
Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease

Abstract
:1. Introduction
2. Mechanisms and Initiators of NET Formation
3. Impact of Neutrophils on Atherogenesis and Atherosclerosis Development
3.1. Neutrophil Migration in Atherosclerosis
3.2. Neutrophil-Mediated Immunomodulation in Atherosclerosis
3.3. Neutrophil-Mediated Vascular Dysfunction in Atherosclerosis
3.4. Neutrophil-Mediated Tissue Damage in Atherosclerosis
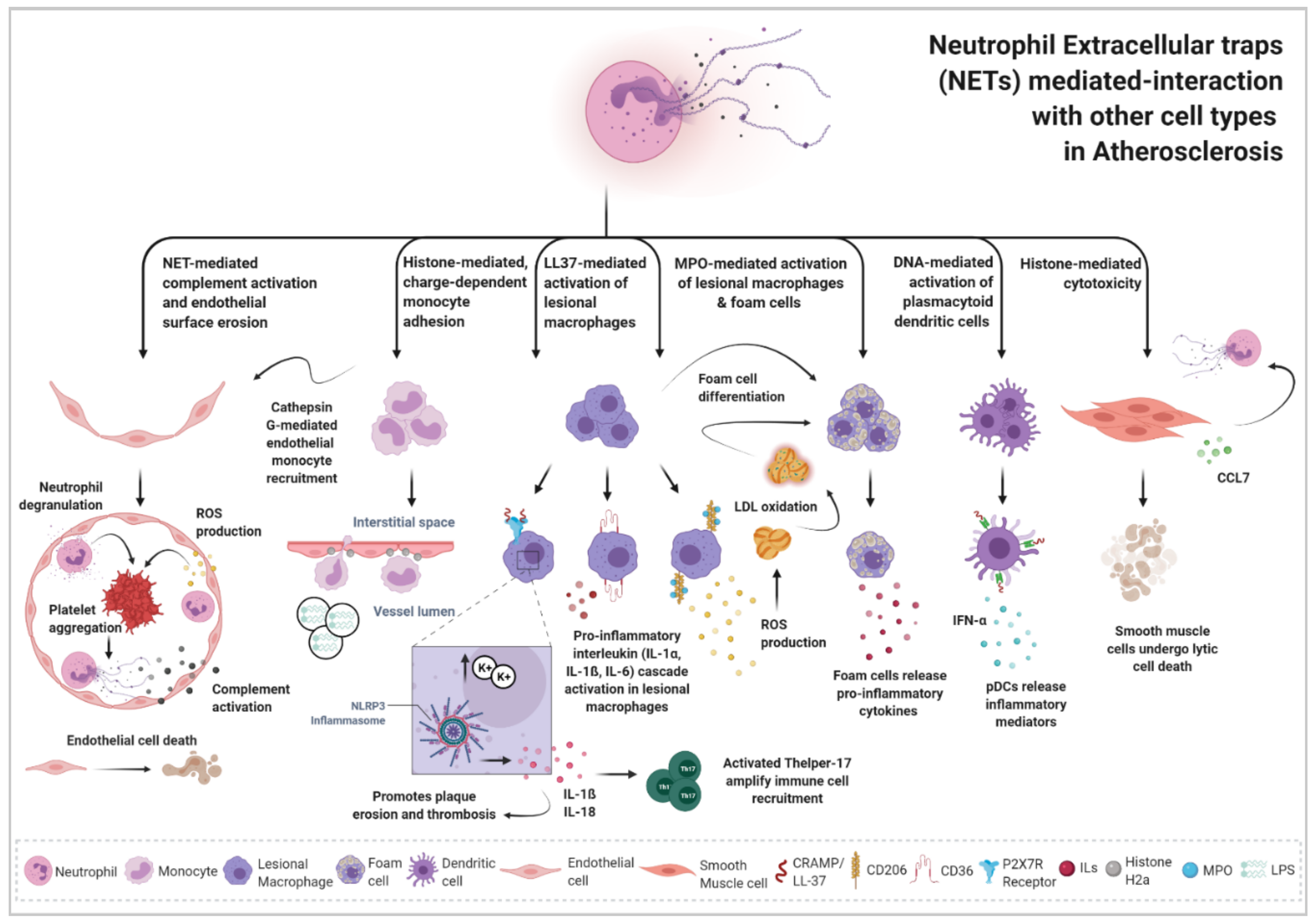
4. Impact of Neutrophil Extracellular Traps in Atherosclerosis Disease: Crosstalk of NETs with Other Cell Types
4.1. NET-Endothelial Cell Interaction
4.2. NET-Monocyte Interaction
4.3. NET-Macrophage Interaction
4.4. NET-DCs Interaction
4.5. NET-SMCs Interaction
4.6. NET-Platelet Interaction
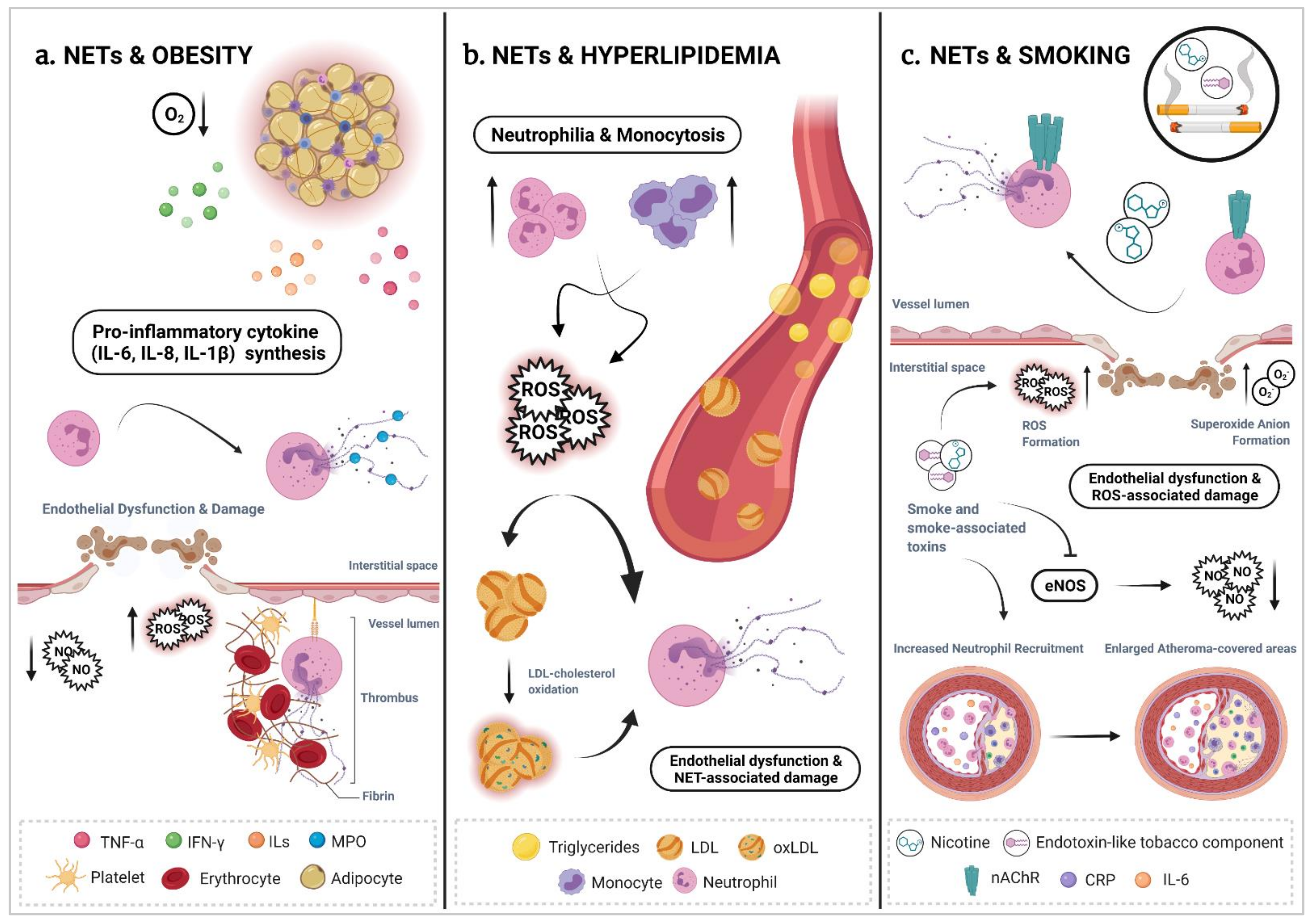
5. The Interplay of Neutrophil Extracellular Traps with Established Risk Factors for Cardiovascular Disease
5.1. NETs & Obesity
5.2. NETs & Hyperlipidemia
5.3. NETs & Hypertension
5.4. NETs & Physical Activity
5.5. NETs & Sleep Health
5.6. NETs & Smoking
6. Therapeutical Approach
NETs as Biomarkers of Cardiovascular Disease
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Noncommunicable Diseases. Available online: https://www.who.int/news-room/fact-sheets/detail/noncommunicable-diseases. (accessed on 20 May 2021).
