
M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines

 , , ,
, , ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Pharmacological Treatment
2.3. Western Blot Analysis
2.4. Flow Cytometry Analysis
2.5. Immunocytochemistry
2.6. M2 Receptor Knock-Down
- (siRNA1129-1) sense, 5′-AUUUACUACUAAAUCCUCCCCC-3′
- antisense 5′-GGGGGAGGAUUUAGUAGUAAAU-3′;
- (siRNA1129-2) sense 5′-AUGUAGCCCAUUUCUUCCCCC-3′
- antisense 5′-GGGGGAAGAAAUGGGCUACUA-3′;
- (siRNA 1129-3) sense 5′-UCCUUUGAGUUUCAGGCUGCCCCC-3′
- antisense 5′- GGGGGCAGCCUGAAACUCAAAGGA-3′;
- (siRNA 1129-4) sense 5′-AGUUACACCUUGACCUAACCCCC-3′
- antisense 5′-GGGGGUUAGGUCAAGGUGUAACU-3′
2.7. Time-Lapse Live Cell Imaging
2.8. Statistical Analysis
3. Results
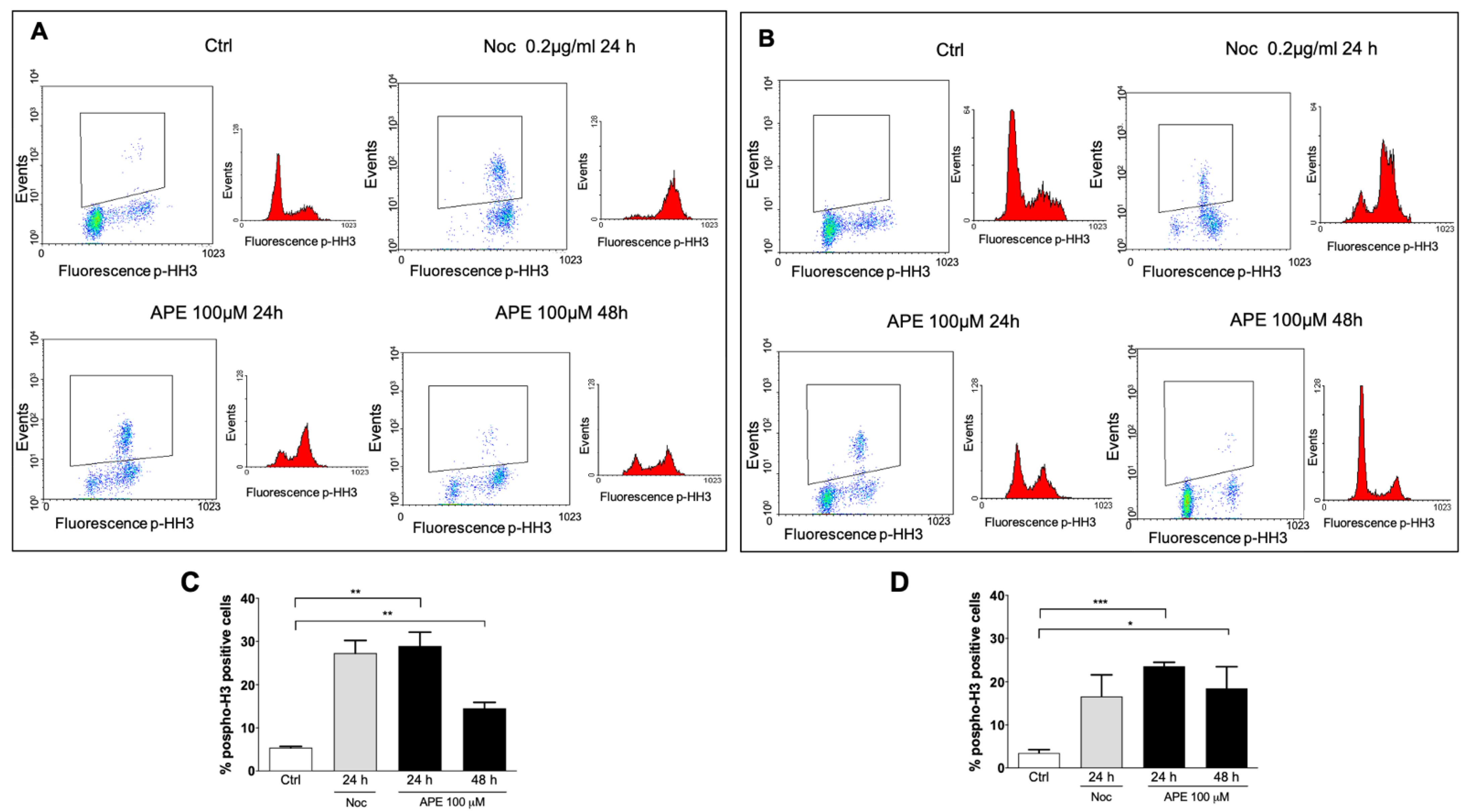
3.1. M2 Receptor Activation Caused Accumulation of the GB Cell Lines in the M Phase
3.2. Analysis of Mitosis Progression
3.3. APE-Arrested Glioblastoma Cells during Prometa-Metaphase Transition and Induced Multipolar Mitotic Spindles
3.4. Modulation of SIRT2 and Acetylated Tubulin Expression after APE Treatment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Rock, K.; McArdle, O.; Forde, P.; Dunne, M.; Fitzpatrick, D.; O’Neill, B.; Faul, C. A clinical review of treatment outcomes in glioblastoma multiforme—The validation in a non-trial population of the results of a randomised Phase III clinical trial: Has a more radical approach improved survival? Br. J. Radiol. 2012, 85, e729–e733. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.; Finklestein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current Trends in Targeted Therapies for Glioblastoma Multiforme. Neurol. Res. Int. 2012, 2012, 878425. [Google Scholar] [CrossRef] [PubMed]
- Tata, A.M. Muscarinic Acetylcholine Receptors: New Potential Therapeutic Targets in Antinociception and in Cancer Therapy. Recent Pat. CNS Drug Discov. 2008, 3, 94–103. [Google Scholar] [CrossRef]
- Jiménez, E.; Montiel, M. Activation of MAP kinase by muscarinic cholinergic receptors induces cell proliferation and protein synthesis in human breast cancer cells. J. Cell. Physiol. 2005, 204, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Oppitz, M.; Möbus, V.; Brock, S.; Drews, U. Muscarinic Receptors in Cell Lines from Ovarian Carcinoma: Negative Correlation with Survival of Patients. Gynecol. Oncol. 2002, 85, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Song, P.; Sekhon, H.S.; Lu, A.; Arredondo, J.; Sauer, D.; Gravett, C.; Mark, G.P.; Grando, S.A.; Spindel, E.R. M3 Muscarinic receptor antagonists inhibit small cell lung carcinoma growth and mitogen-activated protein kinase phosphorylation induced by acetylcholine secretion. Cancer Res. 2007, 67, 3936–3944. [Google Scholar] [CrossRef] [Green Version]
