
The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy

 , ,
, ,  and
and 
Abstract
:1. Introduction: Glycative Stress and Unhealthy Aging
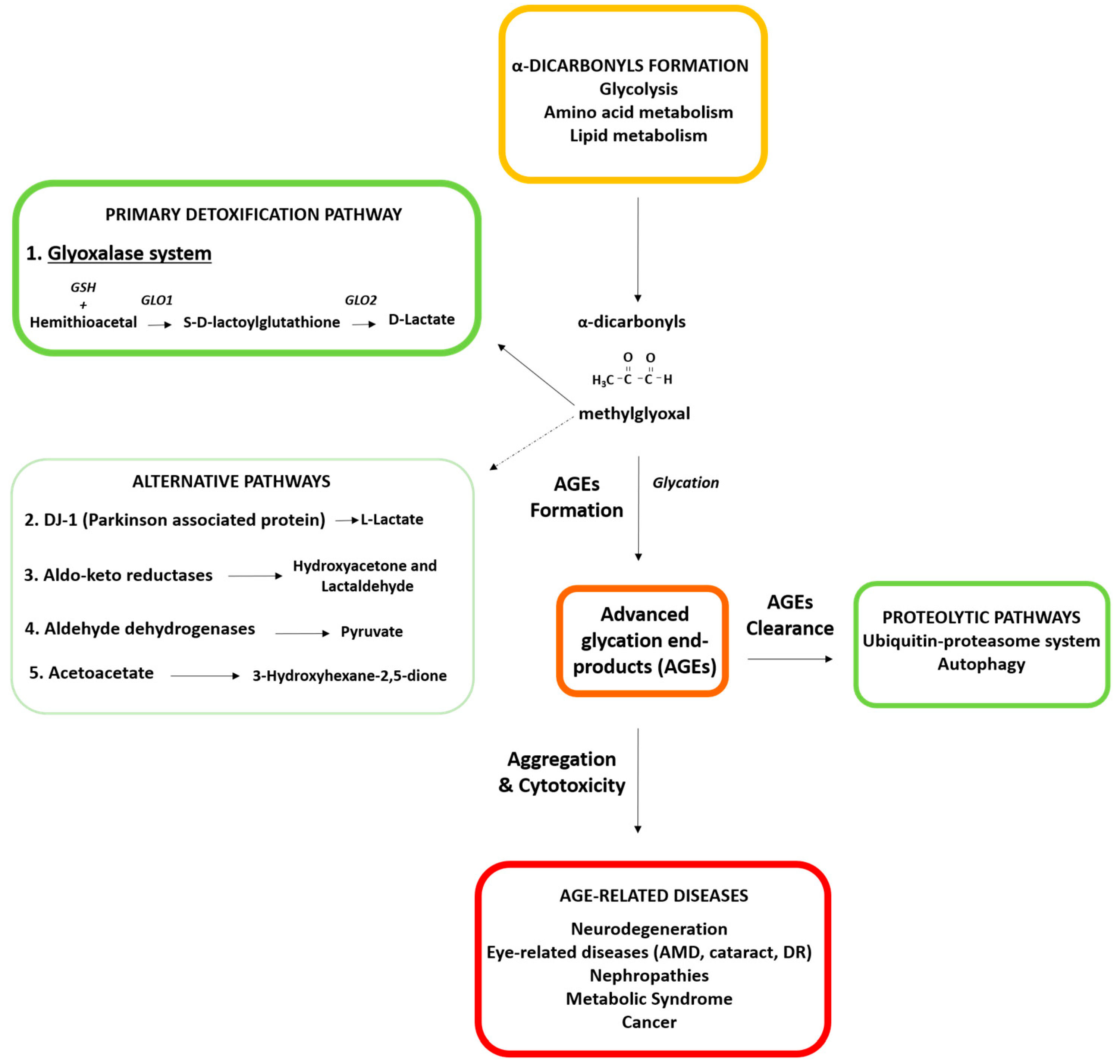
2. Detoxifying Mechanisms against Glycative Stress: Major Role of the Glyoxalase System
2.1. Glyoxalase System: The Major Detoxifying Route for Reactive Dicarbonyls
2.2. Alternative Detoxification Mechanisms as Putative Backup Systems to Compensate the Lack of Glyoxalase Activity
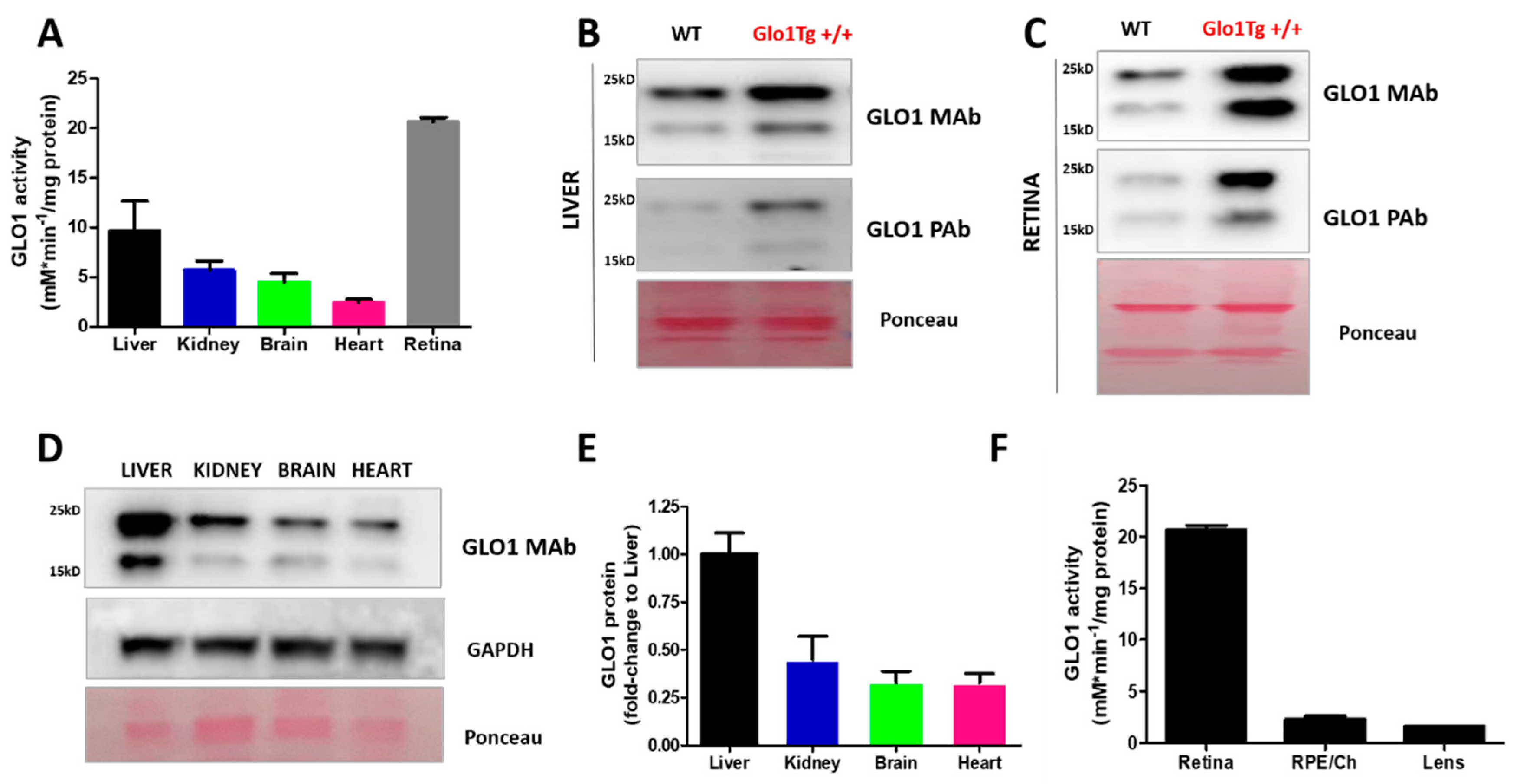
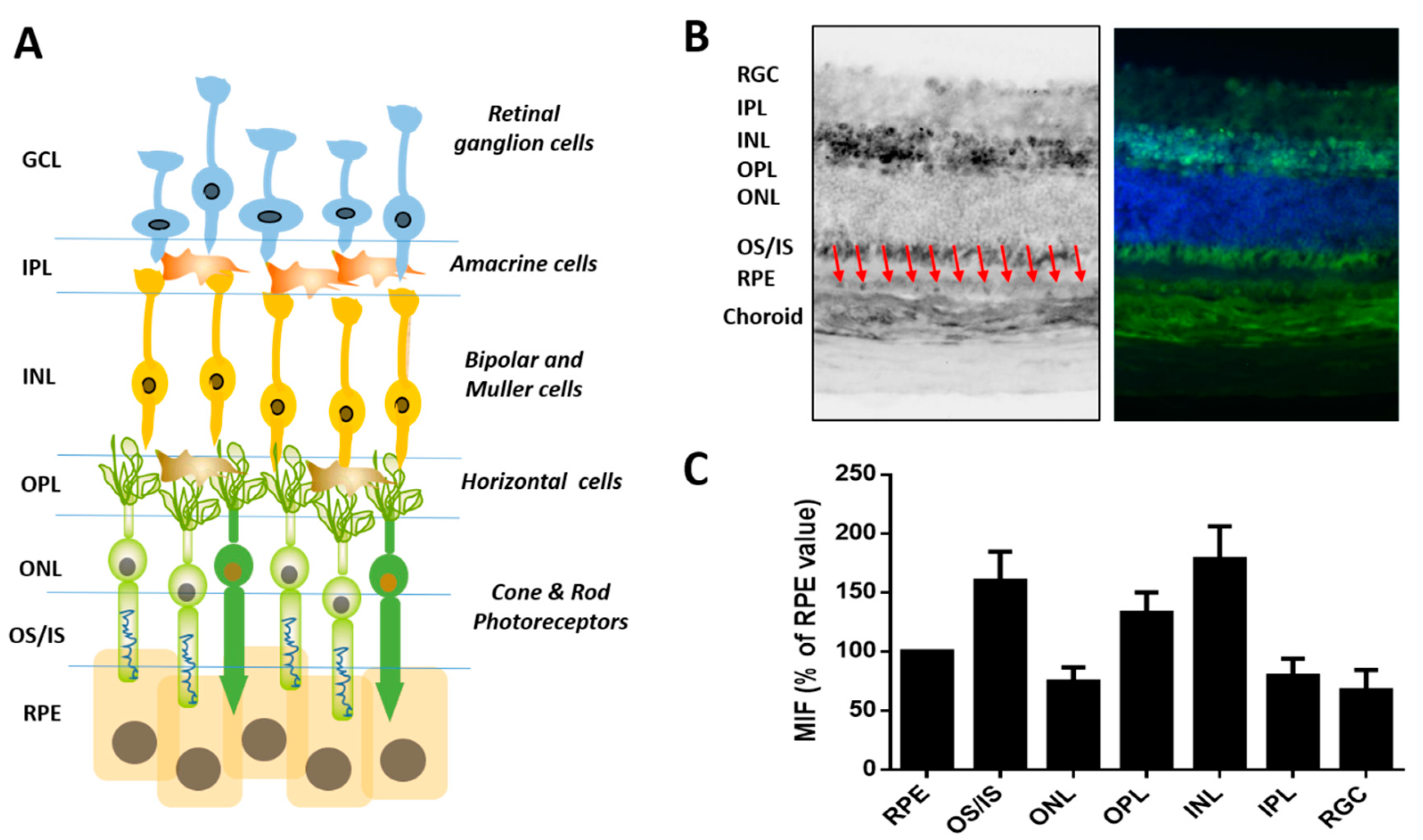
2.3. Tissue-Dependent Activity of Glyoxalase System
3. Biology of the Glyoxalase System in Aging and Age-Related Diseases
3.1. The Involvement of Glyoxalase System in Aging and Age-Related Diseases
3.2. Genetically Modified Models for the Study of Glyoxalase System Biology
4. Nutritional Intervention to Enhance the Glyoxalase System and Decrease Accumulation of AGEs
4.1. Isothiocyanates
4.2. Polyphenols
4.3. Vitamins
4.4. Other Dietary Compounds
5. Concluding Remarks and Pending Questions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prasad, C.; Imrhan, V.; Marotta, F.; Juma, S.; Vijayagopal, P. Lifestyle and Advanced Glycation End Products (AGEs) Burden: Its Relevance to Healthy Aging. Aging Dis. 2014, 5, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Bejarano, E.; Taylor, A. Mechanistic targeting of advanced glycation end-products in age-related diseases. Biochim. Biophys. Acta. Mol. Basis Dis. 2018, 1864, 3631–3643. [Google Scholar] [CrossRef] [PubMed]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef]
- Semba, R.D.; Nicklett, E.J.; Ferrucci, L. Does accumulation of advanced glycation end products contribute to the aging phenotype? J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2010, 65, 963–975. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A. Mechanistically linking age-related diseases and dietary carbohydrate via autophagy and the ubiquitin proteolytic systems. Autophagy 2012, 8, 1404–1406. [Google Scholar] [CrossRef] [Green Version]
- Jahngen, J.H.; Lipman, R.D.; Eisenhauer, D.A.; Jahngen, E.G., Jr.; Taylor, A. Aging and cellular maturation cause changes in ubiquitin-eye lens protein conjugates. Arch. Biochem. Biophys. 1990, 276, 32–37. [Google Scholar] [CrossRef]
- Jahngen-Hodge, J.; Cyr, D.; Laxman, E.; Taylor, A. Ubiquitin and ubiquitin conjugates in human lens. Exp. Eye Res. 1992, 55, 897–902. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Yurek-George, A.; Argirov, O.K. Kinetics and mechanism of the reaction of aminoguanidine with the alpha-oxoaldehydes glyoxal, methylglyoxal, and 3-deoxyglucosone under physiological conditions. Biochem. Pharmacol. 2000, 60, 55–65. [Google Scholar] [CrossRef]
- Uchiki, T.; Weikel, K.A.; Jiao, W.; Shang, F.; Caceres, A.; Pawlak, D.; Handa, J.T.; Brownlee, M.; Nagaraj, R.; Taylor, A. Glycation-altered proteolysis as a pathobiologic mechanism that links dietary glycemic index, aging, and age-related disease (in nondiabetics). Aging Cell 2012, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Vlassara, H.; Striker, G.E. AGE restriction in diabetes mellitus: A paradigm shift. Nat. Rev. Endocrinol. 2011, 7, 526–539. [Google Scholar] [CrossRef] [Green Version]
- Cai, W.; Uribarri, J.; Zhu, L.; Chen, X.; Swamy, S.; Zhao, Z.; Grosjean, F.; Simonaro, C.; Kuchel, G.A.; Schnaider-Beeri, M.; et al. Oral glycotoxins are a modifiable cause of dementia and the metabolic syndrome in mice and humans. Proc. Natl. Acad. Sci. USA 2014, 111, 4940–4945. [Google Scholar] [CrossRef] [Green Version]
- Uribarri, J.; Cai, W.; Ramdas, M.; Goodman, S.; Pyzik, R.; Chen, X.; Zhu, L.; Striker, G.E.; Vlassara, H. Restriction of advanced glycation end products improves insulin resistance in human type 2 diabetes: Potential role of AGER1 and SIRT1. Diabetes Care 2011, 34, 1610–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beeri, M.S.; Moshier, E.; Schmeidler, J.; Godbold, J.; Uribarri, J.; Reddy, S.; Sano, M.; Grossman, H.T.; Cai, W.; Vlassara, H.; et al. Serum concentration of an inflammatory glycotoxin, methylglyoxal, is associated with increased cognitive decline in elderly individuals. Mech. Ageing Dev. 2011, 132, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Sruthi, C.R.; Raghu, K.G. Advanced glycation end products and their adverse effects: The role of autophagy. J. Biochem. Mol. Toxicol. 2021, 35, e22710. [Google Scholar] [CrossRef] [PubMed]
- Bierhaus, A.; Fleming, T.; Stoyanov, S.; Leffler, A.; Babes, A.; Neacsu, C.; Sauer, S.K.; Eberhardt, M.; Schnölzer, M.; Lasitschka, F.; et al. Methylglyoxal modification of Nav1.8 facilitates nociceptive neuron firing and causes hyperalgesia in diabetic neuropathy. Nat. Med. 2012, 18, 926–933. [Google Scholar] [CrossRef] [PubMed]
- Barati, M.T.; Merchant, M.L.; Kain, A.B.; Jevans, A.W.; McLeish, K.R.; Klein, J.B. Proteomic analysis defines altered cellular redox pathways and advanced glycation end-product metabolism in glomeruli of db/db diabetic mice. Am. J. Physiol. Ren. Physiol. 2007, 293, F1157–F1165. [Google Scholar] [CrossRef] [Green Version]
- Palsamy, P.; Subramanian, S. Resveratrol protects diabetic kidney by attenuating hyperglycemia-mediated oxidative stress and renal inflammatory cytokines via Nrf2-Keap1 signaling. Biochim. Biophys. Acta 2011, 1812, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.G.; Tan, G.; Binger, K.J.; Pickering, R.J.; Thomas, M.C.; Nagaraj, R.H.; Cooper, M.E.; Wilkinson-Berka, J.L. Candesartan attenuates diabetic retinal vascular pathology by restoring glyoxalase-I function. Diabetes 2010, 59, 3208–3215. [Google Scholar] [CrossRef] [Green Version]
- Karachalias, N.; Babaei-Jadidi, R.; Rabbani, N.; Thornalley, P.J. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia 2010, 53, 1506–1516. [Google Scholar] [CrossRef] [Green Version]
- Maessen, D.E.; Stehouwer, C.D.; Schalkwijk, C.G. The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. 2015, 128, 839–861. [Google Scholar] [CrossRef]
- Schalkwijk, C.G.; Stehouwer, C.D.A. Methylglyoxal, a Highly Reactive Dicarbonyl Compound, in Diabetes, Its Vascular Complications, and Other Age-Related Diseases. Physiol. Rev. 2020, 100, 407–461. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, N.M.; Wouters, K.; Huijberts, M.S.; Gijbels, M.J.; Sluimer, J.C.; Scheijen, J.L.; Heeneman, S.; Biessen, E.A.; Daemen, M.J.; Brownlee, M.; et al. Higher levels of advanced glycation endproducts in human carotid atherosclerotic plaques are associated with a rupture-prone phenotype. Eur. Heart J. 2014, 35, 1137–1146. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, N.; Thornalley, P.J. Peptide mapping of human serum albumin modified minimally by methylglyoxal in vitro and in vivo. Ann. N. Y. Acad. Sci. 2005, 1043, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Desai, K.; Clausen, J.T.; Wu, L. Increased methylglyoxal and advanced glycation end products in kidney from spontaneously hypertensive rats. Kidney Int. 2004, 66, 2315–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Desai, K.; Chang, T.; Wu, L. Vascular methylglyoxal metabolism and the development of hypertension. J. Hypertens. 2005, 23, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Yuzawa, H.; Nohara, I.; Ohnishi, T.; Obata, N.; Iwayama, Y.; Haga, S.; Toyota, T.; Ujike, H.; Arai, M.; et al. Enhanced carbonyl stress in a subpopulation of schizophrenia. Arch. Gen. Psychiatry 2010, 67, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Ahmed, U.; Thornalley, P.J.; Hager, K.; Fleischer, G.; Münch, G. Protein glycation, oxidation and nitration adduct residues and free adducts of cerebrospinal fluid in Alzheimer’s disease and link to cognitive impairment. J. Neurochem. 2005, 92, 255–263. [Google Scholar] [CrossRef]