- Ng, R.; Sutradhar, R.; Yao, Z.; Wodchis, W.P.; Rosella, L.C. Smoking, drinking, diet and physical activity-Modifiable lifestyle risk factors and their associations with age to first chronic disease. Int. J. Epidemiol. 2020, 49, 113–130. [Google Scholar] [CrossRef]
- Schloss, M.J.; Swirski, F.K.; Nahrendorf, M. Modifiable Cardiovascular Risk, Hematopoiesis, and Innate Immunity. Circ. Res. 2020, 126, 1242–1259. [Google Scholar] [CrossRef] [PubMed]
- Peltzer, K.; Pengpid, S. Prevalence, risk awareness and health beliefs of behavioural risk factors for cardiovascular disease among university students in nine ASEAN countries. BMC Public Health 2018, 18, 237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, G.A.; Huffman, M.D.; Moran, A.E.; Feigin, V.; Mensah, G.A.; Naghavi, M.; Murray, C.J.L. Global and regional patterns in cardiovascular mortality from 1990 to 2013. Circulation 2015, 132, 1667–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Legein, B.; Temmerman, L.; Biessen, E.A.L.; Lutgens, E. Inflammation and immune system interactions in atherosclerosis. Cell. Mol. Life Sci. 2013, 70, 3847–3869. [Google Scholar] [CrossRef]
- Quillard, T.; Araújo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis and detachment: Implications for superficial erosion. Eur. Heart J. 2015, 36, 1394–1404. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Schumski, A.; Ortega-Gómez, A.; Wichapong, K.; Winter, C.; Lemnitzer, P.; Viola, J.R.; Pinilla-Vera, M.; Folco, E.; Solis-Mezarino, V.; Völker-Albert, M.; et al. Endotoxinemia Accelerates Atherosclerosis through Electrostatic Charge-Mediated Monocyte Adhesion. Circulation 2021, 254–266. [Google Scholar] [CrossRef] [PubMed]
- Häger, M.; Cowland, J.B.; Borregaard, N. Neutrophil granules in health and disease. J. Intern. Med. 2010, 268, 25–34. [Google Scholar] [CrossRef]
- Son, G.; Kremer, M.; Hines, I.N. Contribution of Gut Bacteria to Liver Pathobiology. Gastroenterol. Res. Pract. 2010, 2010, 13. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Hossain, M.; Thanabalasuriar, A.; Gunzer, M.; Meininger, C.; Kubes, P. Visualizing the function and fate of neutrophils in sterile injury and repair. Science 2017, 358, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Wang, J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018, 371, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Casanova-Acebes, M.; Nicolás-Ávila, J.A.; Yao Li, J.L.; García-Silva, S.; Balachander, A.; Rubio-Ponce, A.; Weiss, L.A.; Adrover, J.M.; Burrows, K.; A-González, N.; et al. Neutrophils instruct homeostatic and pathological states in naive tissues. J. Exp. Med. 2018, 215, 2778–2795. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Grail, D.; Hodgson, G.; Metcalf, D.; Stanley, E.; Cheers, C.; Fowler, K.J.; Basu, S.; Zhan, Y.F.; Dunn, A.R. Mice lacking granulocyte colony-stimulating factor have chronic neutropenia, granulocyte and macrophage progenitor cell deficiency, and impaired neutrophil mobilization. Blood 1994, 84, 1737–1746. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Sheshachalam, A.; Srivastava, N.; Mitchell, T.; Lacy, P.; Eitzen, G. Granule protein processing and regulated secretion in neutrophils. Front. Immunol. 2014, 5, 448. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Marcos, V.; Zhou, Z.; Yildirim, A.Ö.; Bohla, A.; Hector, A.; Vitkov, L.; Wiedenbauer, E.M.; Krautgartner, W.D.; Stoiber, W.; Belohradsky, B.H.; et al. CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 2010, 16, 1018–1023. [Google Scholar] [CrossRef]
- Slaba, I.; Wang, J.; Kolaczkowska, E.; Mcdonald, B.; Lee, W.Y.; Kubes, P. Imaging the dynamic platelet-neutrophil response in sterile liver injury and repair in mice. Hepatology 2015, 62, 1593–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytom. A 2012, 81, 238–247. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444. [Google Scholar] [CrossRef]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manda, A.; Pruchniak, M.P.; Araźna, M.; Demkow, U.A. Neutrophil extracellular traps in physiology and pathology. Cent. Eur. J. Immunol. 2014, 39, 116–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Kichik, V.; Mondragón-Flores, R.; Mondragón-Castelán, M.; Gonzalez-Pozos, S.; Muñiz-Hernandez, S.; Rojas-Espinosa, O.; Chacón-Salinas, R.; Estrada-Parra, S.; Estrada-García, I. Neutrophil extracellular traps are induced by Mycobacterium tuberculosis. Tuberculosis 2009, 89, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Marin-Esteban, V.; Turbica, I.; Dufour, G.; Semiramoth, N.; Gleizes, A.; Gorges, R.; Beau, I.; Servin, A.L.; Moal, V.L.-L.; Sandré, C.; et al. Afa/Dr diffusely adhering Escherichia coli strain C1845 induces neutrophil extracellular traps that kill bacteria and damage human enterocyte-like cells. Infect. Immun. 2012, 80, 1891–1899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitkov, L.; Klappacher, M.; Hannig, M.; Krautgartner, W.D. Extracellular neutrophil traps in periodontitis. J. Periodontal Res. 2009, 44, 664–672. [Google Scholar] [CrossRef]
- De Buhr, N.; Von Köckritz-Blickwede, M. How Neutrophil Extracellular Traps Become Visible. J. Immunol. Res. 2016, 2016, 4604713. [Google Scholar] [CrossRef] [Green Version]
- Alasmari, S.Z. In Vivo Imaging of Neutrophil Extracellular Traps (NETs): Visualization Methods and Outcomes. BioMed Res. Int. 2020, 2020, 4192745. [Google Scholar] [CrossRef] [Green Version]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.V.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [Green Version]
- Chèvre, R.; González-Granado, J.M.; Megens, R.T.A.; Sreeramkumar, V.; Silvestre-Roig, C.; Molina-Sánchez, P.; Weber, C.; Soehnlein, O.; Hidalgo, A.; Andrés, V. High-resolution imaging of intravascular atherogenic inflammation in live mice. Circ. Res. 2014, 114, 770–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koji, T.; Yuhki, K.; Tadanobu, S.; Masato, O.; Shozo, I.; Yuji, T.; Yoshinaga, O.; Yasuhiro, I.; Toshimitsu, A.; Keiichi, U.; et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS ONE 2014, 9, e111888. [Google Scholar]
- Zhang, S.; Lu, X.; Shu, X.; Tian, X.; Yang, H.; Yang, W.; Zhang, Y.; Wang, G. Elevated plasma cfDNA may be associated with active lupus nephritis and partially attributed to abnormal regulation of neutrophil extracellular traps (NETs) in patients with systemic lupus erythematosus. Intern. Med. 2014, 53, 2763–2771. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Nakazawa, D.; Shida, H.; Miyoshi, A.; Kusunoki, Y.; Tomaru, U.; Ishizu, A. NETosis markers: Quest for specific, objective, and quantitative markers. Clin. Chim. Acta 2016, 459, 89–93. [Google Scholar] [CrossRef]
- Rada, B. Neutrophil extracellular traps. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2019; Volume 1982, pp. 517–528. [Google Scholar]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.J.; Brinkmann, V.; Jenne, D.E. Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef] [PubMed]
- Tatsiy, O.; McDonald, P.P. Physiological Stimuli Induce PAD4-Dependent, ROS-Independent NETosis, With Early and Late Events Controlled by Discrete Signaling Pathways. Front. Immunol. 2018, 9, 2036. [Google Scholar] [CrossRef] [Green Version]
- Thålin, C.; Daleskog, M.; Göransson, S.P.; Schatzberg, D.; Lasselin, J.; Laska, A.C.; Kallner, A.; Helleday, T.; Wallén, H.; Demers, M. Validation of an enzyme-linked immunosorbent assay for the quantification of citrullinated histone H3 as a marker for neutrophil extracellular traps in human plasma. Immunol. Res. 2017, 65, 706–712. [Google Scholar] [CrossRef] [Green Version]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]
