
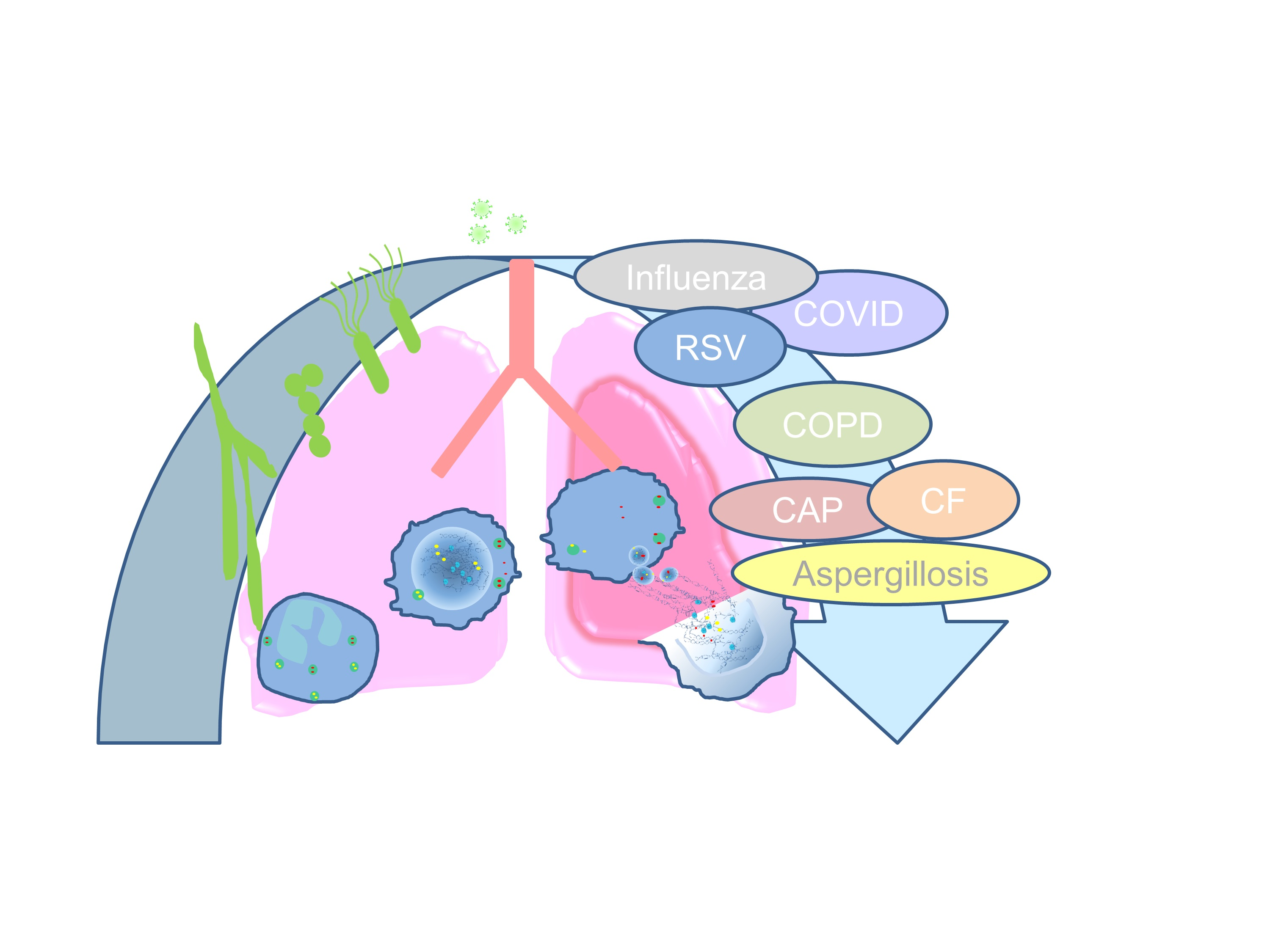
A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression?


Abstract
:
1. Introduction
2. The Role of NETs in Virus-Induced Lung Diseases
2.1. COVID-19
2.2. Respiratory Syncitial Virus
2.3. Influenza
3. The Role of NETs in Bacteria-Associated Lung Diseases
3.1. Streptococcus Pneumoniae
3.2. Staphylococcus Aureus
3.3. Cystic Fibrosis and Pseudomonas Aeruginosa
3.4. Chronic Obstructive Pulmonary Disease
4. Pathogenic Fungal Lung Infection—Aspergillosis
5. NET-Targeting Therapies
5.1. DNase1
5.2. Histones
5.3. Neutrophil Elastase
5.4. Other Treatments
6. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Urban, C.F.; Reichard, U.; Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell. Microbiol. 2006, 8, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, M.; Shan, Q.; D’Ortona, S.; Maurer, R.; Mitchell, R.; Olesen, H.; Thiel, S.; Huebner, J.; Gadjeva, M. Cystic fibrosis sputum DNA has NETosis characteristics and neutrophil extracellular trap release is regulated by macrophage migration-inhibitory factor. J. Innate Immun. 2014, 6, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [Green Version]
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77. [Google Scholar] [CrossRef]
- Stojkov, D.; Amini, P.; Oberson, K.; Sokollik, C.; Duppenthaler, A.; Simon, H.U.; Yousefi, S. ROS and glutathionylation balance cytoskeletal dynamics in neutrophil extracellular trap formation. J. Cell Biol. 2017, 216, 4073–4090. [Google Scholar] [CrossRef]
- Amini, P.; Stojkov, D.; Felser, A.; Jackson, C.B.; Courage, C.; Schaller, A.; Gelman, L.; Soriano, M.E.; Nuoffer, J.M.; Scorrano, L.; et al. Neutrophil extracellular trap formation requires OPA1-dependent glycolytic ATP production. Nat. Commun. 2018, 9, 2958. [Google Scholar] [CrossRef] [Green Version]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Metzler, K.D.; Goosmann, C.; Lubojemska, A.; Zychlinsky, A.; Papayannopoulos, V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell Rep. 2014, 8, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Diaz-Godinez, C.; Fonseca, Z.; Nequiz, M.; Laclette, J.P.; Rosales, C.; Carrero, J.C. Entamoeba histolytica Trophozoites Induce a Rapid Non-classical NETosis Mechanism Independent of NOX2-Derived Reactive Oxygen Species and PAD4 Activity. Front. Cell Infect. Microbiol. 2018, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Witsch, T.; Farley, K.; Gallant, M.; Remold-O’Donnell, E.; Wagner, D.D. Neutrophil elastase-deficient mice form neutrophil extracellular traps in an experimental model of deep vein thrombosis. J. Thromb. Haemost. 2016, 14, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Avondt, K.; van der Linden, M.; Naccache, P.H.; Egan, D.A.; Meyaard, L. Signal Inhibitory Receptor on Leukocytes-1 Limits the Formation of Neutrophil Extracellular Traps, but Preserves Intracellular Bacterial Killing. J. Immunol. 2016, 196, 3686–3694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amulic, B.; Knackstedt, S.L.; Abu Abed, U.; Deigendesch, N.; Harbort, C.J.; Caffrey, B.E.; Brinkmann, V.; Heppner, F.L.; Hinds, P.W.; Zychlinsky, A. Cell-Cycle Proteins Control Production of Neutrophil Extracellular Traps. Dev. Cell 2017, 43, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469. [Google Scholar] [CrossRef]
- McDonald, B.; Urrutia, R.; Yipp, B.G.; Jenne, C.N.; Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 2012, 12, 324–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilsczek, F.H.; Salina, D.; Poon, K.K.; Fahey, C.; Yipp, B.G.; Sibley, C.D.; Robbins, S.M.; Green, F.H.; Surette, M.G.; Sugai, M.; et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to Staphylococcus aureus. J. Immunol. 2010, 185, 7413–7425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Yipp, B.G.; Petri, B.; Salina, D.; Jenne, C.N.; Scott, B.N.; Zbytnuik, L.D.; Pittman, K.; Asaduzzaman, M.; Wu, K.; Meijndert, H.C.; et al. Infection-induced NETosis is a dynamic process involving neutrophil multitasking in vivo. Nat. Med. 2012, 18, 1386–1393. [Google Scholar] [CrossRef] [Green Version]
- Roos, D.; Voetman, A.A.; Meerhof, L.J. Functional activity of enucleated human polymorphonuclear leukocytes. J. Cell Biol. 1983, 97, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, N.; Douda, D.N.; Bruggemann, T.R.; Ricklefs, I.; Duvall, M.G.; Abdulnour, R.E.; Martinod, K.; Tavares, L.; Wang, X.; Cernadas, M.; et al. Neutrophil cytoplasts induce TH17 differentiation and skew inflammation toward neutrophilia in severe asthma. Sci. Immunol. 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Branzk, N.; Lubojemska, A.; Hardison, S.E.; Wang, Q.; Gutierrez, M.G.; Brown, G.D.; Papayannopoulos, V. Neutrophils sense microbe size and selectively release neutrophil extracellular traps in response to large pathogens. Nat. Immunol. 2014, 15, 1017–1025. [Google Scholar] [CrossRef] [Green Version]
- Guimaraes-Costa, A.B.; Nascimento, M.T.; Froment, G.S.; Soares, R.P.; Morgado, F.N.; Conceicao-Silva, F.; Saraiva, E.M. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc. Natl. Acad. Sci. USA 2009, 106, 6748–6753. [Google Scholar] [CrossRef] [Green Version]
- McCormick, A.; Heesemann, L.; Wagener, J.; Marcos, V.; Hartl, D.; Loeffler, J.; Heesemann, J.; Ebel, F. NETs formed by human neutrophils inhibit growth of the pathogenic mold Aspergillus fumigatus. Microbes Infect. 2010, 12, 928–936. [Google Scholar] [CrossRef]
- Saitoh, T.; Komano, J.; Saitoh, Y.; Misawa, T.; Takahama, M.; Kozaki, T.; Uehata, T.; Iwasaki, H.; Omori, H.; Yamaoka, S.; et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe 2012, 12, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Wong, C.H.; Zemp, F.J.; McDonald, B.; Rahman, M.M.; Forsyth, P.A.; McFadden, G.; Kubes, P. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 2013, 13, 169–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS ONE 2012, 7, e32366. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, X.; Pelayo, R.; Monestier, M.; Ammollo, C.T.; Semeraro, F.; Taylor, F.B.; Esmon, N.L.; Lupu, F.; Esmon, C.T. Extracellular histones are major mediators of death in sepsis. Nat. Med. 2009, 15, 1318–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrams, S.T.; Zhang, N.; Manson, J.; Liu, T.; Dart, C.; Baluwa, F.; Wang, S.S.; Brohi, K.; Kipar, A.; Yu, W.; et al. Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 2013, 187, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brill, A.; Fuchs, T.A.; Chauhan, A.K.; Yang, J.J.; De Meyer, S.F.; Kollnberger, M.; Wakefield, T.W.; Lammle, B.; Massberg, S.; Wagner, D.D. von Willebrand factor-mediated platelet adhesion is critical for deep vein thrombosis in mouse models. Blood 2011, 117, 1400–1407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etulain, J.; Martinod, K.; Wong, S.L.; Cifuni, S.M.; Schattner, M.; Wagner, D.D. P-selectin promotes neutrophil extracellular trap formation in mice. Blood 2015, 126, 242–246. [Google Scholar] [CrossRef] [Green Version]