- Guizzetti, M.; Costa, P.; Peters, J.; Costa, L.G. Acetylcholine as a mitogen: Muscarinic receptor-mediated proliferation of rat astrocytes and human astrocytoma cells. Eur. J. Pharmacol. 1996, 297, 265–273. [Google Scholar] [CrossRef]
- Matute, C.; Arellano, R.O.; Conde-Guerri, B.; Miledi, R. mRNA coding for neurotransmitter receptors in a human astrocytoma. Proc. Natl. Acad. Sci. USA 1992, 89, 3399–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurwitz, D.; Razon, N.; Sokolovsky, M.; Soreq, H. Expression of muscarinic binding sites in primary human brain tumors. Dev. Brain Res. 1984, 14, 61–70. [Google Scholar] [CrossRef]
- Pacini, L.; De Falco, E.; Di Bari, M.; Coccia, A.; Siciliano, C.; Ponti, D.; Pastore, A.L.; Petrozza, V.; Carbone, A.; Tata, A.M.; et al. M2muscarinic receptors inhibit cell proliferation and migration in urothelial bladder cancer cells. Cancer Biol. Ther. 2014, 15, 1489–1498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Ponti, D.; Calogero, A.; Tata, A.M. M2 muscarinic receptors inhibit cell proliferation in human glioblastoma cell lines. Life Sci. 2012, 91, 1134–1137. [Google Scholar] [CrossRef]
- Lucianò, A.M.; Perciballi, E.; Fiore, M.; Del Bufalo, D.; Tata, A.M. The Combination of the M2 Muscarinic Receptor Agonist and Chemotherapy Affects Drug Resistance in Neuroblastoma Cells. Int. J. Mol. Sci. 2020, 21, 8433. [Google Scholar] [CrossRef]
- Ferretti, M.; Fabbiano, C.; Di Bari, M.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Ponti, D.; Ruggieri, P.; Raco, A.; Ricordy, R.; et al. M2 receptor activation inhibits cell cycle progression and survival in human glioblastoma cells. J. Cell. Mol. Med. 2013, 17, 552–566. [Google Scholar] [CrossRef]
- Alessandrini, F.; Cristofaro, I.; Di Bari, M.; Zasso, J.; Conti, L.; Tata, A.M. The activation of M2 muscarinic receptor inhibits cell growth and survival in human glioblastoma cancer stem cells. Int. Immunopharmacol. 2015, 29, 105–109. [Google Scholar] [CrossRef]
- Di Bari, M.; Tombolillo, V.; Conte, C.; Castigli, E.; Sciaccaluga, M.; Iorio, E.; Carpinelli, G.; Ricordy, R.; Fiore, M.; Degrassi, F.; et al. Cytotoxic and genotoxic effects mediated by M2 muscarinic receptor activation in human glioblastoma cells. Neurochem. Int. 2015, 90, 261–270. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Vacchelli, E.; Kroemer, G. Cell Death Signaling and Anticancer Therapy. Front. Oncol. 2011, 1. [Google Scholar] [CrossRef] [Green Version]
- Naqvi, A.Z.; Mahjabeen, I.; Ameen, S.; Ahmad, M.W.; Khan, A.U.; Akram, Z.; Kayani, M.A. Genetic and expression variations of cell cycle pathway genes in brain tumor patients. Biosci. Rep. 2020, 40, BSR20190629. [Google Scholar] [CrossRef]
- Bigner, S.H.; Vogelstein, B. Cytogenetics and Molecular Genetics of Malignant Gliomas and Medulloblastoma. Brain Pathol. 1990, 1, 12–18. [Google Scholar] [CrossRef]
- Mc Gee, M.M. Targeting the Mitotic Catastrophe Signaling Pathway in Cancer. Mediat. Inflamm. 2015, 2015, 146282. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef]