- Vicente Miranda, H.; Szego, É.M.; Oliveira, L.M.A.; Breda, C.; Darendelioglu, E.; de Oliveira, R.M.; Ferreira, D.G.; Gomes, M.A.; Rott, R.; Oliveira, M.; et al. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain A J. Neurol. 2017, 140, 1399–1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, E.; Taylor, A. Too sweet: Problems of protein glycation in the eye. Exp. Eye Res. 2019, 178, 255–262. [Google Scholar] [CrossRef]
- Aragonès, G.; Rowan, S.; Francisco, S.G.; Yang, W.; Weinberg, J.; Taylor, A.; Bejarano, E. Glyoxalase System as a Therapeutic Target against Diabetic Retinopathy. Antioxidants 2020, 9, 1062. [Google Scholar] [CrossRef]
- Tessier, F.; Obrenovich, M.; Monnier, V.M. Structure and mechanism of formation of human lens fluorophore LM-1. Relationship to vesperlysine A and the advanced Maillard reaction in aging, diabetes, and cataractogenesis. J. Biol. Chem. 1999, 274, 20796–20804. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Hoerauf, H.; Alt, A.; Schleicher, E.; Clausen, J.T.; Bretzel, R.G.; Laqua, H. N(epsilon)(carboxymethyl)lysin and the AGE receptor RAGE colocalize in age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1855–1859. [Google Scholar]
- Handa, J.T.; Verzijl, N.; Matsunaga, H.; Aotaki-Keen, A.; Lutty, G.A.; te Koppele, J.M.; Miyata, T.; Hjelmeland, L.M. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Investig. Ophthalmol. Vis. Sci. 1999, 40, 775–779. [Google Scholar]
- Weikel, K.A.; Fitzgerald, P.; Shang, F.; Caceres, M.A.; Bian, Q.; Handa, J.T.; Stitt, A.W.; Taylor, A. Natural history of age-related retinal lesions that precede AMD in mice fed high or low glycemic index diets. Investig. Ophthalmol. Vis. Sci. 2012, 53, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Rowan, S.; Jiang, S.; Korem, T.; Szymanski, J.; Chang, M.L.; Szelog, J.; Cassalman, C.; Dasuri, K.; McGuire, C.; Nagai, R.; et al. Involvement of a gut-retina axis in protection against dietary glycemia-induced age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2017, 114, E4472–E4481. [Google Scholar] [CrossRef] [Green Version]
- Rowan, S.; Jiang, S.; Chang, M.L.; Volkin, J.; Cassalman, C.; Smith, K.M.; Streeter, M.D.; Spiegel, D.A.; Moreira-Neto, C.; Rabbani, N.; et al. A low glycemic diet protects disease-prone Nrf2-deficient mice against age-related macular degeneration. Free. Radic. Biol. Med. 2020, 150, 75–86. [Google Scholar] [CrossRef]
- Stitt, A.W. AGEs and diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2010, 51, 4867–4874. [Google Scholar] [CrossRef] [PubMed]
- Engelen, L.; Stehouwer, C.D.; Schalkwijk, C.G. Current therapeutic interventions in the glycation pathway: Evidence from clinical studies. Diabetes Obes. Metab. 2013, 15, 677–689. [Google Scholar] [CrossRef]
- Brings, S.; Fleming, T.; Freichel, M.; Muckenthaler, M.U.; Herzig, S.; Nawroth, P.P. Dicarbonyls and Advanced Glycation End-Products in the Development of Diabetic Complications and Targets for Intervention. Int. J. Mol. Sci. 2017, 18, 984. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; et al. Autophagy Inhibits the Accumulation of Advanced Glycation End Products by Promoting Lysosomal Biogenesis and Function in the Kidney Proximal Tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef] [Green Version]
- Aragonès, G.; Dasuri, K.; Olukorede, O.; Francisco, S.G.; Renneburg, C.; Kumsta, C.; Hansen, M.; Kageyama, S.; Komatsu, M.; Rowan, S.; et al. Autophagic receptor p62 protects against glycation-derived toxicity and enhances viability. Aging Cell 2020, 19, e13257. [Google Scholar] [CrossRef] [PubMed]
- Gavilán, E.; Pintado, C.; Gavilan, M.P.; Daza, P.; Sánchez-Aguayo, I.; Castaño, A.; Ruano, D. Age-related dysfunctions of the autophagy lysosomal pathway in hippocampal pyramidal neurons under proteasome stress. Neurobiol. Aging 2015, 36, 1953–1963. [Google Scholar] [CrossRef] [PubMed]
- Quinet, G.; Gonzalez-Santamarta, M.; Louche, C.; Rodriguez, M.S. Mechanisms Regulating the UPS-ALS Crosstalk: The Role of Proteaphagy. Molecules 2020, 25, 2352. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.H.; Park, J.H.; Chung, K.C. The central regulator p62 between ubiquitin proteasome system and autophagy and its role in the mitophagy and Parkinson’s disease. BMB Rep. 2020, 53, 56–63. [Google Scholar] [CrossRef] [Green Version]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Ji, C.H.; Kwon, Y.T. Crosstalk and Interplay between the Ubiquitin-Proteasome System and Autophagy. Mol. Cells 2017, 40, 441–449. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.T.; Ciechanover, A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem. Sci. 2017, 42, 873–886. [Google Scholar] [CrossRef]
- Makrides, S.C. Protein synthesis and degradation during aging and senescence. Biol. Rev. Camb. Philos. Soc. 1983, 58, 343–422. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef]
- Keller, J.N.; Dimayuga, E.; Chen, Q.; Thorpe, J.; Gee, J.; Ding, Q. Autophagy, proteasomes, lipofuscin, and oxidative stress in the aging brain. Int. J. Biochem. Cell Biol. 2004, 36, 2376–2391. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Macian, F. Autophagy and the immune function in aging. Curr. Opin. Immunol. 2014, 29, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bejarano, E.; Murray, J.W.; Wang, X.; Pampliega, O.; Yin, D.; Patel, B.; Yuste, A.; Wolkoff, A.W.; Cuervo, A.M. Defective recruitment of motor proteins to autophagic compartments contributes to autophagic failure in aging. Aging Cell 2018, 17, e12777. [Google Scholar] [CrossRef]
- Carrard, G.; Bulteau, A.L.; Petropoulos, I.; Friguet, B. Impairment of proteasome structure and function in aging. Int. J. Biochem. Cell Biol. 2002, 34, 1461–1474. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Husom, A.D.; Thompson, L.V. Altered proteasome structure, function, and oxidation in aged muscle. FASEB J. 2005, 19, 644–646. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M. Autophagy and aging: Keeping that old broom working. Trends Genet. TIG 2008, 24, 604–612. [Google Scholar] [CrossRef] [Green Version]
- Shang, F.; Taylor, A. Roles for the ubiquitin-proteasome pathway in protein quality control and signaling in the retina: Implications in the pathogenesis of age-related macular degeneration. Mol. Asp. Med. 2012, 33, 446–466. [Google Scholar] [CrossRef] [Green Version]
- Moaddel, R.; Ubaida-Mohien, C.; Tanaka, T.; Lyashkov, A.; Basisty, N.; Schilling, B.; Semba, R.D.; Franceschi, C.; Gorospe, M.; Ferrucci, L. Proteomics in aging research: A roadmap to clinical, translational research. Aging Cell 2021, 20, e13325. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J. The glyoxalase system: New developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990, 269, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Abordo, E.A.; Minhas, H.S.; Thornalley, P.J. Accumulation of alpha-oxoaldehydes during oxidative stress: A role in cytotoxicity. Biochem. Pharmacol. 1999, 58, 641–648. [Google Scholar] [CrossRef]
