axis That May Sidestep Regulation by pMEK3/6 in C2C12-Mstn−/− Cells
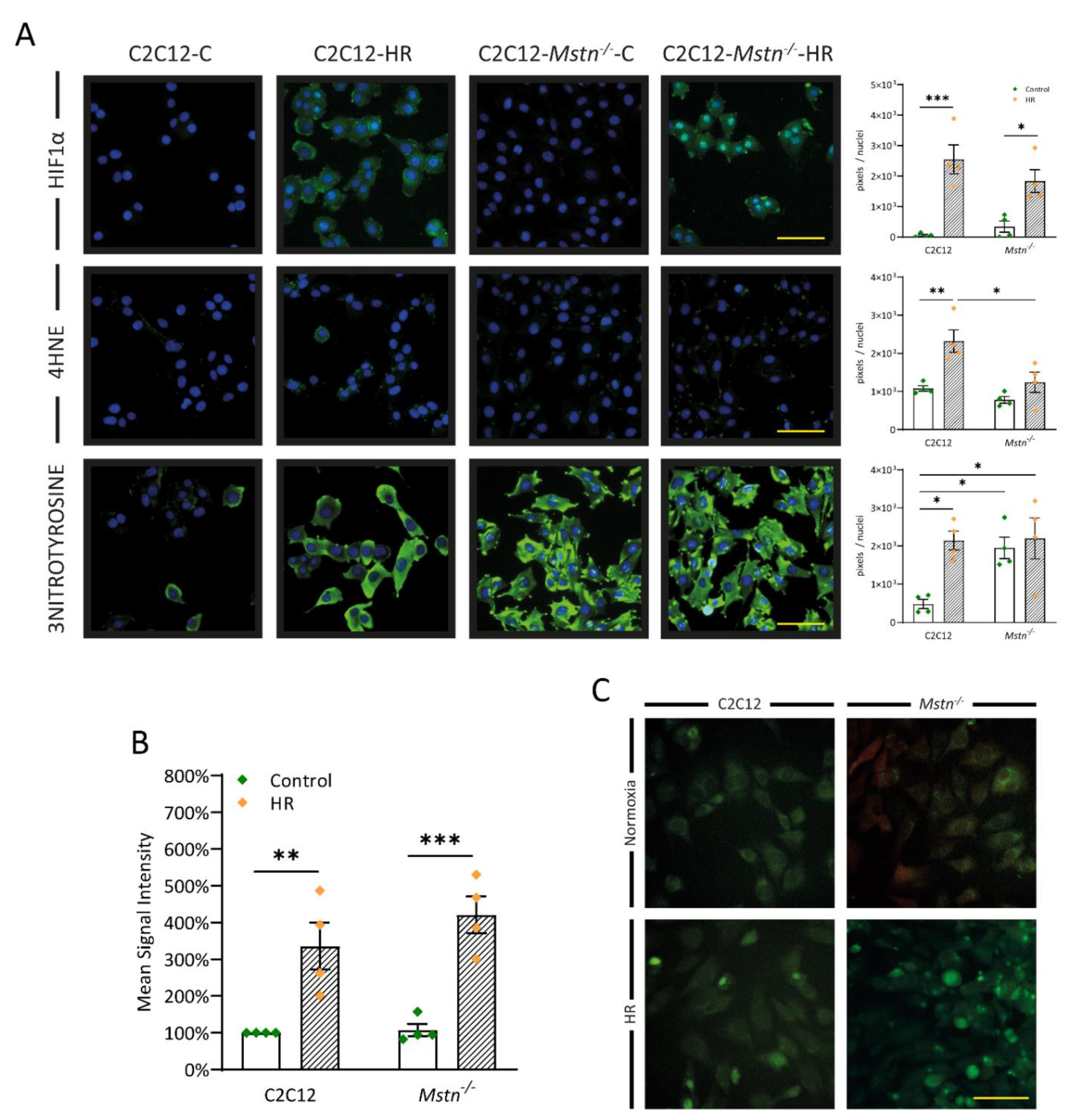
3.5. Inhibiting Myostatin Signaling Lowers Early Lipid Peroxidation and Nitrosative Stress Albeit ROS Generation in General Is Equal in C2C12 and C2C12-Mstn−/− Cells after HR
3.6. C2C12-Mstn−/− Cells Exhibit Increased Cell Migration under Hypoxic Conditions While C2C12 Cells Demonstrate Diminished DNA Replication
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, G.; Thakkar, M.; Robinson, C.; Doré, S. Limb Remote Ischemic Conditioning: Mechanisms, Anesthetics, and the Potential for Expanding Therapeutic Options. Front. Neurol. 2018, 9, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillero, E.; Akashi, H.; Najjar, M.; Ji, R.; Brandstetter, L.M.; Wang, C.; Liao, X.; Zhang, X.; Sperry, A.; Gailes, M.; et al. Activin type II receptor ligand signaling inhibition after experimental ischemic heart failure attenuates cardiac remodeling and prevents fibrosis. Am. J. Physiol. Hear. Circ. Physiol. 2020, 318, H378–H390. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, D.-H.; Shin, S.; Kim, S.J.; Kim, T.L.; Choi, Y.S. Effects of dexmedetomidine on inflammatory mediators after tourniquet-induced ischemia-reperfusion injury: A randomized, double-blinded, controlled study. Minerva Anestesiol. 2018, 85, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Yiang, G.-T.; Liao, W.-T.; Tsai, A.P.-Y.; Cheng, Y.-L.; Cheng, P.-W.; Li, C.-Y.; Li, C.-J. Current Mechanistic Concepts in Ischemia and Reperfusion Injury. Cell. Physiol. Biochem. 2018, 46, 1650–1667. [Google Scholar] [CrossRef]
- Toldo, S.; Mauro, A.G.; Cutter, Z.; Abbate, A. Inflammasome, pyroptosis, and cytokines in myocardial ischemia-reperfusion injury. Am. J. Physiol. Hear. Circ. Physiol. 2018, 315, H1553–H1568. [Google Scholar] [CrossRef] [PubMed]
- Seidlmayer, L.K.; Juettner, V.V.; Kettlewell, S.; Pavlov, E.V.; Blatter, L.A.; Dedkova, E.N. Distinct mPTP activation mechanisms in ischaemia–reperfusion: Contributions of Ca2+, ROS, pH, and inorganic polyphosphate. Cardiovasc. Res. 2015, 106, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef]
- Reed, J.C. Warner-Lambert/Parke Davis award lecture: Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev. 2012, 92, 689–737. [Google Scholar] [CrossRef] [Green Version]
- Irving, E.A.; Bamford, M. Role of mitogen- and stress-activated kinases in ischemic injury. J. Cereb. Blood Flow Metab. 2002, 22, 631–647. [Google Scholar] [CrossRef] [Green Version]