- Rossaint, J.; Kuhne, K.; Skupski, J.; Van Aken, H.; Looney, M.R.; Hidalgo, A.; Zarbock, A. Directed transport of neutrophil-derived extracellular vesicles enables platelet-mediated innate immune response. Nat. Commun. 2016, 7, 13464. [Google Scholar] [CrossRef]
- Von Bruhl, M.L.; Stark, K.; Steinhart, A.; Chandraratne, S.; Konrad, I.; Lorenz, M.; Khandoga, A.; Tirniceriu, A.; Coletti, R.; Kollnberger, M.; et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J. Exp. Med. 2012, 209, 819–835. [Google Scholar] [CrossRef]
- Rossaint, J.; Herter, J.M.; Van Aken, H.; Napirei, M.; Doring, Y.; Weber, C.; Soehnlein, O.; Zarbock, A. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood 2014, 123, 2573–2584. [Google Scholar] [CrossRef]
- Sporn, L.A.; Marder, V.J.; Wagner, D.D. Inducible secretion of large, biologically potent von Willebrand factor multimers. Cell 1986, 46, 185–190. [Google Scholar] [CrossRef]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G activates protease-activated receptor-4 in human platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef] [Green Version]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef]
- Lazzaretto, B.; Fadeel, B. Intra- and Extracellular Degradation of Neutrophil Extracellular Traps by Macrophages and Dendritic Cells. J. Immunol. 2019, 203, 2276–2290. [Google Scholar] [CrossRef] [Green Version]
- Apel, F.; Andreeva, L.; Knackstedt, L.S.; Streeck, R.; Frese, C.K.; Goosmann, C.; Hopfner, K.P.; Zychlinsky, A. The cytosolic DNA sensor cGAS recognizes neutrophil extracellular traps. Sci. Signal. 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Kusunoki, Y.; Miyoshi, A.; Nishio, S.; Tomaru, U.; Atsumi, T.; Ishizu, A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J. Autoimmun. 2016, 67, 19–28. [Google Scholar] [CrossRef]
- Doster, R.S.; Rogers, L.M.; Gaddy, J.A.; Aronoff, D.M. Macrophage Extracellular Traps: A Scoping Review. J. Innate Immun. 2018, 10, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Knopf, J.; Leppkes, M.; Schett, G.; Herrmann, M.; Munoz, L.E. Aggregated NETs Sequester and Detoxify Extracellular Histones. Front. Immunol. 2019, 10, 2176. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Schauer, C.; Czegley, C.; Kling, L.; Petru, L.; Schmid, B.; Weidner, D.; Reinwald, C.; Biermann, M.H.C.; Blunder, S.; et al. Aggregated neutrophil extracellular traps resolve inflammation by proteolysis of cytokines and chemokines and protection from antiproteases. FASEB J. 2019, 33, 1401–1414. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudit, G.Y.; Crackower, M.A.; Backx, P.H.; Penninger, J.M. The role of ACE2 in cardiovascular physiology. Trends Cardiovasc. Med. 2003, 13, 93–101. [Google Scholar] [CrossRef]
- Rafii, S.; Butler, J.M.; Ding, B.S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [Green Version]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Dassler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Barton, L.M.; Duval, E.J.; Stroberg, E.; Ghosh, S.; Mukhopadhyay, S. COVID-19 Autopsies, Oklahoma, USA. Am. J. Clin. Pathol. 2020, 153, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, C.Y.; Zhou, P.; Yue, H.; Du, R. Histopathologic Changes and SARS-CoV-2 Immunostaining in the Lung of a Patient With COVID-19. Ann. Intern. Med. 2020, 173, 324. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk Factors Associated With Acute Respiratory Distress Syndrome and Death in Patients With Coronavirus Disease 2019 Pneumonia in Wuhan, China. JAMA Intern. Med. 2020, 180, 934–943. [Google Scholar] [CrossRef] [Green Version]
- Zuo, Y.; Kanthi, Y.; Knight, J.S.; Kim, A.H.J. The interplay between neutrophils, complement, and microthrombi in COVID-19. Best Pr. Res. Clin. Rheumatol. 2021, 35, 101661. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetite, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Belouzard, S.; Madu, I.; Whittaker, G.R. Elastase-mediated activation of the severe acute respiratory syndrome coronavirus spike protein at discrete sites within the S2 domain. J. Biol. Chem. 2010, 285, 22758–22763. [Google Scholar] [CrossRef] [Green Version]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Joppich, M.; Hoffknecht, M.L.; Gold, C.; Engel, A.; Polewka, V.; Muenchhoff, M.; et al. Vascular neutrophilic inflammation and immunothrombosis distinguish severe COVID-19 from influenza pneumonia. J. Thromb. Haemost. 2021, 19, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Invest. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Lo, M.W.; Kemper, C.; Woodruff, T.M. COVID-19: Complement, Coagulation, and Collateral Damage. J. Immunol. 2020, 205, 1488–1495. [Google Scholar] [CrossRef]
- Ramlall, V.; Thangaraj, P.M.; Meydan, C.; Foox, J.; Butler, D.; Kim, J.; May, B.; De Freitas, J.K.; Glicksberg, B.S.; Mason, C.E.; et al. Immune complement and coagulation dysfunction in adverse outcomes of SARS-CoV-2 infection. Nat. Med. 2020, 26, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Nicolai, L.; Leunig, A.; Brambs, S.; Kaiser, R.; Weinberger, T.; Weigand, M.; Muenchhoff, M.; Hellmuth, J.C.; Ledderose, S.; Schulz, H.; et al. Immunothrombotic Dysregulation in COVID-19 Pneumonia Is Associated With Respiratory Failure and Coagulopathy. Circulation 2020, 142, 1176–1189. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arter. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pieterse, E.; Rother, N.; Yanginlar, C.; Gerretsen, J.; Boeltz, S.; Munoz, L.E.; Herrmann, M.; Pickkers, P.; Hilbrands, L.B.; van der Vlag, J. Cleaved N-terminal histone tails distinguish between NADPH oxidase (NOX)-dependent and NOX-independent pathways of neutrophil extracellular trap formation. Ann. Rheum. Dis. 2018, 77, 1790–1798. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolhnikoff, M.; Duarte-Neto, A.N.; de Almeida Monteiro, R.A.; da Silva, L.F.F.; de Oliveira, E.P.; Saldiva, P.H.N.; Mauad, T.; Negri, E.M. Pathological evidence of pulmonary thrombotic phenomena in severe COVID-19. J. Thromb. Haemost. 2020, 18, 1517–1519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240. [Google Scholar] [CrossRef]
- Mejias, A.; Chavez-Bueno, S.; Jafri, H.S.; Ramilo, O. Respiratory syncytial virus infections: Old challenges and new opportunities. Pediatr. Infect. Dis. J. 2005, 24, S189–S196. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, P.A.; Bueno, S.M.; Carreno, L.J.; Riedel, C.A.; Kalergis, A.M. Respiratory syncytial virus infection and immunity. Rev. Med. Virol. 2012, 22, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Lay, M.K.; Gonzalez, P.A.; Leon, M.A.; Cespedes, P.F.; Bueno, S.M.; Riedel, C.A.; Kalergis, A.M. Advances in understanding respiratory syncytial virus infection in airway epithelial cells and consequential effects on the immune response. Microbes Infect. 2013, 15, 230–242. [Google Scholar] [CrossRef]