- Gavillet, M.; Martinod, K.; Renella, R.; Harris, C.; Shapiro, N.I.; Wagner, D.D.; Williams, D.A. Flow cytometric assay for direct quantification of neutrophil extracellular traps in blood samples. Am. J. Hematol. 2015, 90, 1155–1158. [Google Scholar] [CrossRef] [Green Version]
- Von Köckritz-Blickwede, M.; Chow, O.; Ghochani, M.; Nizet, V. 7-Visualization and Functional Evaluation of Phagocyte Extracellular Traps. In Immunology of Infection; Kabelitz, D., Kaufmann, S.H.E., Eds.; Academic Press: Cambridge, MA, USA, 2010; Volume 37, pp. 139–160. ISBN 0580-9517. [Google Scholar]
- Neumann, A.; Völlger, L.; Berends, E.T.M.; Molhoek, E.M.; Stapels, D.A.C.; Midon, M.; Friães, A.; Pingoud, A.; Rooijakkers, S.H.M.; Gallo, R.L.; et al. Novel role of the antimicrobial peptide LL-37 in the protection of neutrophil extracellular traps against degradation by bacterial nucleases. J. Innate Immun. 2014, 6, 860–868. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.-A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Goosmann, C.; Kühn, L.I.; Zychlinsky, A. Automatic quantification of in vitro NET formation. Front. Immunol. 2012, 3, 413. [Google Scholar] [CrossRef] [Green Version]
- De Buhr, N.; Stehr, M.; Neumann, A.; Naim, H.Y.; Valentin-Weigand, P.; von Köckritz-Blickwede, M.; Baums, C.G. Identification of a novel DNase of Streptococcus suis (EndAsuis) important for neutrophil extracellular trap degradation during exponential growth. Microbiology 2015, 161, 838–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Buhr, N.; Neumann, A.; Jerjomiceva, N.; von Köckritz-Blickwede, M.; Baums, C.G. Streptococcus suis DNase SsnA contributes to degradation of neutrophil extracellular traps (NETs) and evasion of NET-mediated antimicrobial activity. Microbiology 2014, 160, 385–395. [Google Scholar] [CrossRef] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Zaromitidou, M.; Siasos, G.; Papageorgiou, N.; Oikonomou, E.; Tousoulis, D. Atherosclerosis and coronary artery disease: From basics to genetics. In Cardiovascular Diseases: Genetic Susceptibility, Environmental Factors and their Interaction; Elsevier Inc.: Amsterdam, The Netherlands, 2016; pp. 3–24. ISBN 9780128033135. [Google Scholar]
- Bäck, M.; Yurdagul, A.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Li, Y.S.; Chien, S. Shear stress-initiated signaling and its regulation of endothelial function. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2191–2198. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Subramanian, P.; Sevilmis, G.; Globke, B.; Soehnlein, O.; Karshovska, E.; Megens, R.; Heyll, K.; Chun, J.; Saulnier-Blache, J.S.; et al. Lipoprotein-derived lysophosphatidic acid promotes atherosclerosis by releasing CXCL1 from the endothelium. Cell Metab. 2011, 13, 592–600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumski, A.; Winter, C.; Döring, Y.; Soehnlein, O. The Ins and Outs of Myeloid Cells in Atherosclerosis. J. Innate Immun. 2018, 10, 479–486. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nat. Immunol. 2011, 12, 204–212. [Google Scholar] [CrossRef]
- Zernecke, A.; Liehn, E.A.; Gao, J.L.; Kuziel, W.A.; Murphy, P.M.; Weber, C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: Involvement of IL-10. Blood 2006, 107, 4240–4243. [Google Scholar] [CrossRef]
- Kluger, J. Beyond cholesterol. Time 1997, 150, 48. [Google Scholar]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Endothelial barrier and its abnormalities in cardiovascular disease. Front. Physiol. 2015, 6, 365. [Google Scholar] [CrossRef]
- Woollard, K.J. Immunological aspects of atherosclerosis. Clin. Sci. 2013, 125, 221–235. [Google Scholar] [CrossRef]
- Soehnlein, O.; Lindbom, L.; Weber, C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood 2009, 114, 4613–4623. [Google Scholar] [CrossRef]
- Doherty, D.E.; Downey, G.P.; Worthen, G.S.; Haslett, C.; Henson, P.M. Monocyte retention and migration in pulmonary inflammation. Requirement for neutrophils. Lab. Invest. 1988, 59, 200–213. [Google Scholar]
- Janardhan, K.S.; Sandhu, S.K.; Singh, B. Neutrophil depletion inhibits early and late monocyte/macrophage increase in lung inflammation. Front. Biosci. 2006, 11, 1569–1576. [Google Scholar] [CrossRef] [Green Version]
- Fillion, I.; Ouellet, N.; Simard, M.; Bergeron, Y.; Sato, S.; Bergeron, M.G. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J. Immunol. 2001, 166, 7353–7361. [Google Scholar] [CrossRef]
- Voisin, M.-B.; Woodfin, A.; Nourshargh, S. Monocytes and Neutrophils Exhibit Both Distinct and Common Mechanisms in Penetrating the Vascular Basement Membrane In Vivo. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1193–1199. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.V.; Davidson, D.C.; Jackson, J.W.; Singh, V.B.; Silva, J.; Ramirez, S.H.; Maggirwar, S.B. Characterization of platelet-monocyte complexes in HIV-1-infected individuals: Possible role in HIV-associated neuroinflammation. J. Immunol. 2014, 192, 4674–4684. [Google Scholar] [CrossRef] [Green Version]
- Badrnya, S.; Schrottmaier, W.C.; Kral, J.B.; Yaiw, K.-C.; Volf, I.; Schabbauer, G.; Söderberg-Nauclér, C.; Assinger, A. Platelets mediate oxidized low-density lipoprotein-induced monocyte extravasation and foam cell formation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 571–580. [Google Scholar] [CrossRef] [Green Version]
- Gaertner, F.; Ahmad, Z.; Rosenberger, G.; Fan, S.; Nicolai, L.; Busch, B.; Yavuz, G.; Luckner, M.; Ishikawa-Ankerhold, H.; Hennel, R.; et al. Migrating Platelets Are Mechano-scavengers that Collect and Bundle Bacteria. Cell 2017, 171, 1368–1382.e23. [Google Scholar] [CrossRef]
- Jackson, S.P. Arterial thrombosis--insidious, unpredictable and deadly. Nat. Med. 2011, 17, 1423–1436. [Google Scholar] [CrossRef]
- Wong, C.H.Y.; Jenne, C.N.; Petri, B.; Chrobok, N.L.; Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 2013, 14, 785–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soehnlein, O. Multiple roles for neutrophils in atherosclerosis. Circ. Res. 2012, 110, 875–888. [Google Scholar] [CrossRef] [Green Version]
- Ortega-Gomez, A.; Salvermoser, M.; Rossaint, J.; Pick, R.; Brauner, J.; Lemnitzer, P.; Tilgner, J.; De Jong, R.J.; Megens, R.T.A.; Jamasbi, J.; et al. Cathepsin G Controls Arterial but Not Venular Myeloid Cell Recruitment. Circulation 2016, 134, 1176–1188. [Google Scholar] [CrossRef] [Green Version]
- Döring, Y.; Drechsler, M.; Wantha, S.; Kemmerich, K.; Lievens, D.; Vijayan, S.; Gallo, R.L.; Weber, C.; Soehnlein, O. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ. Res. 2012, 110, 1052–1056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molinaro, R.; Yu, M.; Sausen, G.; Bichsel, C.A.; Corbo, C.; Folco, E.J.; Lee, G.Y.; Liu, Y.; Tesmenitsky, Y.; Shvartz, E.; et al. Targeted delivery of protein arginine deiminase-4 inhibitors to limit arterial intimal NETosis and preserve endothelial integrity. Cardiovasc. Res. 2021, cvab074. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Ortega-Gómez, A.; Döring, Y.; Weber, C. Neutrophil-macrophage interplay in atherosclerosis: Protease-mediated cytokine processing versus NET release. Thromb. Haemost. 2015, 114, 866–867. [Google Scholar]
- Poupel, L.; Boissonnas, A.; Hermand, P.; Dorgham, K.; Guyon, E.; Auvynet, C.; Charles, F.S.; Lesnik, P.; Deterre, P.; Combadiere, C. Pharmacological inhibition of the chemokine receptor, CX3CR1, reduces atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2297–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernecke, A.; Shagdarsuren, E.; Weber, C. Chemokines in atherosclerosis an update. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1897–1908. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.; Silvestre-Roig, C.; Ortega-Gomez, A.; Lemnitzer, P.; Poelman, H.; Schumski, A.; Winter, J.; Drechsler, M.; de Jong, R.; Immler, R.; et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab. 2018, 28, 175–182.e5. [Google Scholar] [CrossRef] [Green Version]