- Vander Jagt, D.L.; Han, L.P.; Lehman, C.H. Kinetic evaluation of substrate specificity in the glyoxalase-I-catalyzed disproportionation of -ketoaldehydes. Biochemistry 1972, 11, 3735–3740. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, M.; Thornalley, P.J.; Giardino, I.; Beisswenger, P.; Thorpe, S.R.; Onorato, J.; Brownlee, M. Overexpression of glyoxalase-I in bovine endothelial cells inhibits intracellular advanced glycation endproduct formation and prevents hyperglycemia-induced increases in macromolecular endocytosis. J. Clin. Investig. 1998, 101, 1142–1147. [Google Scholar] [CrossRef]
- Ranganathan, S.; Ciaccio, P.J.; Walsh, E.S.; Tew, K.D. Genomic sequence of human glyoxalase-I: Analysis of promoter activity and its regulation. Gene 1999, 240, 149–155. [Google Scholar] [CrossRef]
- Conboy, C.M.; Spyrou, C.; Thorne, N.P.; Wade, E.J.; Barbosa-Morais, N.L.; Wilson, M.D.; Bhattacharjee, A.; Young, R.A.; Tavaré, S.; Lees, J.A.; et al. Cell cycle genes are the evolutionarily conserved targets of the E2F4 transcription factor. PLoS ONE 2007, 2, e1061. [Google Scholar] [CrossRef] [PubMed]
- Orso, F.; Corà, D.; Ubezio, B.; Provero, P.; Caselle, M.; Taverna, D. Identification of functional TFAP2A and SP1 binding sites in new TFAP2A-modulated genes. BMC Genom. 2010, 11, 355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
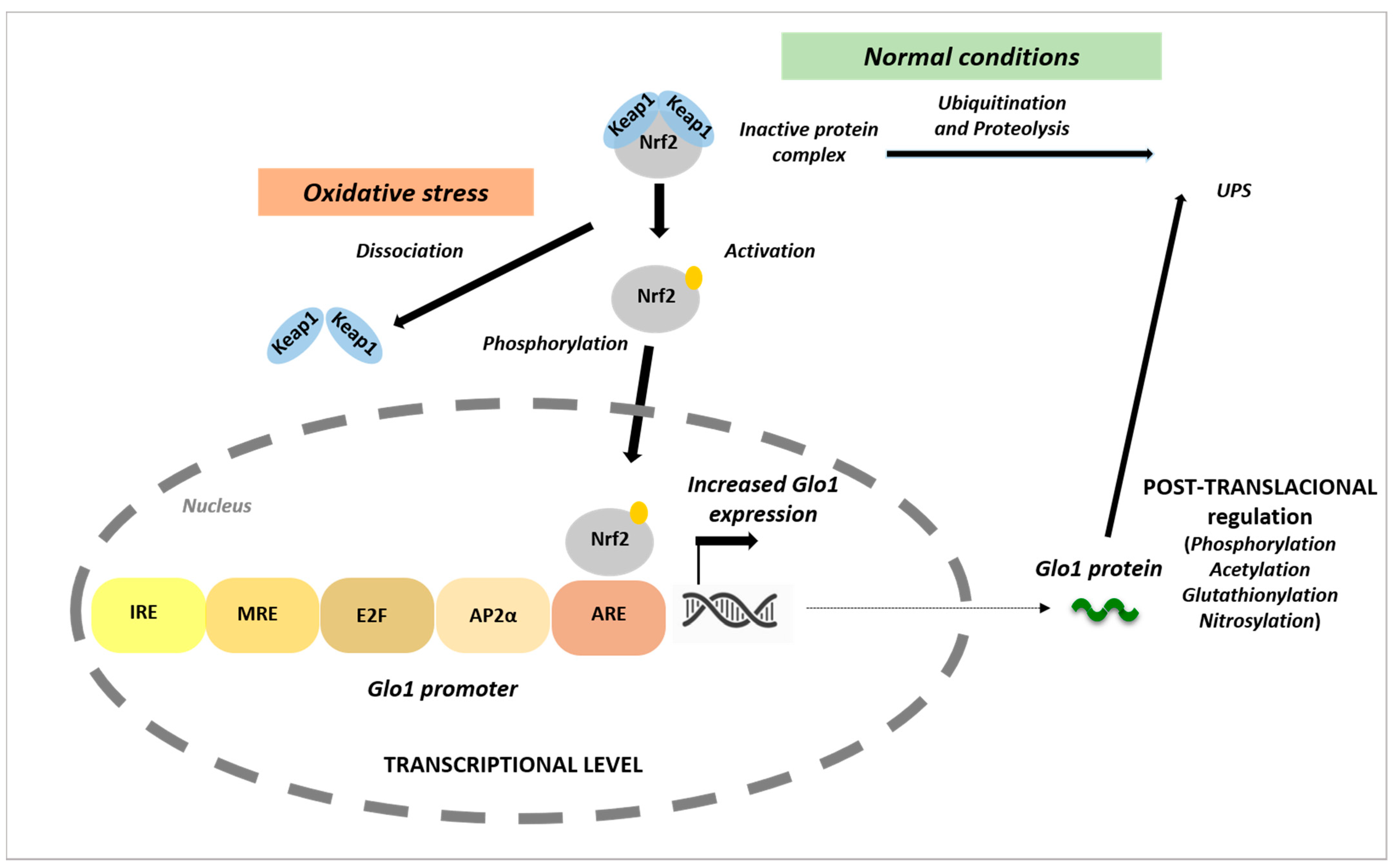
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Ishii, T.; Wakabayashi, N.; Yamamoto, M. Regulatory mechanisms of cellular response to oxidative stress. Free. Radic. Res. 1999, 31, 319–324. [Google Scholar] [CrossRef]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [Green Version]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- James, D.; Devaraj, S.; Bellur, P.; Lakkanna, S.; Vicini, J.; Boddupalli, S. Novel concepts of broccoli sulforaphanes and disease: Induction of phase II antioxidant and detoxification enzymes by enhanced-glucoraphanin broccoli. Nutr. Rev. 2012, 70, 654–665. [Google Scholar] [CrossRef]
- Angeloni, C.; Malaguti, M.; Rizzo, B.; Barbalace, M.C.; Fabbri, D.; Hrelia, S. Neuroprotective effect of sulforaphane against methylglyoxal cytotoxicity. Chem. Res. Toxicol. 2015, 28, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Alfarano, M.; Pastore, D.; Fogliano, V.; Schalkwijk, C.G.; Oliviero, T. The Effect of Sulforaphane on Glyoxalase I Expression and Activity in Peripheral Blood Mononuclear Cells. Nutrients 2018, 10, 1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, A.; Fernandes, R.; Crisóstomo, J.; Seiça, R.M.; Sena, C.M. The Sulforaphane and pyridoxamine supplementation normalize endothelial dysfunction associated with type 2 diabetes. Sci. Rep. 2017, 7, 14357. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.H.; Qu, J.; Shen, X. NF-kappaB/p65 antagonizes Nrf2-ARE pathway by depriving CBP from Nrf2 and facilitating recruitment of HDAC3 to MafK. Biochim. Biophys. Acta 2008, 1783, 713–727. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Li, H.; Xi, H.S.; Li, S. HIF1α is required for survival maintenance of chronic myeloid leukemia stem cells. Blood 2012, 119, 2595–2607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauh, D.; Fischer, F.; Gertz, M.; Lakshminarasimhan, M.; Bergbrede, T.; Aladini, F.; Kambach, C.; Becker, C.F.; Zerweck, J.; Schutkowski, M.; et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 2013, 4, 2327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundby, A.; Lage, K.; Weinert, B.T.; Bekker-Jensen, D.B.; Secher, A.; Skovgaard, T.; Kelstrup, C.D.; Dmytriyev, A.; Choudhary, C.; Lundby, C.; et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012, 2, 419–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiniger, N.; Lau, K.; McCalla, D.; Eby, B.; Cheng, B.; Lu, Y.; Qu, W.; Quadri, N.; Ananthakrishnan, R.; Furmansky, M.; et al. Deletion of the receptor for advanced glycation end products reduces glomerulosclerosis and preserves renal function in the diabetic OVE26 mouse. Diabetes 2010, 59, 2043–2054. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.; Katz, S.; Krebs-Haupenthal, J.; Chen, J.; Saadatmand, A.; Cortizo, F.G.; Moraru, A.; Zemva, J.; Campos, M.C.; Teleman, A.; et al. Phosphorylation of T107 by CamKIIδ Regulates the Detoxification Efficiency and Proteomic Integrity of Glyoxalase 1. Cell Rep. 2020, 32, 108160. [Google Scholar] [CrossRef]
- Kold-Christensen, R.; Johannsen, M. Methylglyoxal Metabolism and Aging-Related Disease: Moving from Correlation toward Causation. Trends Endocrinol. Metab. TEM 2020, 31, 81–92. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lev, N.; Roncevic, D.; Ickowicz, D.; Melamed, E.; Offen, D. Role of DJ-1 in Parkinson’s disease. J. Mol. Neurosci. MN 2006, 29, 215–225. [Google Scholar] [CrossRef]
- Lee, J.Y.; Song, J.; Kwon, K.; Jang, S.; Kim, C.; Baek, K.; Kim, J.; Park, C. Human DJ-1 and its homologs are novel glyoxalases. Hum. Mol. Genet. 2012, 21, 3215–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richarme, G.; Mihoub, M.; Dairou, J.; Bui, L.C.; Leger, T.; Lamouri, A. Parkinsonism-associated protein DJ-1/Park7 is a major protein deglycase that repairs methylglyoxal- and glyoxal-glycated cysteine, arginine, and lysine residues. J. Biol. Chem. 2015, 290, 1885–1897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richarme, G.; Liu, C.; Mihoub, M.; Abdallah, J.; Leger, T.; Joly, N.; Liebart, J.C.; Jurkunas, U.V.; Nadal, M.; Bouloc, P.; et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 2017, 357, 208–211. [Google Scholar] [CrossRef] [Green Version]
- Galligan, J.J.; Wepy, J.A.; Streeter, M.D.; Kingsley, P.J.; Mitchener, M.M.; Wauchope, O.R.; Beavers, W.N.; Rose, K.L.; Wang, T.; Spiegel, D.A.; et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. USA 2018, 115, 9228–9233. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Omans, N.D.; Leicher, R.; Osunsade, A.; Agustinus, A.S.; Finkin-Groner, E.; D’Ambrosio, H.; Liu, B.; Chandarlapaty, S.; Liu, S.; et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. 2019, 10, 1289. [Google Scholar] [CrossRef] [Green Version]
- Pfaff, D.H.; Fleming, T.; Nawroth, P.; Teleman, A.A. Evidence Against a Role for the Parkinsonism-associated Protein DJ-1 in Methylglyoxal Detoxification. J. Biol. Chem. 2017, 292, 685–690. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Ferrari, M.; Ellis, E.M. Human aldo-keto reductase AKR7A2 protects against the cytotoxicity and mutagenicity of reactive aldehydes and lowers intracellular reactive oxygen species in hamster V79-4 cells. Chem.-Biol. Interact. 2012, 195, 25–34. [Google Scholar] [CrossRef]
- Li, D.; Ellis, E.M. Aldo-keto reductase 7A5 (AKR7A5) attenuates oxidative stress and reactive aldehyde toxicity in V79-4 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2014, 28, 707–714. [Google Scholar] [CrossRef]