- Weaver, B.A.; Cleveland, D.W. Decoding the links between mitosis, cancer, and chemotherapy: The mitotic checkpoint, adaptation, and cell death. Cancer Cell 2005, 8, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Vitovcova, B.; Skarkova, V.; Rudolf, K.; Rudolf, E. Biology of Glioblastoma Multiforme—Exploration of Mitotic Catastrophe as a Potential Treatment Modality. Int. J. Mol. Sci. 2020, 21, 5324. [Google Scholar] [CrossRef]
- Bataller, M.; Portugal, J. Mitotic Catastrophe as a Consequence of Chemotherapy. Anti-Cancer Agents Med. Chem. 2006, 6, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- De Brabander, M.J.; Van de Veire, R.M.L.; Aerts, F.E.M.; Borgers, M.; Janssen, P.A.J. The effects of methyl (5-(2-thienylcarbonyl)-1H-benzimidazol-2-yl) carbamate, (R 17934; NSC 238159), a new synthetic antitumoral drug interfering with microtubules, on mammalian cells cultured in vitro. Cancer Res. 1976, 36, 905–916. [Google Scholar]
- Inoue, T.; Hiratsuka, M.; Osaki, M.H.; Yamada, H.; Kishimoto, I.; Yamaguchi, S.; Nakano, S.; Katoh, M.; Ito, H.; Oshimura, M. SIRT2, a tubulin deacetylase, acts to block the entry to chromosome condensation in response to mitotic stress. Oncogene 2007, 26, 945–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Hiratsuka, M.; Osaki, M.; Oshimura, M. The Molecular Biology of Mammalian SIRT Proteins: SIRT2 Functions on Cell Cycle Regulation. Cell Cycle 2007, 6, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- So, E.C.; Huang, Y.-M.; Hsing, C.-H.; Liao, Y.-K.; Wu, S.-N. Arecoline inhibits intermediate-conductance calcium-activated potassium channels in human glioblastoma cell lines. Eur. J. Pharmacol. 2015, 758, 177–187. [Google Scholar] [CrossRef]
- Thompson, E.G.; Sontheimer, H. Acetylcholine Receptor Activation as a Modulator of Glioblastoma Invasion. Cells 2019, 8, 1203. [Google Scholar] [CrossRef] [Green Version]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The Human Sir2 Ortholog, SIRT2, Is an NAD+-Dependent Tubulin Deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Dryden, S.C.; Nahhas, F.A.; Nowak, J.E.; Goustin, A.-S.; Tainsky, M.A. Role for Human SIRT2 NAD-Dependent Deacetylase Activity in Control of Mitotic Exit in the Cell Cycle. Mol. Cell. Biol. 2003, 23, 3173–3185. [Google Scholar] [CrossRef] [Green Version]
- Hiratsuka, M.; Inoue, T.; Toda, T.; Kimura, N.; Shirayoshi, Y.; Kamitani, H.; Watanabe, T.; Ohama, E.; Tahimic, C.G.; Kurimasa, A.; et al. Proteomics-based identification of differentially expressed genes in human gliomas: Down-regulation of SIRT2 gene. Biochem. Biophys. Res. Commun. 2003, 309, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Nguyen, M.; Qin, F.X.-F.; Tong, Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell 2007, 6, 505–514. [Google Scholar] [CrossRef]