- Zarubin, T.; Han, J. Activation and signaling of the p38 MAP kinase pathway. Cell Res. 2005, 15, 11–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philip, B.; Lu, Z.; Gao, Y. Regulation of GDF-8 signaling by the p38 MAPK. Cell. Signal. 2005, 17, 365–375. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA. 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [Green Version]
- Verzola, D.; Barisione, C.; Picciotto, D.; Garibotto, G.; Koppe, L. Emerging role of myostatin and its inhibition in the setting of chronic kidney disease. Kidney Int. 2019, 95, 506–517. [Google Scholar] [CrossRef]
- Wallner, C.; Jaurich, H.; Wagner, J.M.; Becerikli, M.; Harati, K.; Dadras, M.; Lehnhardt, M.; Behr, B. Inhibition of GDF8 (Myostatin) accelerates bone regeneration in diabetes mellitus type 2. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriram, S.; Subramanian, S.; Sathiakumar, D.; Venkatesh, R.; Salerno, M.S.; McFarlane, C.D.; Kambadur, R.; Sharma, M. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-κB. Aging Cell 2011, 10, 931–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallner, C.; Drysch, M.; Becerikli, M.; Schmidt, S.V.; Hahn, S.; Wagner, J.M.; Reinkemeier, F.; Dadras, M.; Sogorski, A.; von Glinski, M.; et al. Deficiency of myostatin protects skeletal muscle cells from ischemia reperfusion injury. Sci. Rep. 2021, 11, 12572. [Google Scholar] [CrossRef]
- Giuliano, C.J.; Lin, A.; Girish, V.; Sheltzer, J.M. Generating Single Cell–Derived Knockout Clones in Mammalian Cells with CRISPR/Cas9. Curr. Protoc. Mol. Biol. 2019, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castillero, E.; Akashi, H.; Wang, C.; Najjar, M.; Ji, R.; Kennel, P.J.; Sweeney, H.L.; Schulze, P.C.; George, I. Cardiac myostatin upregulation occurs immediately after myocardial ischemia and is involved in skeletal muscle activation of atrophy. Biochem. Biophys. Res. Commun. 2015, 457, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Van Loo, G.; Schotte, P.; Van Gurp, M.; Demol, H.; Hoorelbeke, B.; Gevaert, K.; Rodriguez, I.; Ruiz-Carrillo, A.; Vandekerckhove, J.; Declercq, W.; et al. Endonuclease G: A mitochondrial protein released in apoptosis and involved in caspase-independent DNA degradation. Cell Death Differ. 2001, 8, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Wisdom, R.; Johnson, R.S.; Moore, C. c-Jun regulates cell cycle progression and apoptosis by distinct mechanisms. EMBO J. 1999, 18, 188–197. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Q.; Ding, N.; Zeng, Y.F.; Xiang, Y.Y.; Yang, M.W.; Hong, F.F.; Yang, S.L. New progress in roles of nitric oxide during hepatic ischemia reperfusion injury. World J. Gastroenterol. 2017, 23, 2505–2510. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Chen, H.S.; Xu, M.J.; Shen, J.G. Targeting reactive nitrogen species: A promising therapeutic strategy for cerebral ischemia-reperfusion injury. Acta Pharmacol. Sin. 2013, 34, 67–77. [Google Scholar] [CrossRef] [Green Version]