- Baba, S.P.; Barski, O.A.; Ahmed, Y.; O’Toole, T.E.; Conklin, D.J.; Bhatnagar, A.; Srivastava, S. Reductive metabolism of AGE precursors: A metabolic route for preventing AGE accumulation in cardiovascular tissue. Diabetes 2009, 58, 2486–2497. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, J.; Fleming, T.; Schumacher, D.; Eckstein, V.; Freichel, M.; Herzig, S.; Nawroth, P. Loss of Glyoxalase 1 Induces Compensatory Mechanism to Achieve Dicarbonyl Detoxification in Mammalian Schwann Cells. J. Biol. Chem. 2017, 292, 3224–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodd, E.; Wiggenhauser, L.M.; Morgenstern, J.; Fleming, T.H.; Poschet, G.; Büttner, M.; Tabler, C.T.; Wohlfart, D.P.; Nawroth, P.P.; Kroll, J. The combination of loss of glyoxalase1 and obesity results in hyperglycemia. JCI Insight 2019, 4, e126154. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.; Morgenstern, J.; Oguchi, Y.; Volk, N.; Kopf, S.; Groener, J.B.; Nawroth, P.P.; Fleming, T.; Freichel, M. Compensatory mechanisms for methylglyoxal detoxification in experimental & clinical diabetes. Mol. Metab. 2018, 18, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Kalapos, M.P. On the mammalian acetone metabolism: From chemistry to clinical implications. Biochim. Biophys. Acta 2003, 1621, 122–139. [Google Scholar] [CrossRef]
- Salomón, T.; Sibbersen, C.; Hansen, J.; Britz, D.; Svart, M.V.; Voss, T.S.; Møller, N.; Gregersen, N.; Jørgensen, K.A.; Palmfeldt, J.; et al. Ketone Body Acetoacetate Buffers Methylglyoxal via a Non-enzymatic Conversion during Diabetic and Dietary Ketosis. Cell Chem. Biol. 2017, 24, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spencer, P.S.; Chen, X. The Role of Protein Adduction in Toxic Neuropathies of Exogenous and Endogenous Origin. Toxics 2021, 9, 98. [Google Scholar] [CrossRef] [PubMed]
- Boekelheide, K.; Fleming, S.L.; Allio, T.; Embree-Ku, M.E.; Hall, S.J.; Johnson, K.J.; Kwon, E.J.; Patel, S.R.; Rasoulpour, R.J.; Schoenfeld, H.A.; et al. 2,5-hexanedione-induced testicular injury. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 125–147. [Google Scholar] [CrossRef]
- Jain, D.; Jain, R.; Eberhard, D.; Eglinger, J.; Bugliani, M.; Piemonti, L.; Marchetti, P.; Lammert, E. Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J. Mol. Cell Biol. 2012, 4, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Leverenz, J.B.; Baird, G.; et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain A J. Neurol. 2010, 133, 713–726. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Neric, N.J.; Crabb, J.S.; Crabb, J.W.; Bhattacharya, S.K.; Rayborn, M.E.; Hollyfield, J.G.; Bonilha, V.L. Age-related changes in the retinal pigment epithelium (RPE). PLoS ONE 2012, 7, e38673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, M.; Nihonmatsu-Kikuchi, N.; Itokawa, M.; Rabbani, N.; Thornalley, P.J. Measurement of glyoxalase activities. Biochem. Soc. Trans. 2014, 42, 491–494. [Google Scholar] [CrossRef]
- Brouwers, O.; Niessen, P.M.; Ferreira, I.; Miyata, T.; Scheffer, P.G.; Teerlink, T.; Schrauwen, P.; Brownlee, M.; Stehouwer, C.D.; Schalkwijk, C.G. Overexpression of glyoxalase-I reduces hyperglycemia-induced levels of advanced glycation end products and oxidative stress in diabetic rats. J. Biol. Chem. 2011, 286, 1374–1380. [Google Scholar] [CrossRef] [Green Version]
- Brouwers, O.; Niessen, P.M.; Miyata, T.; Østergaard, J.A.; Flyvbjerg, A.; Peutz-Kootstra, C.J.; Sieber, J.; Mundel, P.H.; Brownlee, M.; Janssen, B.J.; et al. Glyoxalase-1 overexpression reduces endothelial dysfunction and attenuates early renal impairment in a rat model of diabetes. Diabetologia 2014, 57, 224–235. [Google Scholar] [CrossRef] [Green Version]
- Distler, M.G.; Plant, L.D.; Sokoloff, G.; Hawk, A.J.; Aneas, I.; Wuenschell, G.E.; Termini, J.; Meredith, S.C.; Nobrega, M.A.; Palmer, A.A. Glyoxalase 1 increases anxiety by reducing GABAA receptor agonist methylglyoxal. J. Clin. Investig. 2012, 122, 2306–2315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.M.; Lin, C.E.; Chen, H.H.; Cheng, Y.F.; Cheng, H.W.; Imai, K. Effect of prednisolone on glyoxalase 1 in an inbred mouse model of aristolochic acid nephropathy using a proteomics method with fluorogenic derivatization-liquid chromatography-tandem mass spectrometry. PLoS ONE 2020, 15, e0227838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.G.; Smith, D.G.; Bhat, M.; Nagaraj, R.H. Glyoxalase I is critical for human retinal capillary pericyte survival under hyperglycemic conditions. J. Biol. Chem. 2006, 281, 11864–11871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berner, A.K.; Brouwers, O.; Pringle, R.; Klaassen, I.; Colhoun, L.; McVicar, C.; Brockbank, S.; Curry, J.W.; Miyata, T.; Brownlee, M.; et al. Protection against methylglyoxal-derived AGEs by regulation of glyoxalase 1 prevents retinal neuroglial and vasodegenerative pathology. Diabetologia 2012, 55, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, N.; Xue, M.; Thornalley, P.J. Dicarbonyls and glyoxalase in disease mechanisms and clinical therapeutics. Glycoconj. J. 2016, 33, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Morcos, M.; Du, X.; Pfisterer, F.; Hutter, H.; Sayed, A.A.; Thornalley, P.; Ahmed, N.; Baynes, J.; Thorpe, S.; Kukudov, G.; et al. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell 2008, 7, 260–269. [Google Scholar] [CrossRef]
- Sharma-Luthra, R.; Kale, R.K. Age related changes in the activity of the glyoxalase system. Mech. Ageing Dev. 1994, 73, 39–45. [Google Scholar] [CrossRef]
- Amicarelli, F.; Di Ilio, C.; Masciocco, L.; Bonfigli, A.; Zarivi, O.; D’Andrea, M.R.; Di Giulio, C.; Miranda, M. Aging and detoxifying enzymes responses to hypoxic or hyperoxic treatment. Mech. Ageing Dev. 1997, 97, 215–226. [Google Scholar] [CrossRef]
- Kirk, J.E. The glyoxalase I activity of arterial tissue in individuals of various ages. J. Gerontol. 1960, 15, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Haik, G.M., Jr.; Lo, T.W.; Thornalley, P.J. Methylglyoxal concentration and glyoxalase activities in the human lens. Exp. Eye Res. 1994, 59, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Mailankot, M.; Padmanabha, S.; Pasupuleti, N.; Major, D.; Howell, S.; Nagaraj, R.H. Glyoxalase I activity and immunoreactivity in the aging human lens. Biogerontology 2009, 10, 711–720. [Google Scholar] [CrossRef] [Green Version]
- McLellan, A.C.; Thornalley, P.J. Glyoxalase activity in human red blood cells fractioned by age. Mech. Ageing Dev. 1989, 48, 63–71. [Google Scholar] [CrossRef]
- Kuhla, B.; Boeck, K.; Lüth, H.J.; Schmidt, A.; Weigle, B.; Schmitz, M.; Ogunlade, V.; Münch, G.; Arendt, T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging 2006, 27, 815–822. [Google Scholar] [CrossRef]
- Chen, F.; Wollmer, M.A.; Hoerndli, F.; Münch, G.; Kuhla, B.; Rogaev, E.I.; Tsolaki, M.; Papassotiropoulos, A.; Götz, J. Role for glyoxalase I in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 7687–7692. [Google Scholar] [CrossRef] [Green Version]
- Kuhla, B.; Boeck, K.; Schmidt, A.; Ogunlade, V.; Arendt, T.; Münch, G.; Lüth, H.J. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol. Aging 2007, 28, 29–41. [Google Scholar] [CrossRef]
- Rankinen, T.; Zuberi, A.; Chagnon, Y.C.; Weisnagel, S.J.; Argyropoulos, G.; Walts, B.; Pérusse, L.; Bouchard, C. The human obesity gene map: The 2005 update. Obesity 2006, 14, 529–644. [Google Scholar] [CrossRef]