- Jaovisidha, P.; Peeples, M.E.; Brees, A.A.; Carpenter, L.R.; Moy, J.N. Respiratory syncytial virus stimulates neutrophil degranulation and chemokine release. J. Immunol. 1999, 163, 2816–2820. [Google Scholar]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.; de Graaff, P.M.; Kimpen, J.L.; Koenderman, L. Respiratory syncytial virus inhibits granulocyte apoptosis through a phosphatidylinositol 3-kinase and NF-kappaB-dependent mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar] [CrossRef] [Green Version]
- Cortjens, B.; de Boer, O.J.; de Jong, R.; Antonis, A.F.; Sabogal Pineros, Y.S.; Lutter, R.; van Woensel, J.B.; Bem, R.A. Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 2016, 238, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef]
- Muraro, S.P.; De Souza, G.F.; Gallo, S.W.; Da Silva, B.K.; De Oliveira, S.D.; Vinolo, M.A.R.; Saraiva, E.M.; Porto, B.N. Respiratory Syncytial Virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018, 8, 14166. [Google Scholar] [CrossRef]
- Rewar, S.; Mirdha, D.; Rewar, P. Treatment and Prevention of Pandemic H1N1 Influenza. Ann. Glob. Health 2015, 81, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Paules, C.; Subbarao, K. Influenza. Lancet 2017, 390, 697–708. [Google Scholar] [CrossRef]
- Perrone, L.A.; Plowden, J.K.; Garcia-Sastre, A.; Katz, J.M.; Tumpey, T.M. H5N1 and 1918 pandemic influenza virus infection results in early and excessive infiltration of macrophages and neutrophils in the lungs of mice. PLoS Pathog. 2008, 4, e1000115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tate, M.D.; Deng, Y.M.; Jones, J.E.; Anderson, G.P.; Brooks, A.G.; Reading, P.C. Neutrophils ameliorate lung injury and the development of severe disease during influenza infection. J. Immunol. 2009, 183, 7441–7450. [Google Scholar] [CrossRef]
- Tate, M.D.; Brooks, A.G.; Reading, P.C. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 2008, 9, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Feng, K.; Wang, Y.C.; Mei, J.J.; Ning, R.T.; Zheng, H.W.; Wang, J.J.; Worthen, G.S.; Wang, X.; Song, J.; et al. Critical role of CXCL4 in the lung pathogenesis of influenza (H1N1) respiratory infection. Mucosal Immunol. 2017, 10, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Aloe, C.; Wilson, N.; Bozinovski, S. G-CSFR antagonism reduces neutrophilic inflammation during pneumococcal and influenza respiratory infections without compromising clearance. Sci. Rep. 2019, 9, 17732. [Google Scholar] [CrossRef]
- Wareing, M.D.; Shea, A.L.; Inglis, C.A.; Dias, P.B.; Sarawar, S.R. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral. Immunol. 2007, 20, 369–378. [Google Scholar] [CrossRef]
- Fujisawa, H. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J. Virol. 2008, 82, 2772–2783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malachowa, N.; Freedman, B.; Sturdevant, D.E.; Kobayashi, S.D.; Nair, V.; Feldmann, F.; Starr, T.; Steele-Mortimer, O.; Kash, J.C.; Taubenberger, J.K.; et al. Differential Ability of Pandemic and Seasonal H1N1 Influenza A Viruses To Alter the Function of Human Neutrophils. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.; Klauschen, F.; Kuchen, S.; Germain, R.N. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell 2013, 154, 197–212. [Google Scholar] [CrossRef] [Green Version]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef] [Green Version]
- Rudd, J.M.; Pulavendran, S.; Ashar, H.K.; Ritchey, J.W.; Snider, T.A.; Malayer, J.R.; Marie, M.; Chow, V.T.K.; Narasaraju, T. Neutrophils Induce a Novel Chemokine Receptors Repertoire During Influenza Pneumonia. Front. Cell. Infect. Microbiol. 2019, 9, 108. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.L.Y.; Nicholls, J.M.; Peiris, J.S.M.; Lau, Y.L.; Chan, M.C.W.; Chan, R.W.Y. Host DNA released by NETosis in neutrophils exposed to seasonal H1N1 and highly pathogenic H5N1 influenza viruses. Respir. Res. 2020, 21, 160. [Google Scholar] [CrossRef]
- Zhu, L.; Liu, L.; Zhang, Y.; Pu, L.; Liu, J.; Li, X.; Chen, Z.; Hao, Y.; Wang, B.; Han, J.; et al. High Level of Neutrophil Extracellular Traps Correlates With Poor Prognosis of Severe Influenza A Infection. J. Infect. Dis. 2018, 217, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Salvatore, M.; Garcia-Sastre, A.; Ruchala, P.; Lehrer, R.I.; Chang, T.; Klotman, M.E. alpha-Defensin inhibits influenza virus replication by cell-mediated mechanism(s). J. Infect. Dis. 2007, 196, 835–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandell, L.A. Community-acquired pneumonia: An overview. Postgrad. Med. 2015, 127, 607–615. [Google Scholar] [CrossRef]
- Liu, N.; Zheng, Z.; Chen, P.; Hou, P.; Wang, X.; Li, H.; Chen, R. Detection of aspiration of nasopharyngeal secretion and the relationship between the aspiration of nasopharyngeal secretion and the incidence of pneumonia. Zhonghua Jie He He Hu Xi Za Zhi 2015, 38, 511–515. [Google Scholar] [PubMed]
- Standish, A.J.; Weiser, J.N. Human neutrophils kill Streptococcus pneumoniae via serine proteases. J. Immunol. 2009, 183, 2602–2609. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yamaguchi, M.; Terao, Y.; Hamada, S.; Ooshima, T.; Kawabata, S. alpha-Enolase of Streptococcus pneumoniae induces formation of neutrophil extracellular traps. J. Biol. Chem. 2012, 287, 10472–10481. [Google Scholar] [CrossRef] [Green Version]
- Hyams, C.; Camberlein, E.; Cohen, J.M.; Bax, K.; Brown, J.S. The Streptococcus pneumoniae capsule inhibits complement activity and neutrophil phagocytosis by multiple mechanisms. Infect. Immun. 2010, 78, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Moorthy, A.N.; Rai, P.; Jiao, H.; Wang, S.; Tan, K.B.; Qin, L.; Watanabe, H.; Zhang, Y.; Teluguakula, N.; Chow, V.T. Capsules of virulent pneumococcal serotypes enhance formation of neutrophil extracellular traps during in vivo pathogenesis of pneumonia. Oncotarget 2016, 7, 19327–19340. [Google Scholar] [CrossRef] [Green Version]
- Ebrahimi, F.; Giaglis, S.; Hahn, S.; Blum, C.A.; Baumgartner, C.; Kutz, A.; van Breda, S.V.; Mueller, B.; Schuetz, P.; Christ-Crain, M.; et al. Markers of neutrophil extracellular traps predict adverse outcome in community-acquired pneumonia: Secondary analysis of a randomised controlled trial. Eur. Respir. J. 2018, 51. [Google Scholar] [CrossRef] [PubMed]
- Beiter, K.; Wartha, F.; Albiger, B.; Normark, S.; Zychlinsky, A.; Henriques-Normark, B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr. Biol. 2006, 16, 401–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Kuang, Z.; Wilson, B.A.; Lau, G.W. Competence-independent activity of pneumococcal EndA [corrected] mediates degradation of extracellular dna and nets and is important for virulence. PLoS ONE 2013, 8, e70363. [Google Scholar] [CrossRef] [Green Version]