- Ionita, M.G.; Van Den Borne, P.; Catanzariti, L.M.; Moll, F.L.; De Vries, J.P.P.M.; Pasterkamp, G.; Vink, A.; De Kleijn, D.P.V. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture-prone lesions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1842–1848. [Google Scholar] [CrossRef]
- Wang, W.; Liu, W.; Fidler, T.; Wang, Y.; Tang, Y.; Woods, B.; Welch, C.; Cai, B.; Silvestre-Roig, C.; Ai, D.; et al. Macrophage inflammation, erythrophagocytosis, and accelerated atherosclerosis in JAK2V617F mice. Circ. Res. 2018, 123, E35–E47. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, F.; Lenglet, S.; Gayet-Ageron, A.; Bertolotto, M.; Pelli, G.; Palombo, D.; Pane, B.; Spinella, G.; Steffens, S.; Raffaghello, L.; et al. Systemic and intraplaque mediators of inflammation are increased in patients symptomatic for ischemic stroke. Stroke 2010, 41, 1394–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naruko, T.; Ueda, M.; Haze, K.; Van der Wal, A.C.; Van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Z.S.; Jeng, J.S.; Wang, C.H.; Yip, P.K.; Wu, T.H.; Lee, T.K. Correlations between peripheral differential leukocyte counts and carotid atherosclerosis in non-smokers. Atherosclerosis 2001, 158, 431–436. [Google Scholar] [CrossRef]
- Leclercq, A.; Houard, X.; Philippe, M.; Ollivier, V.; Sebbag, U.; Meilhac, O.; Michel, J.-B. Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J. Leukoc. Biol. 2007, 82, 1420–1429. [Google Scholar] [CrossRef]
- Rotzius, P.; Thams, S.; Soehnlein, O.; Kenne, E.; Tseng, C.N.; Björkström, N.K.; Malmberg, K.J.; Lindbom, L.; Eriksson, E.E. Distinct infiltration of neutrophils in lesion shoulders in ApoE -/- mice. Am. J. Pathol. 2010, 177, 493–500. [Google Scholar] [CrossRef]
- Hemdahl, A.L.; Gabrielsen, A.; Zhu, C.; Eriksson, P.; Hedin, U.; Kastrup, J.; Thorén, P.; Hansson, G.K. Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 136–142. [Google Scholar] [CrossRef] [Green Version]
- Barnathan, E.S.; Raghunath, P.N.; Tomaszewski, J.E.; Ganz, T.; Cines, D.B.; Higazi, A.A.-R. Immunohistochemical localization of defensin in human coronary vessels. Am. J. Pathol. 1997, 150, 1009–1020. [Google Scholar]
- Edfeldt, K.; Agerberth, B.; Rottenberg, M.E.; Gudmundsson, G.H.; Wang, X.B.; Mandal, K.; Xu, Q.; Yan, Z.Q. Involvement of the antimicrobial peptide LL-37 in human atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1551–1557. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Bot, I.; Djalali-Talab, Y.; Shagdarsuren, E.; Bidzhekov, K.; Meiler, S.; Krohn, R.; Schober, A.; Sperandio, M.; Soehnlein, O.; et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 2008, 102, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Drechsler, M.; Megens, R.T.A.; Van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845. [Google Scholar] [CrossRef]
- Combadière, C.; Potteaux, S.; Gao, J.L.; Esposito, B.; Casanova, S.; Lee, E.J.; Debré, P.; Tedgui, A.; Murphy, P.M.; Mallat, Z. Decreased atherosclerotic lesion formation in CX3CR1/apolipoprotein E double knockout mice. Circulation 2003, 107, 1009–1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combadière, C.; Potteaux, S.; Rodero, M.; Simon, T.; Pezard, A.; Esposito, B.; Merval, R.; Proudfoot, A.; Tedgui, A.; Mallat, Z. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6Chi and Ly6Clo monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation 2008, 117, 1649–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saederup, N.; Chan, L.; Lira, S.A.; Charo, I.F. Fractalkine deficiency markedly reduces macrophage accumulation and atherosclerotic lesion formation in CCR2-/- mice: Evidence for independent chemokine functions in atherogenesis. Circulation 2008, 117, 1642–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subbarao, K.; Jala, V.R.; Mathis, S.; Suttles, J.; Zacharias, W.; Ahamed, J.; Ali, H.; Tseng, M.T.; Haribabu, B. Role of leukotriene B4 receptors in the development of atherosclerosis: Potential mechanisms. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 369–375. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Chung, K.F.; Page, C.P. Platelet-activating factor as a mediator of allergic disease. J. Allergy Clin. Immunol. 1988, 81, 919–934. [Google Scholar] [CrossRef]
- Apostolopoulos, J.; Davenport, P.; Tipping, P.G. Interleukin-8 production by macrophages from atheromatous plaques. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 1007–1012. [Google Scholar] [CrossRef]
- Apostolakis, S.; Vogiatzi, K.; Amanatidou, V.; Spandidos, D.A. Interleukin 8 and cardiovascular disease. Cardiovasc. Res. 2009, 84, 353–360. [Google Scholar] [CrossRef] [Green Version]
- Imhof, B.A.; Jemelin, S.; Emre, Y. Toll-like receptors elicit different recruitment kinetics of monocytes and neutrophils in mouse acute inflammation. Eur. J. Immunol. 2017, 47, 1002–1008. [Google Scholar] [CrossRef]
- Ciornei, C.D.; Tapper, H.; Bjartell, A.; Sternby, N.H.; Bodelsson, M. Human antimicrobial peptide LL-37 is present in atherosclerotic plaques and induces death of vascular smooth muscle cells: A laboratory study. BMC Cardiovasc. Disord. 2006, 6, 49. [Google Scholar] [CrossRef] [Green Version]
- Prame Kumar, K.; Nicholls, A.J.; Wong, C.H.Y. Partners in crime: Neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. 2018, 371, 551–565. [Google Scholar] [CrossRef] [Green Version]
- Alard, J.E.; Ortega-Gomez, A.; Wichapong, K.; Bongiovanni, D.; Horckmans, M.; Megens, R.T.A.; Leoni, G.; Ferraro, B.; Rossaint, J.; Paulin, N.; et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015, 7, 317ra196. [Google Scholar] [CrossRef] [PubMed]
- Abu-Fanne, R.; Maraga, E.; Abd-Elrahman, I.; Hankin, A.; Blum, G.; Abdeen, S.; Hijazi, N.; Cines, D.B.; Higazi, A.A.R. αDefensins induce a post-translational modification of low density lipoprotein (LDL) that promotes atherosclerosis at normal levels of plasma cholesterol. J. Biol. Chem. 2016, 291, 2777–2786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higazi, M.; Abdeen, S.; Abu-Fanne, R.; Heyman, S.N.; Masarwy, A.; Bdeir, K.; Maraga, E.; Cines, D.B.; Al-Roof Higazi, A. Opposing effects of HNP1 (α-defensin-1) on plasma cholesterol and atherogenesis. PLoS ONE 2020, 15, e0231582. [Google Scholar] [CrossRef]
- Heinzelmann, M.; Platz, A.; Flodgaard, H.; Miller, F.N. Heparin binding protein (CAP37) is an opsonin for Staphylococcus aureus and increases phagocytosis in monocytes. Inflammation 1998, 22, 493–507. [Google Scholar] [CrossRef]
- Rasmussen, P.B.; Bjørn, S.; Hastrup, S.; Nielsen, P.F.; Norris, K.; Thim, L.; Wiberg, F.C.; Flodgaard, H. Characterization of recombinant human HBP/CAP37/azurocidin, a pleiotropic mediator of inflammation-enhancing LPS-induced cytokine release from monocytes. FEBS Lett. 1996, 390, 109–112. [Google Scholar] [CrossRef] [Green Version]
- Hartwig, H.; Silvestre Roig, C.; Daemen, M.; Lutgens, E.; Soehnlein, O. Neutrophils in atherosclerosis: A brief overview. Hamostaseologie 2015, 35, 121–127. [Google Scholar]
- Rios, F.J.O.; Ferracini, M.; Pecenin, M.; Koga, M.M.; Wang, Y.; Ketelhuth, D.F.J.; Jancar, S. Uptake of oxLDL and IL-10 Production by Macrophages Requires PAFR and CD36 Recruitment into the Same Lipid Rafts. PLoS ONE 2013, 8, e76893. [Google Scholar] [CrossRef]