- Rabbani, N.; Thornalley, P.J. Glyoxalase 1 Modulation in Obesity and Diabetes. Antioxid. Redox Signal. 2019, 30, 354–374. [Google Scholar] [CrossRef] [PubMed]
- Wuschke, S.; Dahm, S.; Schmidt, C.; Joost, H.G.; Al-Hasani, H. A meta-analysis of quantitative trait loci associated with body weight and adiposity in mice. Int. J. Obes. 2007, 31, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.F.; Elston, R.C.; Tran, L.D.; Siervogel, R.M. Use of the robust sib-pair method to screen for single-locus, multiple-locus, and pleiotropic effects: Application to traits related to hypertension. Am. J. Hum. Genet. 1991, 48, 862–872. [Google Scholar] [PubMed]
- Sanchez, J.C.; Converset, V.; Nolan, A.; Schmid, G.; Wang, S.; Heller, M.; Sennitt, M.V.; Hochstrasser, D.F.; Cawthorne, M.A. Effect of rosiglitazone on the differential expression of diabetes-associated proteins in pancreatic islets of C57Bl/6 lep/lep mice. Mol. Cell. Proteom. MCP 2002, 1, 509–516. [Google Scholar] [CrossRef] [Green Version]
- Wortmann, M.; Peters, A.; Hakimi, M.; Bockler, D.; Dihlmann, S. Glyoxalase I (Glo1) and its metabolites in vascular disease. Biochem. Soc. Trans. 2014, 42, 528–533. [Google Scholar] [CrossRef]
- Maessen, D.; Brouwers, O.; Miyata, T.; Stehouwer, C.; Schalkwijk, C. Glyoxalase-1 overexpression reduces body weight and adipokine expression, and improves insulin sensitivity in high-fat diet-induced obese mice. Diabetologia 2014, 57, 713. [Google Scholar]
- Giacco, F.; Du, X.; D’Agati, V.D.; Milne, R.; Sui, G.; Geoffrion, M.; Brownlee, M. Knockdown of glyoxalase 1 mimics diabetic nephropathy in nondiabetic mice. Diabetes 2014, 63, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, D.; Brownlee, M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (RAGE) and RAGE ligands. Diabetes 2010, 59, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Dobler, D.; Ahmed, N.; Song, L.; Eboigbodin, K.E.; Thornalley, P.J. Increased dicarbonyl metabolism in endothelial cells in hyperglycemia induces anoikis and impairs angiogenesis by RGD and GFOGER motif modification. Diabetes 2006, 55, 1961–1969. [Google Scholar] [CrossRef] [Green Version]
- Phillips, S.A.; Mirrlees, D.; Thornalley, P.J. Modification of the glyoxalase system in streptozotocin-induced diabetic rats. Effect of the aldose reductase inhibitor Statil. Biochem. Pharmacol. 1993, 46, 805–811. [Google Scholar] [CrossRef]
- Atkins, T.W.; Thornally, P.J. Erythrocyte glyoxalase activity in genetically obese (ob/ob) and streptozotocin diabetic mice. Diabetes Res. 1989, 11, 125–129. [Google Scholar] [PubMed]
- Chiu, C.J.; Liu, S.; Willett, W.C.; Wolever, T.M.; Brand-Miller, J.C.; Barclay, A.W.; Taylor, A. Informing food choices and health outcomes by use of the dietary glycemic index. Nutr. Rev. 2011, 69, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Francisco, S.G.; Smith, K.M.; Aragonès, G.; Whitcomb, E.A.; Weinberg, J.; Wang, X.; Bejarano, E.; Taylor, A.; Rowan, S. Dietary Patterns, Carbohydrates, and Age-Related Eye Diseases. Nutrients 2020, 12, 2862. [Google Scholar] [CrossRef]
- Chiu, C.J.; Taylor, A. Dietary hyperglycemia, glycemic index and metabolic retinal diseases. Prog. Retin. Eye Res. 2011, 30, 18–53. [Google Scholar] [CrossRef] [Green Version]
- Rasul, A.; Rashid, A.; Waheed, P.; Khan, S.A. Expression analysis of glyoxalase I gene among patients of diabetic retinopathy. Pak. J. Med Sci. 2018, 34, 139–143. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.; Waheed, P.; Rashid, A.; Khan, S.A. Gene Expression of Glyoxalase II in Diabetic Retinopathy. J. Coll. Physicians Surg. Pak. JCPSP 2018, 28, 523–526. [Google Scholar] [CrossRef]
- Wu, J.C.; Li, X.H.; Peng, Y.D.; Wang, J.B.; Tang, J.F.; Wang, Y.F. Association of two glyoxalase I gene polymorphisms with nephropathy and retinopathy in Type 2 diabetes. J. Endocrinol. Investig. 2011, 34, e343–e348. [Google Scholar] [CrossRef]
- Sachdeva, R.; Schlotterer, A.; Schumacher, D.; Matka, C.; Mathar, I.; Dietrich, N.; Medert, R.; Kriebs, U.; Lin, J.; Nawroth, P.; et al. TRPC proteins contribute to development of diabetic retinopathy and regulate glyoxalase 1 activity and methylglyoxal accumulation. Mol. Metab. 2018, 9, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, V.P.; Civelek, M.; Meng, Q.; Zhang, B.; Zhu, J.; Levian, C.; Huan, T.; Segrè, A.V.; Ghosh, S.; Vivar, J.; et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014, 10, e1004502. [Google Scholar] [CrossRef] [PubMed]
- Kalousová, M.; Jáchymová, M.; Germanová, A.; Kubena, A.A.; Tesar, V.; Zima, T. Genetic predisposition to advanced glycation end products toxicity is related to prognosis of chronic hemodialysis patients. Kidney Blood Press. Res. 2010, 33, 30–36. [Google Scholar] [CrossRef]
- Kalousová, M.; Germanová, A.; Jáchymová, M.; Mestek, O.; Tesar, V.; Zima, T. A419C (E111A) polymorphism of the glyoxalase I gene and vascular complications in chronic hemodialysis patients. Ann. N. Y. Acad. Sci. 2008, 1126, 268–271. [Google Scholar] [CrossRef]
- Tikellis, C.; Pickering, R.J.; Tsorotes, D.; Huet, O.; Cooper, M.E.; Jandeleit-Dahm, K.; Thomas, M.C. Dicarbonyl stress in the absence of hyperglycemia increases endothelial inflammation and atherogenesis similar to that observed in diabetes. Diabetes 2014, 63, 3915–3925. [Google Scholar] [CrossRef] [Green Version]
- Blackburn, N.J.; Vulesevic, B.; Ahmadi, A.; McNeill, B.; Milne, R.W.; Suuronen, E.J. Abstract 14257: Glyoxalase-1 Over-expression Preserves Cardiac Function Post-MI by Enhancing Vascularity and Reducing AGE Accumulation and Cardiomyocyte Apoptosis. Circulation 2013, 128, A14257. [Google Scholar] [CrossRef]
- Ceradini, D.J.; Yao, D.; Grogan, R.H.; Callaghan, M.J.; Edelstein, D.; Brownlee, M.; Gurtner, G.C. Decreasing intracellular superoxide corrects defective ischemia-induced new vessel formation in diabetic mice. J. Biol. Chem. 2008, 283, 10930–10938. [Google Scholar] [CrossRef] [Green Version]
- Angeloni, C.; Zambonin, L.; Hrelia, S. Role of methylglyoxal in Alzheimer’s disease. BioMed Res. Int. 2014, 2014, 238485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciavardelli, D.; Silvestri, E.; Del Viscovo, A.; Bomba, M.; De Gregorio, D.; Moreno, M.; Di Ilio, C.; Goglia, F.; Canzoniero, L.M.; Sensi, S.L. Alterations of brain and cerebellar proteomes linked to Aβ and tau pathology in a female triple-transgenic murine model of Alzheimer’s disease. Cell Death Dis. 2010, 1, e90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurz, A.; Rabbani, N.; Walter, M.; Bonin, M.; Thornalley, P.; Auburger, G.; Gispert, S. Alpha-synuclein deficiency leads to increased glyoxalase I expression and glycation stress. Cell. Mol. Life Sci. CMLS 2011, 68, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.t.; Lim, J.E.; Harr, B.; Wing, C.; Walters, R.; Distler, M.G.; Teschke, M.; Wu, C.; Wiltshire, T.; Su, A.I.; et al. A common and unstable copy number variant is associated with differences in Glo1 expression and anxiety-like behavior. PLoS ONE 2009, 4, e4649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kollmannsberger, L.K.; Gassen, N.C.; Bultmann, A.; Hartmann, J.; Weber, P.; Schmidt, M.V.; Rein, T. Increased glyoxalase-1 levels in Fkbp5 knockout mice caused by glyoxalase-1 gene duplication. G3 2013, 3, 1311–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hovatta, I.; Tennant, R.S.; Helton, R.; Marr, R.A.; Singer, O.; Redwine, J.M.; Ellison, J.A.; Schadt, E.E.; Verma, I.M.; Lockhart, D.J.; et al. Glyoxalase 1 and glutathione reductase 1 regulate anxiety in mice. Nature 2005, 438, 662–666. [Google Scholar] [CrossRef]