- Jhelum, H.; Sori, H.; Sehgal, D. A novel extracellular vesicle-associated endodeoxyribonuclease helps Streptococcus pneumoniae evade neutrophil extracellular traps and is required for full virulence. Sci. Rep. 2018, 8, 7985. [Google Scholar] [CrossRef] [PubMed]
- Janusz, M.J.; Doherty, N.S. Degradation of cartilage matrix proteoglycan by human neutrophils involves both elastase and cathepsin G. J. Immunol. 1991, 146, 3922–3928. [Google Scholar] [PubMed]
- Taylor, S.; Dirir, O.; Zamanian, R.T.; Rabinovitch, M.; Thompson, A.A.R. The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front. Med. 2018, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Domon, H.; Oda, M.; Maekawa, T.; Nagai, K.; Takeda, W.; Terao, Y. Streptococcus pneumoniae disrupts pulmonary immune defence via elastase release following pneumolysin-dependent neutrophil lysis. Sci. Rep. 2016, 6, 38013. [Google Scholar] [CrossRef] [PubMed]
- Mizikova, I.; Ruiz-Camp, J.; Steenbock, H.; Madurga, A.; Vadasz, I.; Herold, S.; Mayer, K.; Seeger, W.; Brinckmann, J.; Morty, R.E. Collagen and elastin cross-linking is altered during aberrant late lung development associated with hyperoxia. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1145–L1158. [Google Scholar] [CrossRef] [Green Version]
- Polverino, E.; Rosales-Mayor, E.; Dale, G.E.; Dembowsky, K.; Torres, A. The Role of Neutrophil Elastase Inhibitors in Lung Diseases. Chest 2017, 152, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiedow, O.; Meyer-Hoffert, U. Neutrophil serine proteases: Potential key regulators of cell signalling during inflammation. J. Intern. Med. 2005, 257, 319–328. [Google Scholar] [CrossRef]
- Van den Berg, C.W.; Tambourgi, D.V.; Clark, H.W.; Hoong, S.J.; Spiller, O.B.; McGreal, E.P. Mechanism of neutrophil dysfunction: Neutrophil serine proteases cleave and inactivate the C5a receptor. J. Immunol. 2014, 192, 1787–1795. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Nagai, K.; Maekawa, T.; Oda, M.; Yonezawa, D.; Takeda, W.; Hiyoshi, T.; Tamura, H.; Yamaguchi, M.; Kawabata, S.; et al. Neutrophil Elastase Subverts the Immune Response by Cleaving Toll-Like Receptors and Cytokines in Pneumococcal Pneumonia. Front. Immunol. 2018, 9, 732. [Google Scholar] [CrossRef] [Green Version]
- Clancy, D.M.; Sullivan, G.P.; Moran, H.B.T.; Henry, C.M.; Reeves, E.P.; McElvaney, N.G.; Lavelle, E.C.; Martin, S.J. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep. 2018, 22, 2937–2950. [Google Scholar] [CrossRef] [Green Version]
- Domon, H.; Maekawa, T.; Isono, T.; Furuta, K.; Kaito, C.; Terao, Y. Proteolytic cleavage of HLA class II by human neutrophil elastase in pneumococcal pneumonia. Sci. Rep. 2021, 11, 2432. [Google Scholar] [CrossRef]
- Jackson, P.L.; Xu, X.; Wilson, L.; Weathington, N.M.; Clancy, J.P.; Blalock, J.E.; Gaggar, A. Human neutrophil elastase-mediated cleavage sites of MMP-9 and TIMP-1: Implications to cystic fibrosis proteolytic dysfunction. Mol. Med. 2010, 16, 159–166. [Google Scholar] [CrossRef]
- Wartha, F.; Beiter, K.; Albiger, B.; Fernebro, J.; Zychlinsky, A.; Normark, S.; Henriques-Normark, B. Capsule and D-alanylated lipoteichoic acids protect Streptococcus pneumoniae against neutrophil extracellular traps. Cell. Microbiol. 2007, 9, 1162–1171. [Google Scholar] [CrossRef]
- Self, W.H.; Wunderink, R.G.; Williams, D.J.; Zhu, Y.; Anderson, E.J.; Balk, R.A.; Fakhran, S.S.; Chappell, J.D.; Casimir, G.; Courtney, D.M.; et al. Staphylococcus aureus Community-acquired Pneumonia: Prevalence, Clinical Characteristics, and Outcomes. Clin. Infect. Dis. 2016, 63, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.S.; Greenberg, J.A.; McNulty, M.C.; Gregg, K.S.; Riddell, J.t.; Mangino, J.E.; Weber, D.M.; Hebert, C.L.; Marzec, N.S.; Barron, M.A.; et al. Bacterial and viral co-infections complicating severe influenza: Incidence and impact among 507 U.S. patients, 2013–2014. J. Clin. Virol. 2016, 80, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Lowy, F.D. Staphylococcus aureus infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.R.; Balasubramanian, D.; Tam, K.; Askenazi, M.; Copin, R.; Shopsin, B.; Torres, V.J.; Ueberheide, B.M. Using Quantitative Spectrometry to Understand the Influence of Genetics and Nutritional Perturbations On the Virulence Potential of Staphylococcus aureus. Mol. Cell. Proteom. 2017, 16, S15–S28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Periasamy, S.; Joo, H.S.; Duong, A.C.; Bach, T.H.; Tan, V.Y.; Chatterjee, S.S.; Cheung, G.Y.; Otto, M. How Staphylococcus aureus biofilms develop their characteristic structure. Proc. Natl. Acad. Sci. USA 2012, 109, 1281–1286. [Google Scholar] [CrossRef] [Green Version]
- Grousd, J.A.; Rich, H.E.; Alcorn, J.F. Host-Pathogen Interactions in Gram-Positive Bacterial Pneumonia. Clin. Microbiol. Rev. 2019, 32. [Google Scholar] [CrossRef]
- Nizet, V. Understanding how leading bacterial pathogens subvert innate immunity to reveal novel therapeutic targets. J. Allergy Clin. Immunol. 2007, 120, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; van Kessel, K.P.; van Strijp, J.A. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Higgins, J.; Loughman, A.; van Kessel, K.P.; van Strijp, J.A.; Foster, T.J. Clumping factor A of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol. Lett. 2006, 258, 290–296. [Google Scholar] [CrossRef] [Green Version]
- Agniswamy, J.; Lei, B.; Musser, J.M.; Sun, P.D. Insight of host immune evasion mediated by two variants of group a Streptococcus Mac protein. J. Biol. Chem. 2004, 279, 52789–52796. [Google Scholar] [CrossRef] [Green Version]
- Cardot-Martin, E.; Casalegno, J.S.; Badiou, C.; Dauwalder, O.; Keller, D.; Prevost, G.; Rieg, S.; Kern, W.V.; Cuerq, C.; Etienne, J.; et al. alpha-Defensins partially protect human neutrophils against Panton-Valentine leukocidin produced by Staphylococcus aureus. Lett. Appl. Microbiol. 2015, 61, 158–164. [Google Scholar] [CrossRef]
- Berends, E.T.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; von Kockritz-Blickwede, M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thammavongsa, V.; Missiakas, D.M.; Schneewind, O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science 2013, 342, 863–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefrancais, E.; Mallavia, B.; Zhuo, H.; Calfee, C.S.; Looney, M.R. Maladaptive role of neutrophil extracellular traps in pathogen-induced lung injury. Jci Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sultan, A.R.; Swierstra, J.W.; Lemmens-den Toom, N.A.; Snijders, S.V.; Hansenova Manaskova, S.; Verbon, A.; van Wamel, W.J.B. Production of Staphylococcal Complement Inhibitor (SCIN) and Other Immune Modulators during the Early Stages of Staphylococcus aureus Biofilm Formation in a Mammalian Cell Culture Medium. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.R.; Hoppenbrouwers, T.; Lemmens-den Toom, N.A.; Snijders, S.V.; van Neck, J.W.; Verbon, A.; de Maat, M.P.M.; van Wamel, W.J.B. During the Early Stages of Staphylococcus aureus Biofilm Formation, Induced Neutrophil Extracellular Traps Are Degraded by Autologous Thermonuclease. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Berends, E.T.M.; Chan, R.; Schwab, E.; Roy, S.; Sen, C.K.; Torres, V.J.; Wozniak, D.J. Staphylococcus aureus biofilms release leukocidins to elicit extracellular trap formation and evade neutrophil-mediated killing. Proc. Natl. Acad. Sci. USA 2018, 115, 7416–7421. [Google Scholar] [CrossRef] [Green Version]