- He, N.; Jia, J.J.; Li, J.H.; Zhou, Y.F.; Lin, B.Y.; Peng, Y.F.; Chen, J.J.; Chen, T.C.; Tong, R.L.; Jiang, L.; et al. Remote ischemic perconditioning prevents liver transplantation-induced ischemia/reperfusion injury in rats: Role of ROS/RNS and eNOS. World J. Gastroenterol. 2017, 23, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Darwish, R.S.; Amiridze, N.; Aarabi, B. Nitrotyrosine as an Oxidative Stress Marker: Evidence for Involvement in Neurologic Outcome in Human Traumatic Brain Injury. J. Trauma Inj. Infect. Crit. Care 2007, 63, 439–442. [Google Scholar] [CrossRef]
- Kristen, A.V.; Ackermann, K.; Buss, S.; Lehmann, L.; Schnabel, P.A.; Haunstetter, A.; Katus, H.A.; Hardt, S.E. Inhibition of apoptosis by the intrinsic but not the extrinsic apoptotic pathway in myocardial ischemia-reperfusion. Cardiovasc. Pathol. 2013, 22, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Mocanu, M.M.; Baxter, G.F.; Yellon, D.M. Caspase inhibition and limitation of myocardial infarct size: Protection against lethal reperfusion injury. Br. J. Pharmacol. 2000, 130, 197–200. [Google Scholar] [CrossRef] [Green Version]
- Scarabelli, T.M.; Stephanou, A.; Pasini, E.; Comini, L.; Raddino, R.; Knight, R.A.; Latchman, D.S. Different signaling pathways induce apoptosis in endothelial cells and cardiac myocytes during ischemia/reperfusion injury. Circ. Res. 2002, 90, 745–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.Y.; Luo, X.; Wang, X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature 2001, 412, 95–99. [Google Scholar] [CrossRef]
- Ashraf, M.; Ebner, M.; Wallner, C.; Haller, M.; Khalid, S.; Schwelberger, H.; Koziel, K.; Enthammer, M.; Hermann, M.; Sickinger, S.; et al. A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury. Cell Commun. Signal. 2014, 12, 6. [Google Scholar] [CrossRef] [Green Version]
- Hanna, A.; Frangogiannis, N.G. The Role of the TGF-β Superfamily in Myocardial Infarction. Front. Cardiovasc. Med. 2019, 6, 140. [Google Scholar] [CrossRef]
- Kumar, S.; Boehm, J.; Lee, J.C. P38 MAP kinases: Key signalling molecules as therapeutic targets for inflammatory diseases. Nat. Rev. Drug Discov. 2003, 2, 717–726. [Google Scholar] [CrossRef]