- Krömer, S.A.; Kessler, M.S.; Milfay, D.; Birg, I.N.; Bunck, M.; Czibere, L.; Panhuysen, M.; Pütz, B.; Deussing, J.M.; Holsboer, F.; et al. Identification of glyoxalase-I as a protein marker in a mouse model of extremes in trait anxiety. J. Neurosci. 2005, 25, 4375–4384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, M.; Uchida, S.; Watanuki, T.; Wakabayashi, Y.; Otsuki, K.; Matsubara, T.; Suetsugi, M.; Funato, H.; Watanabe, Y. Reduced expression of glyoxalase-1 mRNA in mood disorder patients. Neurosci. Lett. 2008, 438, 196–199. [Google Scholar] [CrossRef] [PubMed]
- Szczepanik, J.C.; de Almeida, G.R.L.; Cunha, M.P.; Dafre, A.L. Repeated Methylglyoxal Treatment Depletes Dopamine in the Prefrontal Cortex, and Causes Memory Impairment and Depressive-Like Behavior in Mice. Neurochem. Res. 2020, 45, 354–370. [Google Scholar] [CrossRef]
- Li, H.; Zheng, L.; Chen, C.; Liu, X.; Zhang, W. Brain Senescence Caused by Elevated Levels of Reactive Metabolite Methylglyoxal on D-Galactose-Induced Aging Mice. Front. Neurosci. 2019, 13, 1004. [Google Scholar] [CrossRef]
- Thornalley, P.J.; Rabbani, N. Glyoxalase in tumourigenesis and multidrug resistance. Semin. Cell Dev. Biol. 2011, 22, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.K.; deLeeuw, R.J.; Dosanjh, N.S.; Kimm, L.R.; Cheng, Z.; Horsman, D.E.; MacAulay, C.; Ng, R.T.; Brown, C.J.; Eichler, E.E.; et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007, 80, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Cahan, P.; Li, Y.; Izumi, M.; Graubert, T.A. The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells. Nat. Genet. 2009, 41, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Santarius, T.; Bignell, G.R.; Greenman, C.D.; Widaa, S.; Chen, L.; Mahoney, C.L.; Butler, A.; Edkins, S.; Waris, S.; Thornalley, P.J.; et al. GLO1-A novel amplified gene in human cancer. Genes Chromosomes Cancer 2010, 49, 711–725. [Google Scholar] [CrossRef] [Green Version]
- Loarca, L.; Sassi-Gaha, S.; Artlett, C.M. Two α-dicarbonyls downregulate migration, invasion, and adhesion of liver cancer cells in a p53-dependent manner. Dig. Liver Dis. 2013, 45, 938–946. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Mezzasoma, L.; Fettucciari, K.; Talesa, V.N. A novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int. J. Biochem. Cell Biol. 2013, 45, 836–844. [Google Scholar] [CrossRef]
- Young, T.W.; Mei, F.C.; Yang, G.; Thompson-Lanza, J.A.; Liu, J.; Cheng, X. Activation of antioxidant pathways in ras-mediated oncogenic transformation of human surface ovarian epithelial cells revealed by functional proteomics and mass spectrometry. Cancer Res. 2004, 64, 4577–4584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.X.; Chen, Z.C.; Zhang, G.Y.; Yi, H.; Xiao, Z.Q. A subcelluar proteomic investigation into vincristine-resistant gastric cancer cell line. J. Cell. Biochem. 2008, 104, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Kizaki, A.; Dan, S.; Hashimoto, Y.; Naito, M.; Tsuruo, T. Glyoxalase I is involved in resistance of human leukemia cells to antitumor agent-induced apoptosis. Blood 2000, 95, 3214–3218. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, H.; Mashima, T.; Sato, S.; Hashimoto, Y.; Yamori, T.; Tsuruo, T. Selective activation of apoptosis program by S-p-bromobenzylglutathione cyclopentyl diester in glyoxalase I-overexpressing human lung cancer cells. Clin. Cancer Res. 2001, 7, 2513–2518. [Google Scholar]
- Stratmann, B.; Goldstein, B.; Thornalley, P.J.; Rabbani, N.; Tschoepe, D. Intracellular Accumulation of Methylglyoxal by Glyoxalase 1 Knock Down Alters Collagen Homoeostasis in L6 Myoblasts. Int. J. Mol. Sci. 2017, 18, 480. [Google Scholar] [CrossRef]
- Nigro, C.; Leone, A.; Fiory, F.; Prevenzano, I.; Nicolò, A.; Mirra, P.; Beguinot, F.; Miele, C. Dicarbonyl Stress at the Crossroads of Healthy and Unhealthy Aging. Cells 2019, 8, 749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shafie, A.; Xue, M.; Barker, G.; Zehnder, D.; Thornalley, P.J.; Rabbani, N. Reappraisal of putative glyoxalase 1-deficient mouse and dicarbonyl stress on embryonic stem cells in vitro. Biochem. J. 2016, 473, 4255–4270. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Kwon, D.M.; Kwon, K.; Park, C. Generation and characterization of mouse knockout for glyoxalase 1. Biochem. Biophys. Res. Commun. 2017, 490, 460–465. [Google Scholar] [CrossRef]
- Moraru, A.; Wiederstein, J.; Pfaff, D.; Fleming, T.; Miller, A.K.; Nawroth, P.; Teleman, A.A. Elevated Levels of the Reactive Metabolite Methylglyoxal Recapitulate Progression of Type 2 Diabetes. Cell Metab. 2018, 27, 926–934. [Google Scholar] [CrossRef] [Green Version]
- Vulesevic, B.; McNeill, B.; Giacco, F.; Maeda, K.; Blackburn, N.J.; Brownlee, M.; Milne, R.W.; Suuronen, E.J. Methylglyoxal-Induced Endothelial Cell Loss and Inflammation Contribute to the Development of Diabetic Cardiomyopathy. Diabetes 2016, 65, 1699–1713. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; O’Meara, M.; Zhang, X.; Zhang, K.; Seyoum, B.; Yi, Z.; Kaufman, R.J.; Monks, T.J.; Wang, J.M. Ameliorating Methylglyoxal-Induced Progenitor Cell Dysfunction for Tissue Repair in Diabetes. Diabetes 2019, 68, 1287–1302. [Google Scholar] [CrossRef]
- Qian, S.; Qian, Y.; Huo, D.; Wang, S.; Qian, Q. Tanshinone IIa protects retinal endothelial cells against mitochondrial fission induced by methylglyoxal through glyoxalase 1. Eur. J. Pharmacol. 2019, 857, 172419. [Google Scholar] [CrossRef]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Zhou, C.; Huang, M.; Tang, C.; Liu, X.; Yue, Y.; Diao, Q.; Zheng, Z.; Liu, D. Glyoxalase system: A systematic review of its biological activity, related-diseases, screening methods and small molecule regulators. Biomed. Pharmacother. 2020, 131, 110663. [Google Scholar] [CrossRef]
- Liu, Y.W.; Liu, X.L.; Kong, L.; Zhang, M.Y.; Chen, Y.J.; Zhu, X.; Hao, Y.C. Neuroprotection of quercetin on central neurons against chronic high glucose through enhancement of Nrf2/ARE/glyoxalase-1 pathway mediated by phosphorylation regulation. Biomed. Pharmacother. 2019, 109, 2145–2154. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Turroni, S.; Bianchi, L.; Fabbri, D.; Motori, E.; Malaguti, M.; Leoncini, E.; Maraldi, T.; Bini, L.; Brigidi, P.; et al. Novel targets of sulforaphane in primary cardiomyocytes identified by proteomic analysis. PLoS ONE 2013, 8, e83283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santel, T.; Pflug, G.; Hemdan, N.Y.; Schäfer, A.; Hollenbach, M.; Buchold, M.; Hintersdorf, A.; Lindner, I.; Otto, A.; Bigl, M.; et al. Curcumin inhibits glyoxalase 1: A possible link to its anti-inflammatory and anti-tumor activity. PLoS ONE 2008, 3, e3508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takasawa, R.; Takahashi, S.; Saeki, K.; Sunaga, S.; Yoshimori, A.; Tanuma, S. Structure-activity relationship of human GLO I inhibitory natural flavonoids and their growth inhibitory effects. Bioorganic Med. Chem. 2008, 16, 3969–3975. [Google Scholar] [CrossRef]