- Herzog, S.; Dach, F.; de Buhr, N.; Niemann, S.; Schlagowski, J.; Chaves-Moreno, D.; Neumann, C.; Goretzko, J.; Schwierzeck, V.; Mellmann, A.; et al. High Nuclease Activity of Long Persisting Staphylococcus aureus Isolates Within the Airways of Cystic Fibrosis Patients Protects Against NET-Mediated Killing. Front. Immunol. 2019, 10, 2552. [Google Scholar] [CrossRef] [Green Version]
- Knowles, M.R.; Durie, P.R. What is cystic fibrosis? N. Engl. J. Med. 2002, 347, 439–442. [Google Scholar] [CrossRef]
- Regamey, N.; Tsartsali, L.; Hilliard, T.N.; Fuchs, O.; Tan, H.L.; Zhu, J.; Qiu, Y.S.; Alton, E.W.; Jeffery, P.K.; Bush, A.; et al. Distinct patterns of inflammation in the airway lumen and bronchial mucosa of children with cystic fibrosis. Thorax 2012, 67, 164–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullen, L.; McClean, S. Bacterial Adaptation during Chronic Respiratory Infections. Pathogens 2015, 4, 66–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, D.G.; Floyd, M.; Winn, M.; Moskowitz, S.M.; Rada, B. NET formation induced by Pseudomonas aeruginosa cystic fibrosis isolates measured as release of myeloperoxidase-DNA and neutrophil elastase-DNA complexes. Immunol. Lett. 2014, 160, 186–194. [Google Scholar] [CrossRef]
- Yoo, D.G.; Winn, M.; Pang, L.; Moskowitz, S.M.; Malech, H.L.; Leto, T.L.; Rada, B. Release of cystic fibrosis airway inflammatory markers from Pseudomonas aeruginosa-stimulated human neutrophils involves NADPH oxidase-dependent extracellular DNA trap formation. J. Immunol. 2014, 192, 4728–4738. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.; Greene, C.M.; McElvaney, N.G. Targeting neutrophil elastase in cystic fibrosis. Expert Opin. Targets 2008, 12, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Bruce, K.D. The cystic fibrosis airway microbiome. Cold Spring Harb. Perspect. Med. 2013, 3, a009738. [Google Scholar] [CrossRef] [PubMed]
- Gray, R.D.; Hardisty, G.; Regan, K.H.; Smith, M.; Robb, C.T.; Duffin, R.; Mackellar, A.; Felton, J.M.; Paemka, L.; McCullagh, B.N.; et al. Delayed neutrophil apoptosis enhances NET formation in cystic fibrosis. Thorax 2018, 73, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKeon, D.J.; Condliffe, A.M.; Cowburn, A.S.; Cadwallader, K.C.; Farahi, N.; Bilton, D.; Chilvers, E.R. Prolonged survival of neutrophils from patients with Delta F508 CFTR mutations. Thorax 2008, 63, 660–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzenreiter, R.; Kienberger, F.; Marcos, V.; Schilcher, K.; Krautgartner, W.D.; Obermayer, A.; Huml, M.; Stoiber, W.; Hector, A.; Griese, M.; et al. Ultrastructural characterization of cystic fibrosis sputum using atomic force and scanning electron microscopy. J. Cyst. Fibros. 2012, 11, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Aleman, S.R.; Campos-Garcia, L.; Palma-Nicolas, J.P.; Hernandez-Bello, R.; Gonzalez, G.M.; Sanchez-Gonzalez, A. Understanding the Entanglement: Neutrophil Extracellular Traps (NETs) in Cystic Fibrosis. Front. Cell. Infect. Microbiol. 2017, 7, 104. [Google Scholar] [CrossRef] [Green Version]
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PLoS ONE 2011, 6, e28526. [Google Scholar] [CrossRef]
- Dubois, A.V.; Gauthier, A.; Brea, D.; Varaigne, F.; Diot, P.; Gauthier, F.; Attucci, S. Influence of DNA on the activities and inhibition of neutrophil serine proteases in cystic fibrosis sputum. Am. J. Respir. Cell. Mol. Biol. 2012, 47, 80–86. [Google Scholar] [CrossRef]
- Marcos, V.; Zhou-Suckow, Z.; Onder Yildirim, A.; Bohla, A.; Hector, A.; Vitkov, L.; Krautgartner, W.D.; Stoiber, W.; Griese, M.; Eickelberg, O.; et al. Free DNA in cystic fibrosis airway fluids correlates with airflow obstruction. Mediat. Inflamm. 2015, 2015, 408935. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kinsella, M.G.; Wight, T.N. Degradation of endothelial cell matrix heparan sulfate proteoglycan by elastase and the myeloperoxidase-H2O2-chloride system. Am. J. Pathol. 1993, 143, 907–917. [Google Scholar]
- Farrera, C.; Fadeel, B. Macrophage clearance of neutrophil extracellular traps is a silent process. J. Immunol. 2013, 191, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, A.; Brown, M.E.; Deriy, L.V.; Li, C.; Szeto, F.L.; Chen, Y.; Huang, P.; Tong, J.; Naren, A.P.; Bindokas, V.; et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat. Cell. Biol. 2006, 8, 933–944. [Google Scholar] [CrossRef]
- Young, R.L.; Malcolm, K.C.; Kret, J.E.; Caceres, S.M.; Poch, K.R.; Nichols, D.P.; Taylor-Cousar, J.L.; Saavedra, M.T.; Randell, S.H.; Vasil, M.L.; et al. Neutrophil extracellular trap (NET)-mediated killing of Pseudomonas aeruginosa: Evidence of acquired resistance within the CF airway, independent of CFTR. PLoS ONE 2011, 6, e23637. [Google Scholar] [CrossRef]
- Mulcahy, H.; Charron-Mazenod, L.; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 2008, 4, e1000213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatua, B.; Bhattacharya, K.; Mandal, C. Sialoglycoproteins adsorbed by Pseudomonas aeruginosa facilitate their survival by impeding neutrophil extracellular trap through siglec-9. J. Leukoc. Biol. 2012, 91, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.N.; Mutharasan, R. A cantilever biosensor-based assay for toxin-producing cyanobacteria Microcystis aeruginosa using 16S rRNA. Environ. Sci. Technol. 2013, 47, 12333–12341. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.; Mulcahy, H.; Kanevets, U.; Shi, Y.; Lewenza, S. Surface-localized spermidine protects the Pseudomonas aeruginosa outer membrane from antibiotic treatment and oxidative stress. J. Bacteriol. 2012, 194, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Halverson, T.W.; Wilton, M.; Poon, K.K.; Petri, B.; Lewenza, S. DNA is an antimicrobial component of neutrophil extracellular traps. PLoS Pathog. 2015, 11, e1004593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabe, K.F.; Watz, H. Chronic obstructive pulmonary disease. Lancet 2017, 389, 1931–1940. [Google Scholar] [CrossRef]
- MacNee, W.; Wiggs, B.; Belzberg, A.S.; Hogg, J.C. The effect of cigarette smoking on neutrophil kinetics in human lungs. N. Engl. J. Med. 1989, 321, 924–928. [Google Scholar] [CrossRef]