- Lougheed, M.; Lum, C.M.; Ling, W.; Suzuki, H.; Kodama, T.; Steinbrecher, U. High affinity saturable uptake of oxidized low density lipoprotein by macrophages from mice lacking the scavenger receptor class A type I/II. J. Biol. Chem. 1997, 272, 12938–12944. [Google Scholar] [CrossRef] [Green Version]
- Napoli, C.; de Nigris, F.; Williams-Ignarro, S.; Pignalosa, O.; Sica, V.; Ignarro, L.J. Nitric oxide and atherosclerosis: An update. Nitric Oxide Biol. Chem. 2006, 15, 265–279. [Google Scholar] [CrossRef]
- Manda-Handzlik, A.; Demkow, U. Neutrophils: The Role of Oxidative and Nitrosative Stress in Health and Disease. Adv. Exp. Med. Biol. 2015, 857, 51–60. [Google Scholar] [PubMed]
- Nicholls, S.J.; Hazen, S.L. Myeloperoxidase and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1102–1111. [Google Scholar] [CrossRef] [Green Version]
- Kougias, P.; Chai, H.; Lin, P.H.; Yao, Q.; Lumsden, A.B.; Chen, C. Neutrophil antimicrobial peptide α-defensin causes endothelial dysfunction in porcine coronary arteries. J. Vasc. Surg. 2006, 43, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Channon, K.M.; Qian, H.; George, S.E. Nitric oxide synthase in atherosclerosis and vascular injury: Insights from experimental gene therapy. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1873–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poljsak, B.; Šuput, D.; Milisav, I. Achieving the Balance between ROS and Antioxidants: When to Use the Synthetic Antioxidants. Oxid. Med. Cell. Longev. 2013, 2013, 956792. [Google Scholar] [CrossRef] [PubMed]
- Gryglewski, R.J.; Palmer, R.M.; Moncada, S. Superoxide anion is involved in the breakdown of endothelium-derived vascular relaxing factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef]
- White, C.R.; Brock, T.A.; Chang, L.Y.; Crapo, J.; Briscoe, P.; Ku, D.; Bradley, W.A.; Gianturco, S.H.; Gore, J.; Freeman, B.A. Superoxide and peroxynitrite in atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 1044–1048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verbeuren, T.J.; Jordaens, F.H.; Zonnekeyn, L.L.; Van Hove, C.E.; Coene, M.C.; Herman, A.G. Effect of hypercholesterolemia on vascular reactivity in the rabbit. I. Endothelium-dependent and endothelium-independent contractions and relaxations in isolated arteries of control and hypercholesterolemic rabbits. Circ. Res. 1986, 58, 552–564. [Google Scholar] [CrossRef] [Green Version]
- Aji, W.; Ravalli, S.; Szabolcs, M.; Jiang, X.C.; Sciacca, R.R.; Michler, R.E.; Cannon, P.J. L-arginine prevents xanthoma development and inhibits atherosclerosis in LDL receptor knockout mice. Circulation 1997, 95, 430–437. [Google Scholar] [CrossRef]
- Patel, S.; Kumar, S.; Jyoti, A.; Srinag, B.S.; Keshari, R.S.; Saluja, R.; Verma, A.; Mitra, K.; Barthwal, M.K.; Krishnamurthy, H.; et al. Nitric oxide donors release extracellular traps from human neutrophils by augmenting free radical generation. Nitric Oxide Biol. Chem. 2010, 22, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Lenglet, S.; Thomas, A.; Soehnlein, O.; Montecucco, F.; Burger, F.; Pelli, G.; Galan, K.; Cravatt, B.; Staub, C.; Steffens, S. Fatty acid amide hydrolase deficiency enhances intraplaque neutrophil recruitment in atherosclerotic mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Laxton, R.C.; Hu, Y.; Duchene, J.; Zhang, F.; Zhang, Z.; Leung, K.Y.; Xiao, Q.; Scotland, R.S.; Hodgkinson, C.P.; Smith, K.; et al. A role of matrix metalloproteinase-8 in atherosclerosis. Circ. Res. 2009, 105, 921–929. [Google Scholar] [CrossRef]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Knight, J.S.; Luo, W.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Subramanian, V.; Guo, C.; Grenn, R.C.; Thompson, P.R.; Eitzman, D.T.; et al. Peptidylarginine deiminase inhibition reduces vascular damage and modulates innate immune responses in murine models of atherosclerosis. Circ. Res. 2014, 114, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Franck, G.; Mawson, T.L.; Folco, E.J.; Molinaro, R.; Ruvkun, V.; Engelbertsen, D.; Liu, X.; Tesmenitsky, Y.; Shvartz, E.; Sukhova, G.K.; et al. Roles of PAD4 and netosis in experimental atherosclerosis and arterial injury implications for superfcial erosion. Circ. Res. 2018, 123, 33–42. [Google Scholar] [CrossRef]
- Wang, H.; Wang, C.; Zhao, M.H.; Chen, M. Neutrophil extracellular traps can activate alternative complement pathways. Clin. Exp. Immunol. 2015, 181, 518–527. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.K.; Joshi, M.B.; Philippova, M.; Erne, P.; Hasler, P.; Hahn, S.; Resink, T.J. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010, 584, 3193–3197. [Google Scholar] [CrossRef] [Green Version]
- Wantha, S.; Alard, J.-E.; Megens, R.T.A.; van der Does, A.M.; Döring, Y.; Drechsler, M.; Pham, C.T.N.; Wang, M.-W.; Wang, J.-M.; Gallo, R.L.; et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ. Res. 2013, 112, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef]
- Josefs, T.; Barrett, T.J.; Brown, E.J.; Quezada, A.; Wu, X.; Voisin, M.; Amengual, J.; Fisher, E.A. Neutrophil extracellular traps promote macrophage inflammation and impair atherosclerosis resolution in diabetic mice. JCI Insight 2020, 5, e134796. [Google Scholar] [CrossRef] [Green Version]
- Döring, Y.; Manthey, H.D.; Drechsler, M.; Lievens, D.; Megens, R.T.A.; Soehnlein, O.; Busch, M.; Manca, M.; Koenen, R.R.; Pelisek, J.; et al. Auto-antigenic protein-DNA complexes stimulate plasmacytoid dendritic cells to promote atherosclerosis. Circulation 2012, 125, 1673–1683. [Google Scholar] [CrossRef] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D.J.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- McDonald, B.; Davis, R.P.; Kim, S.-J.; Tse, M.; Esmon, C.T.; Kolaczkowska, E.; Jenne, C.N. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood 2017, 129, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Qi, H.; Kan, K.; Chen, J.; Xie, H.; Guo, X.; Zhang, L. Neutrophil Extracellular Traps Promote Hypercoagulability in Patients With Sepsis. Shock 2017, 47, 132–139. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Bhandari, A.A.; Wagner, D.D. Histones induce rapid and profound thrombocytopenia in mice. Blood 2011, 118, 3708–3714. [Google Scholar] [CrossRef] [Green Version]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.-L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Von Brühl, M.-L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Köllnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Oklu, R.; Albadawi, H.; Watkins, M.T.; Monestier, M.; Sillesen, M.; Wicky, S. Detection of extracellular genomic DNA scaffold in human thrombus: Implications for the use of deoxyribonuclease enzymes in thrombolysis. J. Vasc. Interv. Radiol. 2012, 23, 712–718. [Google Scholar] [CrossRef]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.; Philippi, V.; Stockhausen, S.; Busse, J.; Antonelli, A.; Miller, M.; Schubert, I.; Hoseinpour, P.; Chandraratne, S.; von Brühl, M.-L.; et al. Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 2016, 128, 2435–2449. [Google Scholar] [CrossRef]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Döring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [Green Version]
- Nahrendorf, M.; Swirski, F.K. Lifestyle effects on hematopoiesis and atherosclerosis. Circ. Res. 2015, 116, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Huang, T.; Hu, F.B. Gene-environment interactions and obesity: Recent developments and future directions. BMC Med. Genom. 2015, 8, S2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Cho, Y.A. Gene-environment interaction and obesity. Nutr. Rev. 2008, 66, 684–694. [Google Scholar] [CrossRef]
- WHO. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight. (accessed on 20 May 2021).