- Qudjani, E.; Iman, M.; Davood, A.; Ramandi, M.F.; Shafiee, A. Design and Synthesis of Curcumin-Like Diarylpentanoid Analogues as Potential Anticancer Agents. Recent Pat. Anti-Cancer Drug Discov. 2016, 11, 342–351. [Google Scholar] [CrossRef]
- Zhang, H.; Zhai, J.; Zhang, L.; Li, C.; Zhao, Y.; Chen, Y.; Li, Q.; Hu, X.P. In Vitro Inhibition of Glyoxalase I by Flavonoids: New Insights from Crystallographic Analysis. Curr. Top. Med. Chem. 2016, 16, 460–466. [Google Scholar] [CrossRef]
- Kumar, R.; Bhan Tiku, A. Naringenin Suppresses Chemically Induced Skin Cancer in Two-Stage Skin Carcinogenesis Mouse Model. Nutr. Cancer 2020, 72, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Folgado, S.L.; Pourbagher-Shahri, A.M.; Ashrafizadeh, M.; Samarghandian, S. The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway. Biomed. Pharmacother. 2020, 127, 110234. [Google Scholar] [CrossRef]
- Cheng, A.S.; Cheng, Y.H.; Chiou, C.H.; Chang, T.L. Resveratrol upregulates Nrf2 expression to attenuate methylglyoxal-induced insulin resistance in Hep G2 cells. J. Agric. Food Chem. 2012, 60, 9180–9187. [Google Scholar] [CrossRef] [PubMed]
- Antognelli, C.; Moretti, S.; Frosini, R.; Puxeddu, E.; Sidoni, A.; Talesa, V.N. Methylglyoxal Acts as a Tumor-Promoting Factor in Anaplastic Thyroid Cancer. Cells 2019, 8, 547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santini, S.J.; Cordone, V.; Mijit, M.; Bignotti, V.; Aimola, P.; Dolo, V.; Falone, S.; Amicarelli, F. SIRT1-Dependent Upregulation of Antiglycative Defense in HUVECs Is Essential for Resveratrol Protection against High Glucose Stress. Antioxidants 2019, 8, 346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.H.; Hwang, Y.; Heo, S.J.; Jun, H.S. Diphlorethohydroxycarmalol Attenuates Methylglyoxal-Induced Oxidative Stress and Advanced Glycation End Product Formation in Human Kidney Cells. Oxidative Med. Cell. Longev. 2018, 2018, 3654095. [Google Scholar] [CrossRef]
- Ávila, F.; Theoduloz, C.; López-Alarcón, C.; Dorta, E.; Schmeda-Hirschmann, G. Cytoprotective Mechanisms Mediated by Polyphenols from Chilean Native Berries against Free Radical-Induced Damage on AGS Cells. Oxidative Med. Cell. Longev. 2017, 2017, 9808520. [Google Scholar] [CrossRef]
- Habtemariam, S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules 2019, 24, 1519. [Google Scholar] [CrossRef] [Green Version]
- Arbo, B.D.; André-Miral, C.; Nasre-Nasser, R.G.; Schimith, L.E.; Santos, M.G.; Costa-Silva, D.; Muccillo-Baisch, A.L.; Hort, M.A. Resveratrol Derivatives as Potential Treatments for Alzheimer’s and Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 103. [Google Scholar] [CrossRef]
- Suantawee, T.; Thilavech, T.; Cheng, H.; Adisakwattana, S. Cyanidin Attenuates Methylglyoxal-Induced Oxidative Stress and Apoptosis in INS-1 Pancreatic β-Cells by Increasing Glyoxalase-1 Activity. Nutrients 2020, 12, 1319. [Google Scholar] [CrossRef]
- Frandsen, J.R.; Narayanasamy, P. Neuroprotection through flavonoid: Enhancement of the glyoxalase pathway. Redox Biol. 2018, 14, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Dargusch, R.; Ehren, J.L.; Okada, S.; Sharma, K.; Schubert, D. Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS ONE 2011, 6, e21226. [Google Scholar] [CrossRef]
- Frandsen, J.; Narayanasamy, P. Flavonoid Enhances the Glyoxalase Pathway in Cerebellar Neurons to Retain Cellular Functions. Sci. Rep. 2017, 7, 5126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Cheng, Y.Q.; Lu, Q.; Du, L.; Yin, X.X.; Liu, Y.W. Enhancement of glyoxalase 1, a polyfunctional defense enzyme, by quercetin in the brain in streptozotocin-induced diabetic rats. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynde, M.D.G.; Geleijnse, J.M.; Scheijen, J.; Hanssen, N.M.J.; Dower, J.I.; Afman, L.A.; Stehouwer, C.D.A.; Hollman, P.C.H.; Schalkwijk, C.G. Quercetin, but Not Epicatechin, Decreases Plasma Concentrations of Methylglyoxal in Adults in a Randomized, Double-Blind, Placebo-Controlled, Crossover Trial with Pure Flavonoids. J. Nutr. 2018, 148, 1911–1916. [Google Scholar] [CrossRef]
- Frandsen, J.; Choi, S.R.; Narayanasamy, P. Neural Glyoxalase Pathway Enhancement by Morin Derivatives in an Alzheimer’s Disease Model. ACS Chem. Neurosci. 2020, 11, 356–366. [Google Scholar] [CrossRef]
- Chen, Y.J.; Kong, L.; Tang, Z.Z.; Zhang, Y.M.; Liu, Y.; Wang, T.Y.; Liu, Y.W. Hesperetin ameliorates diabetic nephropathy in rats by activating Nrf2/ARE/glyoxalase 1 pathway. Biomed. Pharmacother. 2019, 111, 1166–1175. [Google Scholar] [CrossRef]
- Xue, M.; Weickert, M.O.; Qureshi, S.; Kandala, N.B.; Anwar, A.; Waldron, M.; Shafie, A.; Messenger, D.; Fowler, M.; Jenkins, G.; et al. Improved Glycemic Control and Vascular Function in Overweight and Obese Subjects by Glyoxalase 1 Inducer Formulation. Diabetes 2016, 65, 2282–2294. [Google Scholar] [CrossRef] [Green Version]
- Irshad, Z.; Xue, M.; Ashour, A.; Larkin, J.R.; Thornalley, P.J.; Rabbani, N. Activation of the unfolded protein response in high glucose treated endothelial cells is mediated by methylglyoxal. Sci. Rep. 2019, 9, 7889. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Wang, P.; Sang, S. Dietary Genistein Inhibits Methylglyoxal-Induced Advanced Glycation End Product Formation in Mice Fed a High-Fat Diet. J. Nutr. 2019, 149, 776–787. [Google Scholar] [CrossRef]
- Suh, K.S.; Chon, S.; Choi, E.M. Cytoprotective effects of xanthohumol against methylglyoxal-induced cytotoxicity in MC3T3-E1 osteoblastic cells. J. Appl. Toxicol. 2018, 38, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wang, J.; Chen, X.; Liu, P.; Wang, S.; Song, F.; Zhang, Z.; Zhu, F.; Huang, X.; Liu, J.; et al. The Prenylflavonoid Xanthohumol Reduces Alzheimer-Like Changes and Modulates Multiple Pathogenic Molecular Pathways in the Neuro2a/APP(swe) Cell Model of AD. Front. Pharmacol. 2018, 9, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.W.; Zhu, X.; Yang, Q.Q.; Lu, Q.; Wang, J.Y.; Li, H.P.; Wei, Y.Q.; Yin, J.L.; Yin, X.X. Suppression of methylglyoxal hyperactivity by mangiferin can prevent diabetes-associated cognitive decline in rats. Psychopharmacology 2013, 228, 585–594. [Google Scholar] [CrossRef]
- Liu, Y.W.; Cheng, Y.Q.; Liu, X.L.; Hao, Y.C.; Li, Y.; Zhu, X.; Zhang, F.; Yin, X.X. Mangiferin Upregulates Glyoxalase 1 Through Activation of Nrf2/ARE Signaling in Central Neurons Cultured with High Glucose. Mol. Neurobiol. 2017, 54, 4060–4070. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.W.; Zhu, X.; Zhang, L.; Lu, Q.; Wang, J.Y.; Zhang, F.; Guo, H.; Yin, J.L.; Yin, X.X. Up-regulation of glyoxalase 1 by mangiferin prevents diabetic nephropathy progression in streptozotocin-induced diabetic rats. Eur. J. Pharmacol. 2013, 721, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Nagaraj, R.H.; Sarkar, P.; Mally, A.; Biemel, K.M.; Lederer, M.O.; Padayatti, P.S. Effect of pyridoxamine on chemical modification of proteins by carbonyls in diabetic rats: Characterization of a major product from the reaction of pyridoxamine and methylglyoxal. Arch. Biochem. Biophys. 2002, 402, 110–119. [Google Scholar] [CrossRef]