- Quint, J.K.; Wedzicha, J.A. The neutrophil in chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2007, 119, 1065–1071. [Google Scholar] [CrossRef] [PubMed]
- Di Stefano, A.; Caramori, G.; Gnemmi, I.; Contoli, M.; Bristot, L.; Capelli, A.; Ricciardolo, F.L.; Magno, F.; D’Anna, S.E.; Zanini, A.; et al. Association of increased CCL5 and CXCL7 chemokine expression with neutrophil activation in severe stable COPD. Thorax 2009, 64, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aaron, S.D.; Angel, J.B.; Lunau, M.; Wright, K.; Fex, C.; Le Saux, N.; Dales, R.E. Granulocyte inflammatory markers and airway infection during acute exacerbation of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2001, 163, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Chu, F.; Utokaparch, S.; Woods, R.; Elliott, W.M.; Buzatu, L.; Cherniack, R.M.; Rogers, R.M.; Sciurba, F.C.; Coxson, H.O.; et al. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N. Engl. J. Med. 2004, 350, 2645–2653. [Google Scholar] [CrossRef]
- Beeh, K.M.; Kornmann, O.; Buhl, R.; Culpitt, S.V.; Giembycz, M.A.; Barnes, P.J. Neutrophil chemotactic activity of sputum from patients with COPD: Role of interleukin 8 and leukotriene B4. Chest 2003, 123, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Zhu, J.; Bandi, V.; Atmar, R.L.; Hattotuwa, K.; Guntupalli, K.K.; Jeffery, P.K. Biopsy neutrophilia, neutrophil chemokine and receptor gene expression in severe exacerbations of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2003, 168, 968–975. [Google Scholar] [CrossRef]
- Chung, K.F. Inflammatory mediators in chronic obstructive pulmonary disease. Curr. Drug Targets Inflamm. Allergy 2005, 4, 619–625. [Google Scholar] [CrossRef]
- Pedersen, F.; Marwitz, S.; Holz, O.; Kirsten, A.; Bahmer, T.; Waschki, B.; Magnussen, H.; Rabe, K.F.; Goldmann, T.; Uddin, M.; et al. Neutrophil extracellular trap formation and extracellular DNA in sputum of stable COPD patients. Respir. Med. 2015, 109, 1360–1362. [Google Scholar] [CrossRef] [Green Version]
- Wright, T.K.; Gibson, P.G.; Simpson, J.L.; McDonald, V.M.; Wood, L.G.; Baines, K.J. Neutrophil extracellular traps are associated with inflammation in chronic airway disease. Respirology 2016, 21, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabcanovic-Musija, F.; Obermayer, A.; Stoiber, W.; Krautgartner, W.D.; Steinbacher, P.; Winterberg, N.; Bathke, A.C.; Klappacher, M.; Studnicka, M. Neutrophil extracellular trap (NET) formation characterises stable and exacerbated COPD and correlates with airflow limitation. Respir. Res. 2015, 16, 59. [Google Scholar] [CrossRef] [Green Version]
- Dicker, A.J.; Crichton, M.L.; Pumphrey, E.G.; Cassidy, A.J.; Suarez-Cuartin, G.; Sibila, O.; Furrie, E.; Fong, C.J.; Ibrahim, W.; Brady, G.; et al. Neutrophil extracellular traps are associated with disease severity and microbiota diversity in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 2018, 141, 117–127. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.P.; Coombs, N.A.; Johnson, M.J.; Josephs, L.K.; Rigge, L.A.; Staples, K.J.; Thomas, M.; Wilkinson, T.M. Seasonality, risk factors and burden of community-acquired pneumonia in COPD patients: A population database study using linked health care records. Int. J. Chron. Obs. Pulmon. Dis. 2017, 12, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, A.; Khan, M.A.; Bade, G.; Talwar, A. Orchestration of Neutrophil Extracellular Traps (Nets), a Unique Innate Immune Function during Chronic Obstructive Pulmonary Disease (COPD) Development. Biomedicines 2021, 9, 53. [Google Scholar] [CrossRef]
- Latge, J.P. Aspergillus fumigatus and aspergillosis. Clin. Microbiol. Rev. 1999, 12, 310–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, Z.; Liao, W. Fungal respiratory disease. Curr. Opin. Pulm. Med. 2006, 12, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.; Cunha, C.; Bistoni, F.; Romani, L. Immunotherapy of aspergillosis. Clin. Microbiol. Infect. 2012, 18, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chayakulkeeree, M.; Perfect, J.R. Cryptococcosis. Infect. Dis. Clin. N. Am. 2006, 20, 507–544. [Google Scholar] [CrossRef]
- Kronstad, J.W.; Attarian, R.; Cadieux, B.; Choi, J.; D’Souza, C.A.; Griffiths, E.J.; Geddes, J.M.; Hu, G.; Jung, W.H.; Kretschmer, M.; et al. Expanding fungal pathogenesis: Cryptococcus breaks out of the opportunistic box. Nat. Rev. Microbiol. 2011, 9, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.G.; Perfect, J.R. Cryptococcosis in the era of AIDS--100 years after the discovery of Cryptococcus neoformans. Clin. Microbiol. Rev. 1995, 8, 515–548. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Doshi, A.V.; Bhaduri, A.S. Pneumocystis pneumonia. N. Engl. J. Med. 2004, 351, 1262–1263. [Google Scholar]
- Lortholary, O.; Denning, D.W.; Dupont, B. Endemic mycoses: A treatment update. J. Antimicrob. Chemother. 1999, 43, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheat, J. Endemic mycoses in AIDS: A clinical review. Clin. Microbiol. Rev. 1995, 8, 146–159. [Google Scholar] [CrossRef] [PubMed]
- Yaguchi, T. The genus Aspergillus. Med. Mycol. J. 2011, 52, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Mircescu, M.M.; Lipuma, L.; van Rooijen, N.; Pamer, E.G.; Hohl, T.M. Essential role for neutrophils but not alveolar macrophages at early time points following Aspergillus fumigatus infection. J. Infect. Dis. 2009, 200, 647–656. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim-Granet, O.; Philippe, B.; Boleti, H.; Boisvieux-Ulrich, E.; Grenet, D.; Stern, M.; Latge, J.P. Phagocytosis and intracellular fate of Aspergillus fumigatus conidia in alveolar macrophages. Infect. Immun. 2003, 71, 891–903. [Google Scholar] [CrossRef] [Green Version]
- Mouyna, I.; Sarfati, J.; Recco, P.; Fontaine, T.; Henrissatz, B.; Latge, J.P. Molecular characterization of a cell wall-associated beta(1-3)endoglucanase of Aspergillus fumigatus. Med. Mycol. 2002, 40, 455–464. [Google Scholar] [CrossRef]
- Bernard, M.; Mouyna, I.; Dubreucq, G.; Debeaupuis, J.P.; Fontaine, T.; Vorgias, C.; Fuglsang, C.; Latge, J.P. Characterization of a cell-wall acid phosphatase (PhoAp) in Aspergillus fumigatus. Microbiology 2002, 148, 2819–2829. [Google Scholar] [CrossRef] [Green Version]