- Szabó, A.; Oláh, J.; Szunyogh, S.; Lehotzky, A.; Szenasi, T.; Csaplár, M.; Schiedel, M.; Lőw, P.; Jung, M.; Ovádi, J. Modulation of Microtubule Acetylation by the Interplay of TPPP/p25, SIRT2 and New Anticancer Agents with Anti-SIRT2 Potency. Sci. Rep. 2017, 7, 17070. [Google Scholar] [CrossRef] [Green Version]
- Cristofaro, I.; Alessandrini, F.; Spinello, Z.; Guerriero, C.; Fiore, M.; Caffarelli, E.; Laneve, P.; Dini, L.; Conti, L.; Tata, A.M. Cross Interaction between M2 Muscarinic Receptor and Notch1/EGFR Pathway in Human Glioblastoma Cancer Stem Cells: Effects on Cell Cycle Progression and Survival. Cells 2020, 9, 657. [Google Scholar] [CrossRef] [Green Version]
- Cristofaro, I.; Spinello, Z.; Matera, C.; Fiore, M.; Conti, L.; De Amici, M.; Dallanoce, C.; Tata, A.M. Activation of M2 muscarinic acetylcholine receptors by a hybrid agonist enhances cytotoxic effects in GB7 glioblastoma cancer stem cells. Neurochem. Int. 2018, 118, 52–60. [Google Scholar] [CrossRef]
- Lucianò, A.M.; Mattei, F.; Damo, E.; Panzarini, E.; Dini, L.; Tata, A.M. Effects mediated by M2 muscarinic orthosteric agonist on cell growth in human neuroblastoma cell lines. Pure Appl. Chem. 2019, 91, 1641–1650. [Google Scholar] [CrossRef]
- Español, A.J.; Salem, A.; Di Bari, M.; Cristofaro, I.; Sanchez, Y.; Tata, A.M.; Sales, M.E. The metronomic combination of paclitaxel with cholinergic agonists inhibits triple negative breast tumor progression. Participation of M2 receptor subtype. PLoS ONE 2020, 15, e0226450. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Bari, M.; Tombolillo, V.; Alessandrini, F.; Guerriero, C.; Fiore, M.; Asteriti, I.A.; Castigli, E.; Sciaccaluga, M.; Guarguaglini, G.; Degrassi, F.; et al. M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines. Cells 2021, 10, 1727. https://doi.org/10.3390/cells10071727
Di Bari M, Tombolillo V, Alessandrini F, Guerriero C, Fiore M, Asteriti IA, Castigli E, Sciaccaluga M, Guarguaglini G, Degrassi F, et al. M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines. Cells. 2021; 10(7):1727. https://doi.org/10.3390/cells10071727
Chicago/Turabian StyleDi Bari, Maria, Vanessa Tombolillo, Francesco Alessandrini, Claudia Guerriero, Mario Fiore, Italia Anna Asteriti, Emilia Castigli, Miriam Sciaccaluga, Giulia Guarguaglini, Francesca Degrassi, and et al. 2021. "M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines" Cells 10, no. 7: 1727. https://doi.org/10.3390/cells10071727
APA StyleDi Bari, M., Tombolillo, V., Alessandrini, F., Guerriero, C., Fiore, M., Asteriti, I. A., Castigli, E., Sciaccaluga, M., Guarguaglini, G., Degrassi, F., & Tata, A. M. (2021). M2 Muscarinic Receptor Activation Impairs Mitotic Progression and Bipolar Mitotic Spindle Formation in Human Glioblastoma Cell Lines. Cells, 10(7), 1727. https://doi.org/10.3390/cells10071727