- Wetzels, S.; Wouters, K.; Miyata, T.; Scheijen, J.; Hendriks, J.J.A.; Schalkwijk, C.G.; Vanmierlo, T. Advanced Glycation Endproducts Are Increased in the Animal Model of Multiple Sclerosis but Cannot Be Reduced by Pyridoxamine Treatment or Glyoxalase 1 Overexpression. Int. J. Mol. Sci. 2018, 19, 1311. [Google Scholar] [CrossRef] [Green Version]
- Abouzed, T.K.; Munesue, S.; Harashima, A.; Masuo, Y.; Kato, Y.; Khailo, K.; Yamamoto, H.; Yamamoto, Y. Preventive Effect of Salicylate and Pyridoxamine on Diabetic Nephropathy. J. Diabetes Res. 2016, 2016, 1786789. [Google Scholar] [CrossRef] [Green Version]
- Oh, S.; Ahn, H.; Park, H.; Lee, J.I.; Park, K.Y.; Hwang, D.; Lee, S.; Son, K.H.; Byun, K. The attenuating effects of pyridoxamine on adipocyte hypertrophy and inflammation differ by adipocyte location. J. Nutr. Biochem. 2019, 72, 108173. [Google Scholar] [CrossRef] [PubMed]
- Kuricova, K.; Pleskacova, A.; Pacal, L.; Kankova, K. 1,25-Dihydroxyvitamin D increases the gene expression of enzymes protecting from glucolipotoxicity in peripheral blood mononuclear cells and human primary endothelial cells. Food Funct. 2016, 7, 2537–2543. [Google Scholar] [CrossRef]
- Omidian, M.; Djalali, M.; Javanbakht, M.H.; Eshraghian, M.R.; Abshirini, M.; Omidian, P.; Alvandi, E.; Mahmoudi, M. Effects of vitamin D supplementation on advanced glycation end products signaling pathway in T2DM patients: A randomized, placebo-controlled, double blind clinical trial. Diabetol. Metab. Syndr. 2019, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, K.; Watanabe, T.; Takenaka, A. Effect of dietary vitamin E on oxidative stress-related gene-mediated differences in anxiety-like behavior in inbred strains of mice. Physiol. Behav. 2019, 207, 64–72. [Google Scholar] [CrossRef]
- Derakhshanian, H.; Djalali, M.; Mohammad Hassan, M.H.; Alvandi, E.; Eshraghian, M.R.; Mirshafiey, A.; Nadimi, H.; Jahanabadi, S.; Zarei, M.; Djazayery, A. Vitamin D suppresses cellular pathways of diabetes complication in liver. Iran. J. Basic Med Sci. 2019, 22, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Hsu, C.C.; Huang, C.N.; Yin, M.C. Anti-glycative effects of oleanolic acid and ursolic acid in kidney of diabetic mice. Eur. J. Pharmacol. 2010, 628, 255–260. [Google Scholar] [CrossRef]
- Hsu, W.H.; Lee, B.H.; Chang, Y.Y.; Hsu, Y.W.; Pan, T.M. A novel natural Nrf2 activator with PPARγ-agonist (monascin) attenuates the toxicity of methylglyoxal and hyperglycemia. Toxicol. Appl. Pharmacol. 2013, 272, 842–851. [Google Scholar] [CrossRef]
- Truong, C.S.; Seo, E.; Jun, H.S. Psoralea corylifolia L. Seed Extract Attenuates Methylglyoxal-Induced Insulin Resistance by Inhibition of Advanced Glycation End Product Formation. Oxidative Med. Cell. Longev. 2019, 2019, 4310319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cha, S.H.; Hwang, Y.; Heo, S.J.; Jun, H.S. Indole-4-carboxaldehyde Isolated from Seaweed, Sargassum thunbergii, Attenuates Methylglyoxal-Induced Hepatic Inflammation. Mar. Drugs 2019, 17, 486. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Zhang, J.; Chen, L.; Li, J.; Zhang, H.; Guo, X. Glycine Suppresses AGE/RAGE Signaling Pathway and Subsequent Oxidative Stress by Restoring Glo1 Function in the Aorta of Diabetic Rats and in HUVECs. Oxidative Med. Cell. Longev. 2019, 2019, 4628962. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, D.; Chen, L.; Li, J.; Yuan, G.; Yang, G.; Zhang, H.; Guo, X.; Zhang, J. Glycine increases glyoxalase-1 function by promoting nuclear factor erythroid 2-related factor 2 translocation into the nucleus of kidney cells of streptozotocin-induced diabetic rats. J. Diabetes Investig. 2019, 10, 1189–1198. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Organism/Cells | Phenotype | Reference |
|---|---|---|---|
| Glo1 KD | L6 myoblasts | Intracellular accumulation of MG, GO and MG-related AGEs. | Stratmann et al. |
| Mouse Aortic Endothelial Cells | Accumulation of MG, MG modified proteins and impaired angiogenesis. | Nigro et al. | |
| Non-Diabetic mice | Increases in AGEs, oxidative stress, and mimics diabetic nephropathy. | Giacco et al. | |
| Glo1 KO | CRISPR/Cas9 Mouse line | No increases in MG and MGH-1 under hyperglycemic condition. Aldose reductase activity increased in the liver and kidney of Glo1KO mice. | Schumacher et al. |
| CRISPR/Cas9 HEK293 Cells | Glo1KO cells were more sensitive to MG toxicity than wild-type cells. DJ-1 protects histones from adduction by MG. | Galligan et al. | |
| CRISPR/Cas9 Mouse Schwann Cells | No accumulation of MG and MG modified proteins increased catalytic efficiency of aldose reductase. | Morgenstern et al | |
| Drosophila Melanogaster | Increases in MG and MGH-1 in tissues, decreases in insulin sensitivity, and lipid accumulation and hyperglycemia later in life. | Moraru et al. | |
| CRISPR/Cas9 Danio Renio | Under high nutrient intake, high MG levels, impaired glucose tolerance, and retinal blood vessel impairment. | Lodd et al. | |
| Glo1 OE | Caenorhabditis Elegans | Reduce levels of G-H1, CEL and MG-H1, and MG modifications of mitochondrial proteins; decrease in markers of oxidative damage; increase mean and maximum lifespan. | Morcos et al. |
| Diabetic rat (STZ) | Less levels of MG, GO, AGEs and oxidative stress markers, and hyperglycemia reduction. | Brouwers et al. | |
| Diabetic rat (STZ) | Ameliorate CEL and MG-H1 accumulation in the diabetic retina and prevent alterations in retinal neuroglia and vascular cells. | Berner et al. | |
| Diabetic rat (STZ) | Decrease in MG levels, less AGE formation, and reduced renal and endothelial dysfunction in response to induced diabetes. | Brouwers et al. | |
| Bovine Retinal Endothelial Cells | Improvement of cell viability induced by MG and prevent mitochondrial protein glycated modification. | Qian et al. | |
| Diabetic mice (STZ) | Restored the circulating levels of inflammatory markers (E-selectin, V-CAM), reduced RAGE expression and MG-induced endothelial cell loss. | Vulesevic et al. | |
| Mouse Bone Marrow-derived Progenitor Cells (db/db) | Prevent MG-impairment of IRE1α expression and activity in diabetic mice. | Hainan et al. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragonès, G.; Rowan, S.; Francisco, S.G.; Whitcomb, E.A.; Yang, W.; Perini-Villanueva, G.; Schalkwijk, C.G.; Taylor, A.; Bejarano, E. The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells 2021, 10, 1852. https://doi.org/10.3390/cells10081852
Aragonès G, Rowan S, Francisco SG, Whitcomb EA, Yang W, Perini-Villanueva G, Schalkwijk CG, Taylor A, Bejarano E. The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells. 2021; 10(8):1852. https://doi.org/10.3390/cells10081852
Chicago/Turabian StyleAragonès, Gemma, Sheldon Rowan, Sarah G. Francisco, Elizabeth A. Whitcomb, Wenxin Yang, Giuliana Perini-Villanueva, Casper G. Schalkwijk, Allen Taylor, and Eloy Bejarano. 2021. "The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy" Cells 10, no. 8: 1852. https://doi.org/10.3390/cells10081852
APA StyleAragonès, G., Rowan, S., Francisco, S. G., Whitcomb, E. A., Yang, W., Perini-Villanueva, G., Schalkwijk, C. G., Taylor, A., & Bejarano, E. (2021). The Glyoxalase System in Age-Related Diseases: Nutritional Intervention as Anti-Ageing Strategy. Cells, 10(8), 1852. https://doi.org/10.3390/cells10081852