- Hotamisligil, G.S.; Shargill, N.S.; Spiegelman, B.M. Adipose expression of tumor necrosis factor-α: Direct role in obesity-linked insulin resistance. Science 1993, 259, 87–91. [Google Scholar] [CrossRef]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Invest. 2005, 115, 1111–1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.C.; Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 446–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [Green Version]
- Vecchié, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Frühbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Trellakis, S.; Rydleuskaya, A.; Fischer, C.; Canbay, A.; Tagay, S.; Scherag, A.; Bruderek, K.; Schuler, P.J.; Brandau, S. Low adiponectin, high levels of apoptosis and increased peripheral blood neutrophil activity in healthy obese subjects. Obes. Facts 2012, 5, 305–318. [Google Scholar] [CrossRef]
- Bonaventura, A.; Vecchié, A.; Abbate, A.; Montecucco, F. Neutrophil Extracellular Traps and Cardiovascular Diseases: An Update. Cells 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moorthy, A.N.; Tan, K.B.; Wang, S.; Narasaraju, T.; Chow, V.T. Effect of high-fat diet on the formation of pulmonary neutrophil extracellular traps during influenza pneumonia in BALB/c mice. Front. Immunol. 2016, 7, 289. [Google Scholar] [CrossRef] [Green Version]
- Keshari, R.S.; Jyoti, A.; Dubey, M.; Kothari, N.; Kohli, M.; Bogra, J.; Barthwal, M.K.; Dikshit, M. Cytokines Induced Neutrophil Extracellular Traps Formation: Implication for the Inflammatory Disease Condition. PLoS ONE 2012, 7, e48111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Q.; Venugopal, J.; Wang, J.; Kleiman, K.; Guo, C.; Eitzman, D.T. Obesity-induced Endothelial Dysfunction is Prevented by Neutrophil Extracellular Trap Inhibition. Sci. Rep. 2018, 8, 4881. [Google Scholar] [CrossRef]
- Stenlöf, K.; Wernstedt, I.; Fjällman, T.; Wallenius, V.; Wallenius, K.; Jansson, J.O. Interleukin-6 levels in the central nervous system are negatively correlated with fat mass in overweight/obese subjects. J. Clin. Endocrinol. Metab. 2003, 88, 4379–4383. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Liu, F. Transcriptional and post-translational regulation of adiponectin. Biochem. J. 2010, 425, 41–52. [Google Scholar] [CrossRef]
- Talukdar, S.; Oh, D.Y.; Bandyopadhyay, G.; Li, D.; Xu, J.; McNelis, J.; Lu, M.; Li, P.; Yan, Q.; Zhu, Y.; et al. Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat. Med. 2012, 18, 1407–1412. [Google Scholar] [CrossRef] [Green Version]
- Elgazar-Carmon, V.; Rudich, A.; Hadad, N.; Levy, R. Neutrophils transiently infiltrate intra-abdominal fat early in the course of high-fat feeding. J. Lipid Res. 2008, 49, 1894–1903. [Google Scholar] [CrossRef] [Green Version]
- D’Abbondanza, M.; Martorelli, E.E.; Ricci, M.A.; De Vuono, S.; Migliola, E.N.; Godino, C.; Corradetti, S.; Siepi, D.; Paganelli, M.T.; Maugeri, N.; et al. Increased plasmatic NETs by-products in patients in severe obesity. Sci. Rep. 2019, 9, 14678. [Google Scholar] [CrossRef] [Green Version]
- Cichon, I.; Ortmann, W.; Santocki, M.; Opydo-Chanek, M.; Kolaczkowska, E. Scrutinizing Mechanisms of the “Obesity Paradox in Sepsis”: Obesity Is Accompanied by Diminished Formation of Neutrophil Extracellular Traps (NETs) Due to Restricted Neutrophil-Platelet Interactions. Cells 2021, 10, 384. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Braster, Q.; Silvestre Roig, C.; Hartwig, H.; Beckers, L.; den Toom, M.; Döring, Y.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Inhibition of NET Release Fails to Reduce Adipose Tissue Inflammation in Mice. PLoS ONE 2016, 11, e0163922. [Google Scholar] [CrossRef]
- Grebe, A.; Latz, E. Cholesterol crystals and inflammation. Curr. Rheumatol. Rep. 2013, 15, 313. [Google Scholar] [CrossRef] [Green Version]
- Ho-Tin-Noé, B.; Vo, S.; Bayles, R.; Ferrière, S.; Ladjal, H.; Toumi, S.; Deschildre, C.; Ollivier, V.; Michel, J.-B. Cholesterol crystallization in human atherosclerosis is triggered in smooth muscle cells during the transition from fatty streak to fibroatheroma. J. Pathol. 2017, 241, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Abela, G.S.; Aziz, K. Cholesterol crystals cause mechanical damage to biological membranes: A proposed mechanism of plaque rupture and erosion leading to arterial thrombosis. Clin. Cardiol. 2005, 28, 413–420. [Google Scholar] [CrossRef]
- Alipour, A.; van Oostrom, A.J.H.H.M.; Izraeljan, A.; Verseyden, C.; Collins, J.M.; Frayn, K.N.; Plokker, T.W.M.; Elte, J.W.F.; Castro Cabezas, M. Leukocyte activation by triglyceride-rich lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Babio, N.; Ibarrola-Jurado, N.; Bulló, M.; Martínez-González, M.Á.; Wärnberg, J.; Salaverría, I.; Ortega-Calvo, M.; Estruch, R.; Serra-Majem, L.; Covas, M.I.; et al. White blood cell counts as risk markers of developing metabolic syndrome and its components in the PREDIMED study. PLoS ONE 2013, 8, e58354. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carmona-Rivera, C.; Moore, E.; Seto, N.L.; Knight, J.S.; Pryor, M.; Yang, Z.H.; Hemmers, S.; Remaley, A.T.; Mowen, K.A.; et al. Myeloid-specific deletion of peptidylarginine deiminase 4 mitigates atherosclerosis. Front. Immunol. 2018, 9, 1680. [Google Scholar] [CrossRef] [Green Version]
- Jeremic, I.; Schorn, C.; Munoz, L.; Terzic, T.; Schett, G.; Herrmann, M. A9.7 Cholesterol crystals induce neutrophil extracellular traps formation. Ann. Rheum. Dis. 2014, 73, A94. [Google Scholar] [CrossRef]
- Chow, O.A.; von Köckritz-Blickwede, M.; Bright, A.T.; Hensler, M.E.; Zinkernagel, A.S.; Cogen, A.L.; Gallo, R.L.; Monestier, M.; Wang, Y.; Glass, C.K.; et al. Statins enhance formation of phagocyte extracellular traps. Cell Host Microbe 2010, 8, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.; Brogden, G.; Jerjomiceva, N.; Brodesser, S.; Naim, H.Y.; Von Köckritz-Blickwede, M. Lipid alterations in human blood-derived neutrophils lead to formation of neutrophil extracellular traps. Eur. J. Cell Biol. 2014, 93, 347–354. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nũez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Tall, A.R.; Westerterp, M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J. Lipid Res. 2019, 60, 721–727. [Google Scholar] [CrossRef] [Green Version]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; La Bastide-Van Gemert, S.; Wang, N.; et al. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsukawa, Y.; Hsu, W.L.; Yamada, M.; Cologne, J.B.; Suzuki, G.; Yamamoto, H.; Yamane, K.; Akahoshi, M.; Fujiwara, S.; Kohno, N. White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens. Res. 2008, 31, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Zhang, Q.; Wu, H.; Du, H.; Liu, L.; Shi, H.; Wang, C.; Xia, Y.; Guo, X.; Li, C.; et al. Blood Neutrophil to Lymphocyte Ratio as a Predictor of Hypertension. Am. J. Hypertens. 2015, 28, 1339–1346. [Google Scholar] [CrossRef] [Green Version]
- Araos, P.; Figueroa, S.; Amador, C.A. The role of neutrophils in hypertension. Int. J. Mol. Sci. 2020, 21, 8536. [Google Scholar] [CrossRef]
- Hofbauer, T.; Scherz, T.; Müller, J.; Heidari, H.; Staier, N.; Panzenböck, A.; Mangold, A.; Lang, I.M. Arterial hypertension enhances neutrophil extracellular trap formation via an angiotensin-II-dependent pathway. Atherosclerosis 2017, 263, e67–e68. [Google Scholar] [CrossRef]
- Li, J.-H.; Tong, D.-X.; Wang, Y.; Gao, L.; Liu, Y.; Zhang, X.-H.; Chen, W.-J.; Chi, J.-Y.; Liu, N.; Yang, K.; et al. Neutrophil extracellular traps exacerbate coagulation and endothelial damage in patients with essential hypertension and hyperhomocysteinemia. Thromb. Res. 2021, 197, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Lentz, S.R. Mechanisms of homocysteine-induced atherothrombosis. J. Thromb. Haemost. 2005, 3, 1646–1654. [Google Scholar] [CrossRef] [PubMed]
- Korai, M.; Purcell, J.; Kamio, Y.; Mitsui, K.; Furukawa, H.; Yokosuka, K.; Miyamoto, T.; Sato, H.; Sato, H.; Eguchi, S.; et al. Neutrophil Extracellular Traps Promote the Development of Intracranial Aneurysm Rupture. Hypertension 2021, 77, 2084–2093. [Google Scholar] [CrossRef]
- Smith, J.K. Exercise and Atherogenesis. Exerc. Sport Sci. Rev. 2001, 29, 49–53. [Google Scholar]