- Latge, J.P. The pathobiology of Aspergillus fumigatus. Trends Microbiol. 2001, 9, 382–389. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruns, S.; Kniemeyer, O.; Hasenberg, M.; Aimanianda, V.; Nietzsche, S.; Thywissen, A.; Jeron, A.; Latge, J.P.; Brakhage, A.A.; Gunzer, M. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010, 6, e1000873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaillon, S.; Peri, G.; Delneste, Y.; Fremaux, I.; Doni, A.; Moalli, F.; Garlanda, C.; Romani, L.; Gascan, H.; Bellocchio, S.; et al. The humoral pattern recognition receptor PTX3 is stored in neutrophil granules and localizes in extracellular traps. J. Exp. Med. 2007, 204, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Rohm, M.; Grimm, M.J.; D’Auria, A.C.; Almyroudis, N.G.; Segal, B.H.; Urban, C.F. NADPH oxidase promotes neutrophil extracellular trap formation in pulmonary aspergillosis. Infect. Immun. 2014, 82, 1766–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alflen, A.; Aranda Lopez, P.; Hartmann, A.K.; Maxeiner, J.; Bosmann, M.; Sharma, A.; Platten, J.; Ries, F.; Beckert, H.; Ruf, W.; et al. Neutrophil extracellular traps impair fungal clearance in a mouse model of invasive pulmonary aspergillosis. Immunobiology 2020, 225, 151867. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van de Geer, A.; Roos, D.; van den Berg, T.K.; Kuijpers, T.W. How neutrophils kill fungi. Immunol. Rev. 2016, 273, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Gazendam, R.P.; van Hamme, J.L.; Tool, A.T.; Hoogenboezem, M.; van den Berg, J.M.; Prins, J.M.; Vitkov, L.; van de Veerdonk, F.L.; van den Berg, T.K.; Roos, D.; et al. Human Neutrophils Use Different Mechanisms To Kill Aspergillus fumigatus Conidia and Hyphae: Evidence from Phagocyte Defects. J. Immunol. 2016, 196, 1272–1283. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Segal, B.H.; Holland, S.M.; Miller, G.F.; Kwon-Chung, K.J. Virulence of catalase-deficient aspergillus nidulans in p47(phox)-/- mice. Implications for fungal pathogenicity and host defense in chronic granulomatous disease. J. Clin. Invest. 1998, 101, 1843–1850. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, D.E.; Gifford, M.A.; Li, L.L.; Doerschuk, C.M.; Dinauer, M.C. Absence of respiratory burst in X-linked chronic granulomatous disease mice leads to abnormalities in both host defense and inflammatory response to Aspergillus fumigatus. J. Exp. Med. 1997, 185, 207–218. [Google Scholar] [CrossRef]
- Henriet, S.S.; Hermans, P.W.; Verweij, P.E.; Simonetti, E.; Holland, S.M.; Sugui, J.A.; Kwon-Chung, K.J.; Warris, A. Human leukocytes kill Aspergillus nidulans by reactive oxygen species-independent mechanisms. Infect. Immun. 2011, 79, 767–773. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.; Hakkim, A.; Brinkmann, V.; Siler, U.; Seger, R.A.; Zychlinsky, A.; Reichenbach, J. Restoration of NET formation by gene therapy in CGD controls aspergillosis. Blood 2009, 114, 2619–2622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, M.; Niemiec, M.J.; Siler, U.; Urban, C.F.; Reichenbach, J. Restoration of anti-Aspergillus defense by neutrophil extracellular traps in human chronic granulomatous disease after gene therapy is calprotectin-dependent. J. Allergy Clin. Immunol. 2011, 127, 1243–1252.e1247. [Google Scholar] [CrossRef] [PubMed]
- Loussert, C.; Schmitt, C.; Prevost, M.C.; Balloy, V.; Fadel, E.; Philippe, B.; Kauffmann-Lacroix, C.; Latge, J.P.; Beauvais, A. In vivo biofilm composition of Aspergillus fumigatus. Cell. Microbiol. 2010, 12, 405–410. [Google Scholar] [CrossRef]
- Fontaine, T.; Delangle, A.; Simenel, C.; Coddeville, B.; van Vliet, S.J.; van Kooyk, Y.; Bozza, S.; Moretti, S.; Schwarz, F.; Trichot, C.; et al. Galactosaminogalactan, a new immunosuppressive polysaccharide of Aspergillus fumigatus. PLoS Pathog. 2011, 7, e1002372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Geller, A.M.; Bamford, N.C.; Liu, H.; Gravelat, F.N.; Snarr, B.D.; Le Mauff, F.; Chabot, J.; Ralph, B.; Ostapska, H.; et al. Deacetylation of Fungal Exopolysaccharide Mediates Adhesion and Biofilm Formation. mBio 2016, 7, e00252-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravelat, F.N.; Beauvais, A.; Liu, H.; Lee, M.J.; Snarr, B.D.; Chen, D.; Xu, W.; Kravtsov, I.; Hoareau, C.M.; Vanier, G.; et al. Aspergillus galactosaminogalactan mediates adherence to host constituents and conceals hyphal beta-glucan from the immune system. PLoS Pathog. 2013, 9, e1003575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.J.; Liu, H.; Barker, B.M.; Snarr, B.D.; Gravelat, F.N.; Al Abdallah, Q.; Gavino, C.; Baistrocchi, S.R.; Ostapska, H.; Xiao, T.; et al. The Fungal Exopolysaccharide Galactosaminogalactan Mediates Virulence by Enhancing Resistance to Neutrophil Extracellular Traps. PLoS Pathog. 2015, 11, e1005187. [Google Scholar] [CrossRef]
- Nasr, S.Z.; Strouse, P.J.; Soskolne, E.; Maxvold, N.J.; Garver, K.A.; Rubin, B.K.; Moler, F.W. Efficacy of recombinant human deoxyribonuclease I in the hospital management of respiratory syncytial virus bronchiolitis. Chest 2001, 120, 203–208. [Google Scholar] [CrossRef]
- Hodson, M.E.; McKenzie, S.; Harms, H.K.; Koch, C.; Mastella, G.; Navarro, J.; Strandvik, B.; Investigators of the Epidemiologic Registry of Cystic Fibrosis. Dornase alfa in the treatment of cystic fibrosis in Europe: A report from the Epidemiologic Registry of Cystic Fibrosis. Pediatr. Pulmonol. 2003, 36, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Frederiksen, B.; Pressler, T.; Hansen, A.; Koch, C.; Hoiby, N. Effect of aerosolized rhDNase (Pulmozyme) on pulmonary colonization in patients with cystic fibrosis. Acta Paediatr. 2006, 95, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.G.; Chau, A.S.; Egeblad, M.; Barnes, B.J.; Janowitz, T. Nebulized in-line endotracheal dornase alfa and albuterol administered to mechanically ventilated COVID-19 patients: A case series. Mol. Med. 2020, 26, 91. [Google Scholar] [CrossRef] [PubMed]
- Desilles, J.P.; Gregoire, C.; Le Cossec, C.; Lambert, J.; Mophawe, O.; Losser, M.R.; Lambiotte, F.; Le Tacon, S.; Cantier, M.; Engrand, N.; et al. Efficacy and safety of aerosolized intra-tracheal dornase alfa administration in patients with SARS-CoV-2-induced acute respiratory distress syndrome (ARDS): A structured summary of a study protocol for a randomised controlled trial. Trials 2020, 21, 548. [Google Scholar] [CrossRef]
- Tsourouktsoglou, T.D.; Warnatsch, A.; Ioannou, M.; Hoving, D.; Wang, Q.; Papayannopoulos, V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [Google Scholar] [CrossRef]
- Wygrecka, M.; Kosanovic, D.; Wujak, L.; Reppe, K.; Henneke, I.; Frey, H.; Didiasova, M.; Kwapiszewska, G.; Marsh, L.M.; Baal, N.; et al. Antihistone Properties of C1 Esterase Inhibitor Protect against Lung Injury. Am. J. Respir. Crit. Care Med. 2017, 196, 186–199. [Google Scholar] [CrossRef]
- Chirivi, R.G.S.; van Rosmalen, J.W.G.; van der Linden, M.; Euler, M.; Schmets, G.; Bogatkevich, G.; Kambas, K.; Hahn, J.; Braster, Q.; Soehnlein, O.; et al. Therapeutic ACPA inhibits NET formation: A potential therapy for neutrophil-mediated inflammatory diseases. Cell. Mol. Immunol. 2021, 18, 1528–1544. [Google Scholar] [CrossRef] [Green Version]
- Caliezi, C.; Wuillemin, W.A.; Zeerleder, S.; Redondo, M.; Eisele, B.; Hack, C.E. C1-Esterase inhibitor: An anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharm. Rev. 2000, 52, 91–112. [Google Scholar] [PubMed]