- Ploquin, C.; Chabi, B.; Fouret, G.; Vernus, B.; Feillet-Coudray, C.; Coudray, C.; Bonnieu, A.; Ramonatxo, C. Lack of myostatin alters intermyofibrillar mitochondria activity, unbalances redox status, and impairs tolerance to chronic repetitive contractions in muscle. Am. J. Physiol. Endocrinol. Metab. 2012, 302. [Google Scholar] [CrossRef] [PubMed]
- Baati, N.; Feillet-Coudray, C.; Fouret, G.; Vernus, B.; Goustard, B.; Coudray, C.; Lecomte, J.; Blanquet, V.; Magnol, L.; Bonnieu, A.; et al. Myostatin deficiency is associated with lipidomic abnormalities in skeletal muscles. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1044–1055. [Google Scholar] [CrossRef] [Green Version]
- Rahman, I.A.; Mascaro, J.G.; Steeds, R.P.; Frenneaux, M.P.; Nightingale, P.; Gosling, P.; Townsend, P.; Townend, J.N.; Green, D.; Bonser, R.S. Remote ischemic preconditioning in human coronary artery bypass surgery: From promise to disappointment? Circulation 2010, 122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linkermann, A.; Hackl, M.J.; Kunzendorf, U.; Walczak, H.; Krautwald, S.; Jevnikar, A.M. Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant. 2013, 13, 2797–2804. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drysch, M.; Schmidt, S.V.; Becerikli, M.; Reinkemeier, F.; Dittfeld, S.; Wagner, J.M.; Dadras, M.; Sogorski, A.; von Glinski, M.; Lehnhardt, M.; et al. Myostatin Deficiency Protects C2C12 Cells from Oxidative Stress by Inhibiting Intrinsic Activation of Apoptosis. Cells 2021, 10, 1680. https://doi.org/10.3390/cells10071680
Drysch M, Schmidt SV, Becerikli M, Reinkemeier F, Dittfeld S, Wagner JM, Dadras M, Sogorski A, von Glinski M, Lehnhardt M, et al. Myostatin Deficiency Protects C2C12 Cells from Oxidative Stress by Inhibiting Intrinsic Activation of Apoptosis. Cells. 2021; 10(7):1680. https://doi.org/10.3390/cells10071680
Chicago/Turabian StyleDrysch, Marius, Sonja Verena Schmidt, Mustafa Becerikli, Felix Reinkemeier, Stephanie Dittfeld, Johannes Maximilian Wagner, Mehran Dadras, Alexander Sogorski, Maxi von Glinski, Marcus Lehnhardt, and et al. 2021. "Myostatin Deficiency Protects C2C12 Cells from Oxidative Stress by Inhibiting Intrinsic Activation of Apoptosis" Cells 10, no. 7: 1680. https://doi.org/10.3390/cells10071680
APA StyleDrysch, M., Schmidt, S. V., Becerikli, M., Reinkemeier, F., Dittfeld, S., Wagner, J. M., Dadras, M., Sogorski, A., von Glinski, M., Lehnhardt, M., Behr, B., & Wallner, C. (2021). Myostatin Deficiency Protects C2C12 Cells from Oxidative Stress by Inhibiting Intrinsic Activation of Apoptosis. Cells, 10(7), 1680. https://doi.org/10.3390/cells10071680