- Lee, I.M.; Shiroma, E.J.; Lobelo, F.; Puska, P.; Blair, S.N.; Katzmarzyk, P.T.; Alkandari, J.R.; Andersen, L.B.; Bauman, A.E.; Brownson, R.C.; et al. Effect of physical inactivity on major non-communicable diseases worldwide: An analysis of burden of disease and life expectancy. Lancet 2012, 380, 219–229. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Liu, B.; Liang, C.; Li, Y.; Song, Y.H. Cytokine Signaling in Skeletal Muscle Wasting. Trends Endocrinol. Metab. 2016, 27, 335–347. [Google Scholar] [CrossRef]
- Filgueira, T.O.; Castoldi, A.; Santos, L.E.R.; de Amorim, G.J.; de Sousa Fernandes, M.S.; de Lima do Nascimento, A.; Campos, E.Z.; Santos, T.M.; Souto, F.O. The Relevance of a Physical Active Lifestyle and Physical Fitness on Immune Defense: Mitigating Disease Burden, With Focus on COVID-19 Consequences. Front. Immunol. 2021, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Duggal, N.A.; Niemiro, G.; Harridge, S.D.R.; Simpson, R.J.; Lord, J.M. Can physical activity ameliorate immunosenescence and thereby reduce age-related multi-morbidity? Nat. Rev. Immunol. 2019, 19, 563–572. [Google Scholar] [CrossRef]
- Lambert, C.P.; Wright, N.R.; Finck, B.N.; Villareal, D.T. Exercise but not diet-induced weight loss decreases skeletal muscle inflammatory gene expression in frail obese elderly persons. J. Appl. Physiol. 2008, 105, 473–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michishita, R.; Shono, N.; Inoue, T.; Tsuruta, T.; Node, K. Effect of exercise therapy on monocyte and neutrophil counts in overweight women. Am. J. Med. Sci. 2010, 339, 152–156. [Google Scholar]
- Timmerman, K.L.; Flynn, M.G.; Coen, P.M.; Markofski, M.M.; Pence, B.D. Exercise training-induced lowering of inflammatory (CD14+CD16+) monocytes: A role in the anti-inflammatory influence of exercise? J. Leukoc. Biol. 2008, 84, 1271–1278. [Google Scholar] [CrossRef]
- Palmefors, H.; DuttaRoy, S.; Rundqvist, B.; Börjesson, M. The effect of physical activity or exercise on key biomarkers in atherosclerosis—A systematic review. Atherosclerosis 2014, 235, 150–161. [Google Scholar] [CrossRef]
- Noz, M.P.; Hartman, Y.A.W.; Hopman, M.T.E.; Willems, P.H.G.M.; Tack, C.J.; Joosten, L.A.B.; Netea, M.G.; Thijssen, D.H.J.; Riksen, N.P. Sixteen-Week Physical Activity Intervention in Subjects With Increased Cardiometabolic Risk Shifts Innate Immune Function Towards a Less Proinflammatory State. J. Am. Heart Assoc. 2019, 8, e013764. [Google Scholar] [CrossRef]
- Burini, R.C.; Anderson, E.; Durstine, J.L.; Carson, J.A. Inflammation, physical activity, and chronic disease: An evolutionary perspective. Sport. Med. Health Sci. 2020, 2, 1–6. [Google Scholar] [CrossRef]
- Nieman, D.C.; Wentz, L.M. The compelling link between physical activity and the body’s defense system. J. Sport Health Sci. 2019, 8, 201–217. [Google Scholar] [CrossRef]
- Abebayehu, D.; Spence, A.J.; Qayum, A.A.; Taruselli, M.T.; McLeod, J.J.A.; Caslin, H.L.; Chumanevich, A.P.; Kolawole, E.M.; Paranjape, A.; Baker, B.; et al. Lactic Acid Suppresses IL-33–Mediated Mast Cell Inflammatory Responses via Hypoxia-Inducible Factor-1α–Dependent miR-155 Suppression. J. Immunol. 2016, 197, 2909–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Shi, H.; Nieman, D.C.; Hu, Q.; Yang, L.; Liu, T.; Zhu, X.; Wei, H.; Wu, D.; Li, F.; et al. Lactic acid accumulation during exhaustive exercise impairs release of neutrophil extracellular traps in mice. Front. Physiol. 2019, 10, 709. [Google Scholar] [CrossRef]
- Laufs, U.; Wassmann, S.; Czech, T.; Münzel, T.; Eisenhauer, M.; Böhm, M.; Nickenig, G. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 809–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hafner, M.; Stepanek, M.; Taylor, J.; Troxel, W.; Stolk, C. Why Sleep Matters—The Economic Costs of Insufficient Sleep: A Cross-Country Comparative Analysis; RAND Corporation: Santa Monica, CA, USA, 2017; Volume 6. [Google Scholar]
- Grandner, M.A.; Sands-Lincoln, M.R.; Pak, V.M.; Garland, S.N. Sleep duration, cardiovascular disease, and proinflammatory biomarkers. Nat. Sci. Sleep 2013, 5, 93–107. [Google Scholar] [CrossRef] [Green Version]
- Carreras, A.; Zhang, S.X.; Peris, E.; Qiao, Z.; Gileles-Hillel, A.; Li, R.C.; Wang, Y.; Gozal, D. Chronic sleep fragmentation induces endothelial dysfunction and structural vascular changes in mice. Sleep 2014, 37, 1817–1824. [Google Scholar] [CrossRef] [Green Version]
- Vallat, R.; Shah, V.D.; Redline, S.; Attia, P.; Walker, M.P. Broken sleep predicts hardened blood vessels. PLoS Biol. 2020, 18, e3000726. [Google Scholar] [CrossRef]
- Beiter, T.; Fragasso, A.; Hudemann, J.; Schild, M.; Steinacker, J.; Mooren, F.C.; Niess, A.M. Neutrophils release extracellular DNA traps in response to exercise. J. Appl. Physiol. 2014, 117, 325–333. [Google Scholar] [CrossRef] [Green Version]
- Besedovsky, L.; Lange, T.; Haack, M. The sleep-immune crosstalk in health and disease. Physiol. Rev. 2019, 99, 1325–1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Domínguez, F.; Fuster, V.; Fernández-Alvira, J.M.; Fernández-Friera, L.; López-Melgar, B.; Blanco-Rojo, R.; Fernández-Ortiz, A.; García-Pavía, P.; Sanz, J.; Mendiguren, J.M.; et al. Association of Sleep Duration and Quality With Subclinical Atherosclerosis. J. Am. Coll. Cardiol. 2019, 73, 134–144. [Google Scholar] [CrossRef]
- McAlpine, C.S.; Kiss, M.G.; Rattik, S.; He, S.; Vassalli, A.; Valet, C.; Anzai, A.; Chan, C.T.; Mindur, J.E.; Kahles, F.; et al. Sleep modulates haematopoiesis and protects against atherosclerosis. Nature 2019, 566, 383–387. [Google Scholar] [CrossRef]
- Tall, A.R.; Jelic, S. How broken sleep promotes cardiovascular disease. Nature 2019, 566, 329–330. [Google Scholar] [CrossRef]
- Christoffersson, G.; Vågesjö, E.; Pettersson, U.S.; Massena, S.; Nilsson, E.K.; Broman, J.-E.; Schiöth, H.B.; Benedict, C.; Phillipson, M. Acute sleep deprivation in healthy young men: Impact on population diversity and function of circulating neutrophils. Brain. Behav. Immun. 2014, 41, 162–172. [Google Scholar] [CrossRef]
- Adrover, J.M.; Aroca-Crevillén, A.; Crainiciuc, G.; Ostos, F.; Rojas-Vega, Y.; Rubio-Ponce, A.; Cilloniz, C.; Bonzón-Kulichenko, E.; Calvo, E.; Rico, D.; et al. Programmed “disarming” of the neutrophil proteome reduces the magnitude of inflammation. Nat. Immunol. 2020, 21, 135–144. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, G.; Manwani, D.; Mortha, A.; Xu, C.; Faith, J.J.; Burk, R.D.; Kunisaki, Y.; Jang, J.-E.; Scheiermann, C.; et al. Neutrophil ageing is regulated by the microbiome. Nature 2015, 525, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Said, E.A.; Al-Abri, M.A.; Al-Saidi, I.; Al-Balushi, M.S.; Al-Busaidi, J.Z.; Al-Reesi, I.; Koh, C.Y.; Idris, M.A.; Al-Jabri, A.A.; Habbal, O. Sleep deprivation alters neutrophil functions and levels of Th1-related chemokines and CD4+ T cells in the blood. Sleep Breath. 2019, 23, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Celermajer, D.S.; Adams, M.R.; Clarkson, P.; Robinson, J.; McCredie, R.; Donald, A.; Deanfield, J.E. Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. N. Engl. J. Med. 1996, 334, 150–154. [Google Scholar] [CrossRef]
- Howard, G.; Wagenknecht, L.E.; Burke, G.L.; Diez-Roux, A.; Evans, G.W.; McGovern, P.; Nieto, F.J.; Tell, G.S. Cigarette Smoking and Progression of AtherosclerosisThe Atherosclerosis Risk in Communities (ARIC) Study. JAMA 1998, 279, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Zhu, B.Q.; Sun, Y.P.; Sievers, R.E.; Isenberg, W.M.; Glantz, S.A.; Parmley, W.W. Passive smoking increases experimental atherosclerosis in cholesterol-fed rabbits. J. Am. Coll. Cardiol. 1993, 21, 225–232. [Google Scholar] [CrossRef] [Green Version]
- Raij, L.; DeMaster, E.G.; Jaimes, E.A. Cigarette smoke-induced endothelium dysfunction: Role of superoxide anion. J. Hypertens. 2001, 19, 891–897. [Google Scholar] [CrossRef]
- Pryor, W.A.; Stone, K. Oxidants in cigarette smoke. Radicals, hydrogen peroxide, peroxynitrate, and peroxynitrite. Ann. N. Y. Acad. Sci. 1993, 686, 12–18. [Google Scholar] [CrossRef]
- Jaimes, E.A.; DeMaster, E.G.; Tian, R.-X.; Raij, L. Stable compounds of cigarette smoke induce endothelial superoxide anion production via NADPH oxidase activation. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1031–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bermudez, E.A.; Rifai, N.; Buring, J.E.; Manson, J.E.; Ridker, P.M. Relation between markers of systemic vascular inflammation and smoking in women. Am. J. Cardiol. 2002, 89, 1117–1119. [Google Scholar] [CrossRef]