- Liu, D.; Cai, S.; Gu, X.; Scafidi, J.; Wu, X.; Davis, A.E., 3rd. C1 inhibitor prevents endotoxin shock via a direct interaction with lipopolysaccharide. J. Immunol. 2003, 171, 2594–2601. [Google Scholar] [CrossRef] [Green Version]
- Zeerleder, S.; Caliezi, C.; van Mierlo, G.; Eerenberg-Belmer, A.; Sulzer, I.; Hack, C.E.; Wuillemin, W.A. Administration of C1 inhibitor reduces neutrophil activation in patients with sepsis. Clin. Diagn. Lab. Immunol. 2003, 10, 529–535. [Google Scholar] [CrossRef] [Green Version]
- Igonin, A.A.; Protsenko, D.N.; Galstyan, G.M.; Vlasenko, A.V.; Khachatryan, N.N.; Nekhaev, I.V.; Shlyapnikov, S.A.; Lazareva, N.B.; Herscu, P. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit. Care Med. 2012, 40, 770–777. [Google Scholar] [CrossRef]
- Deng, Q.; Pan, B.; Alam, H.B.; Liang, Y.; Wu, Z.; Liu, B.; Mor-Vaknin, N.; Duan, X.; Williams, A.M.; Tian, Y.; et al. Citrullinated Histone H3 as a Therapeutic Target for Endotoxic Shock in Mice. Front. Immunol. 2019, 10, 2957. [Google Scholar] [CrossRef] [PubMed]
- Hagio, T.; Matsumoto, S.; Nakao, S.; Matsuoka, S.; Kawabata, K. Sivelestat, a specific neutrophil elastase inhibitor, prevented phorbol myristate acetate-induced acute lung injury in conscious rabbits. Pulm. Pharm. 2005, 18, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Okayama, N.; Kakihana, Y.; Setoguchi, D.; Imabayashi, T.; Omae, T.; Matsunaga, A.; Kanmura, Y. Clinical effects of a neutrophil elastase inhibitor, sivelestat, in patients with acute respiratory distress syndrome. J. Anesth. 2006, 20, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Tamakuma, S.; Ogawa, M.; Aikawa, N.; Kubota, T.; Hirasawa, H.; Ishizaka, A.; Taenaka, N.; Hamada, C.; Matsuoka, S.; Abiru, T. Relationship between neutrophil elastase and acute lung injury in humans. Pulm. Pharm. 2004, 17, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, S.; Okayama, Y.; Shime, N.; Kimura, A.; Funakoshi, Y.; Kawabata, K.; Ishizaka, A.; Amaya, F. Neutrophil elastase activity in acute lung injury and respiratory distress syndrome. Respirology 2008, 13, 581–584. [Google Scholar] [CrossRef] [PubMed]
- Tagami, T.; Tosa, R.; Omura, M.; Fukushima, H.; Kaneko, T.; Endo, T.; Rinka, H.; Murai, A.; Yamaguchi, J.; Yoshikawa, K.; et al. Effect of a selective neutrophil elastase inhibitor on mortality and ventilator-free days in patients with increased extravascular lung water: A post hoc analysis of the PiCCO Pulmonary Edema Study. J. Intensive Care 2014, 2, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, S.; Hamada, H.; Ito, R.; Katayama, H.; Irifune, K.; Suwaki, T.; Nakanishi, N.; Kanematsu, T.; Dote, K.; Aibiki, M.; et al. Usefulness of a selective neutrophil elastase inhibitor, sivelestat, in acute lung injury patients with sepsis. Drug Des. Devel. 2013, 7, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Lapponi, M.J.; Carestia, A.; Landoni, V.I.; Rivadeneyra, L.; Etulain, J.; Negrotto, S.; Pozner, R.G.; Schattner, M. Regulation of neutrophil extracellular trap formation by anti-inflammatory drugs. J. Pharm. Exp. 2013, 345, 430–437. [Google Scholar] [CrossRef]
- Hirose, T.; Hamaguchi, S.; Matsumoto, N.; Irisawa, T.; Seki, M.; Tasaki, O.; Hosotsubo, H.; Yamamoto, N.; Yamamoto, K.; Akeda, Y.; et al. Presence of neutrophil extracellular traps and citrullinated histone H3 in the bloodstream of critically ill patients. PLoS ONE 2014, 9, e111755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamphier, M.; Zheng, W.; Latz, E.; Spyvee, M.; Hansen, H.; Rose, J.; Genest, M.; Yang, H.; Shaffer, C.; Zhao, Y.; et al. Novel small molecule inhibitors of TLR7 and TLR9: Mechanism of action and efficacy in vivo. Mol. Pharm. 2014, 85, 429–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The antidiabetic drug metformin blunts NETosis in vitro and reduces circulating NETosis biomarkers in vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Gregoire, M.; Uhel, F.; Lesouhaitier, M.; Gacouin, A.; Guirriec, M.; Mourcin, F.; Dumontet, E.; Chalin, A.; Samson, M.; Berthelot, L.L.; et al. Impaired efferocytosis and neutrophil extracellular trap clearance by macrophages in ARDS. Eur. Respir. J. 2018, 52. [Google Scholar] [CrossRef]
- Vargas, A.; Boivin, R.; Cano, P.; Murcia, Y.; Bazin, I.; Lavoie, J.P. Neutrophil extracellular traps are downregulated by glucocorticosteroids in lungs in an equine model of asthma. Respir. Res. 2017, 18, 207. [Google Scholar] [CrossRef] [PubMed]
- Yost, C.C.; Schwertz, H.; Cody, M.J.; Wallace, J.A.; Campbell, R.A.; Vieira-de-Abreu, A.; Araujo, C.V.; Schubert, S.; Harris, E.S.; Rowley, J.W.; et al. Neonatal NET-inhibitory factor and related peptides inhibit neutrophil extracellular trap formation. J. Clin. Invest. 2016, 126, 3783–3798. [Google Scholar] [CrossRef]
- Leaker, B.R.; Barnes, P.J.; O’Connor, B. Inhibition of LPS-induced airway neutrophilic inflammation in healthy volunteers with an oral CXCR2 antagonist. Respir. Res. 2013, 14, 137. [Google Scholar] [CrossRef] [Green Version]
- Wolf, D.; Anto-Michel, N.; Blankenbach, H.; Wiedemann, A.; Buscher, K.; Hohmann, J.D.; Lim, B.; Bauml, M.; Marki, A.; Mauler, M.; et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat. Commun. 2018, 9, 525. [Google Scholar] [CrossRef]
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET:current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Compound | Target | Application | Reference |
|---|---|---|---|
| Dornase Alfa/DNase | DNA | Bronchiolitis Cystic fibrosis | [211,212,213,214,215] |
| Clinical Phase 2 study: COVID19 | NCT04359654 | ||
| C1 esterase inhibitor | Histones | Sepsis patients | [219,220,221,222] |
| tACPA α-H3-cit | Citrullinated Histones | Inflammatory murine disease models | [218,223] |
| Sivelestat | Neutrophil elastase | ARDS patients ALI patients | [225,226,227,228,229] |
| Clinical phase 4 study: ARDS | NCT00036062 | ||
| Aspirin αCLEC Glucocorticoids NET-inhibiting factors | Inhibition of NET formation | Critically ill patients Inflammatory murine disease models | [230,231,232,236] |
| Metformin | HMGB/NET clearance | Diabetes patients | [233,234] |
| CXCR2 antagonist | Neutrophil recruitment | LPS-challenged humans | [237] |
| CD40L-M7 | Mac1 | Inflammatory murine disease models | [238] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Block, H.; Zarbock, A. A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells 2021, 10, 1932. https://doi.org/10.3390/cells10081932
Block H, Zarbock A. A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells. 2021; 10(8):1932. https://doi.org/10.3390/cells10081932
Chicago/Turabian StyleBlock, Helena, and Alexander Zarbock. 2021. "A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression?" Cells 10, no. 8: 1932. https://doi.org/10.3390/cells10081932
APA StyleBlock, H., & Zarbock, A. (2021). A Fragile Balance: Does Neutrophil Extracellular Trap Formation Drive Pulmonary Disease Progression? Cells, 10(8), 1932. https://doi.org/10.3390/cells10081932