- Qiu, S.-L.; Zhang, H.; Tang, Q.; Bai, J.; He, Z.-Y.; Zhang, J.-Q.; Li, M.-H.; Deng, J.-M.; Liu, G.-N.; Zhong, X.-N. Neutrophil extracellular traps induced by cigarette smoke activate plasmacytoid dendritic cells. Thorax 2017, 72, 1084–1093. [Google Scholar] [CrossRef] [Green Version]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Luria, A.; Rhodes, C.; Raghu, H.; Lingampalli, N.; Sharpe, O.; Rada, B.; Sohn, D.H.; Robinson, W.H.; Sokolove, J. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatology 2017, 56, 644–653. [Google Scholar] [CrossRef] [Green Version]
- Hosseinzadeh, A.; Thompson, P.R.; Segal, B.H.; Urban, C.F. Nicotine induces neutrophil extracellular traps. J. Leukoc. Biol. 2016, 100, 1105–1112. [Google Scholar] [CrossRef]
- Li, T.; Zhang, Z.; Li, X.; Dong, G.; Zhang, M.; Xu, Z.; Yang, J. Neutrophil Extracellular Traps: Signaling Properties and Disease Relevance. Mediat. Inflamm. 2020, 2020, 9254087. [Google Scholar] [CrossRef]
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenböck, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ. Res. 2015, 116, 1182–1192. [Google Scholar] [CrossRef] [Green Version]
- Manfredi, A.A.; Rovere-Querini, P.; D’Angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol. Res. 2017, 123, 146–156. [Google Scholar] [CrossRef]
- Liu, J.; Yang, D.; Wang, X.; Zhu, Z.; Wang, T.; Ma, A.; Liu, P. Neutrophil extracellular traps and dsDNA predict outcomes among patients with ST-elevation myocardial infarction. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef] [Green Version]
- Vallés, J.; Lago, A.; Santos, M.T.; Latorre, A.M.; Tembl, J.I.; Salom, J.B.; Nieves, C.; Moscardó, A. Neutrophil extracellular traps are increased in patients with acute ischemic stroke: Prognostic significance. Thromb. Haemost. 2017, 117, 1919–1929. [Google Scholar] [CrossRef]
- Pertiwi, K.R.; Van Der Wal, A.C.; Pabittei, D.R.; Mackaaij, C.; Van Leeuwen, M.B.; Li, X.; De Boer, O.J. Neutrophil Extracellular Traps Participate in All Different Types of Thrombotic and Haemorrhagic Complications of Coronary Atherosclerosis. Thromb. Haemost. 2018, 118, 1078–1087. [Google Scholar] [CrossRef]
- Döring, Y.; Libby, P.; Soehnlein, O. Neutrophil Extracellular Traps Participate in Cardiovascular Diseases: Recent Experimental and Clinical Insights. Circ. Res. 2020, 126, 1228–1241. [Google Scholar] [CrossRef]
- Thålin, C.; Hisada, Y.; Lundström, S.; Mackman, N.; Wallén, H. Neutrophil Extracellular Traps: Villains and Targets in Arterial, Venous, and Cancer-Associated Thrombosis. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 1724–1738. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Identified Target | Technique | Methodology | Dye | Sample | Strengths | Weaknesses | Ref. |
|---|---|---|---|---|---|---|---|
| NETs | Spinning disk confocal microscopy | Live-cell imaging to visualize morphology, dynamics, behavior of NETs and interactions between NET formation and live pathogens | Fluorescent Bacteria and NET markers | Animal Model | Evaluation of NET formation in situ and in vivo Allows NET dynamics evaluation and kinetic studies | Invasive technique that can induce unwanted inflammatory responses Poor temporal resolution Cardiac and Respiratory movement might compromise imaging Expensive, costly, time-consuming and technically challenging Long acquisition times | [41,42]. |
| NETs | Multi-/ Two-photon microscopy | Live-cell imaging to visualize morphology, dynamics, behavior of NETs and interactions between NET formation and live pathogens | Fluorescent Bacteria and NET markers | Animal Model | Evaluation of NET formation in situ and in vivo Allows NET dynamics evaluation and kinetic studies | Invasive technique that can induce unwanted inflammatory responses Poor temporal resolution Cardiac and respiratory movement might compromise imaging Expensive, costly, time-consuming and technically challenging Long acquisition times | [19,42,43,44]. |
| NETs | Intravital Microscopy | Live-cell imaging to visualize morphology, dynamics, behavior of NETs and interactions between NET formation and live pathogens | Fluorescent Bacteria and NET markers | Animal Model | Evaluation of NET formation in situ and in vivo Allows NET dynamics evaluation and kinetic studies | Invasive Technique that can induce unwanted inflammatory responses Poor temporal resolution Cardiac and Respiratory movement might compromise imaging Expensive, costly, time-consuming and technically challenging Long acquisition times | [41,42]. |
| Soluble NET remnants cf-DNA | Fluorescent Reader | PicoGreen® | Plasma Serum Fluids | Objective Quantitative Can potentially detect in vivo NET formation | Low Specificity Cell-free DNA can reflect lytic cell death mechanisms (Necrosis) | [45,46]. | |
| DNA-MPO Complexes | ELISA | Plasma Serum Fluids | Objective Quantitative | Low Specificity MPO (high-cationic nature) can bind to negatively charged cf-DNA MPO can reflect on neutrophil and macrophage activation not related to NET release | [47,48,49,50] | ||
| DNA-NE Complexes | ELISA | Plasma Serum Fluids | Objective Quantitative | NE can reflect on neutrophil activation and degranulation not related to NET release | [47,48,50]. | ||
| Citrullinated Histone H3 | Refined ELISA | Human Plasma | Objective Quantitative Citrullination is the gold hallmark of NET formation | No consensus on a standard ELISA assay to monitor NETosis | [51,52]. | ||
| MPO citH3 | Flow Cytometry | Detection of NET components attached to the neutrophil cell surface | Fluorescent antibodies against MPO and citrullinated histones | Primary cells Cell lines | Objective Unbiased Automated Can be combined with cell-sorting techniques | Does not detect citH3-independent events Potentially able to report live NETosis events but misses lysed cells that underwent NETosis previously | [46,53]. |
| DNA backbone | IF | Co-localization of extracellular DNA, neutrophil markers, neutrophil-derived granule proteins and modified histones | DNA-intercalating dyes (DAPI, PI, SYTOX® Green) | Tissue Sections | Can differentiate between necrosis and NETosis | Biased selection of the field of view might affect results Clump of NETs derived from multiple cells count as a single event | [26,54,55,56,57,58,59,60,61] |
| Neutrophils | IF | Co-localization of extracellular DNA, neutrophil markers, neutrophil-derived granule proteins and modified histones | Fluorescent antibodies against Ly6G (in mice) and CD66b, NE or CD177 (in humans) | Tissue Sections | Can differentiate between necrosis and NETosis | Biased selection of the field of view might affect results Clump of NETs derived from multiple cells count as a single event | [26,54,55,56,57,58,59,60,61] |
| Neutrophilderived Granule proteins | IF | Co-localization of extracellular DNA, neutrophil markers, neutrophil-derived granule proteins and modified histones | Fluorescent antibodies against MPO, NE or LL37 | Tissue Sections | Can differentiate between necrosis and NETosis | Biased selection of the field of view might affect results Clump of NETs derived from multiple cells count as a single event | [26,54,55,56,57,58,59,60,61] |
| Citrullinated Histone | IF | Co-localization of extracellular DNA, neutrophil markers, neutrophil-derived granule proteins and modified histones | Fluorescent antibodies against citH3, citH4 | Tissue Sections | Can differentiate between necrosis and NETosis | Biased selection of the field of view might affect results Clump of NETs derived from multiple cells count as a single event | [26,54,55,56,57,58,59,60,61] |
| NETs | IF | NET detection reagents that are based on fluorescent, chromatin-binding polymers | PlaNET reagents | Primary cells | High Specificity Stain NETs only in activated cells Undetectable in necrotic cells | Non-specific signals reported Not suitable for NETosis kinetic studies | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pérez-Olivares, L.; Soehnlein, O. Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease. Cells 2021, 10, 1985. https://doi.org/10.3390/cells10081985
Pérez-Olivares L, Soehnlein O. Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease. Cells. 2021; 10(8):1985. https://doi.org/10.3390/cells10081985
Chicago/Turabian StylePérez-Olivares, Laura, and Oliver Soehnlein. 2021. "Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease" Cells 10, no. 8: 1985. https://doi.org/10.3390/cells10081985
APA StylePérez-Olivares, L., & Soehnlein, O. (2021). Contemporary Lifestyle and Neutrophil Extracellular Traps: An Emerging Link in Atherosclerosis Disease. Cells, 10(8), 1985. https://doi.org/10.3390/cells10081985