
Disruption of RING and PHD Domains of TRIM28 Evokes Differentiation in Human iPSCs

 , , ,
, , ,
Abstract
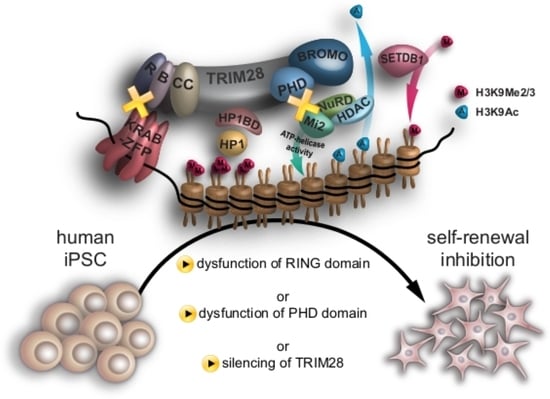
:
1. Introduction
2. Materials and Methods
2.1. Organisms
2.2. Cell Culture
2.3. Cloning Procedures
2.4. Lentiviral Vectors Production, Purification, and Titration
2.5. Reprogramming of Human Fibroblasts towards iPSC
2.6. Generation of TRIM28-Depleted or Mutated iPSC Populations
2.7. Immunofluorescence Staining
2.8. cDNA Samples Preparation
2.9. Real-Time PCR Quantification
2.10. Flow Cytometry
2.11. RT-PCR
2.12. Proliferation Assay
2.13. Spontaneous In Vitro Differentiation Potential Assessment (Embryoid Bodies Formation)
2.14. Western Blot
2.15. Teratoma Formation
2.16. Karyotyping of Generated iPSC Lines
2.17. Bisulfite Sequencing for Promoter Methylation Analysis
2.18. Proteomic Profiling–Reverse Phase Protein Array (RPPA)
2.19. Principal Component Analysis
2.20. Differential Expression Analysis
2.21. Gene Set Enrichment Analysis (GSEA)
2.22. Over-Representation Enrichment Analysis (ORA)
3. Results
3.1. Generating Human iPSC with Doxycycline-Inducible System Results in Repression of Transgene Expression in Established Clones
3.2. Silencing of Endogenous TRIM28 Induces Downregulation of Pluripotency Markers and Differentiation of Human iPSC
3.3. iPSC with Silenced TRIM28 Display Metabolic Changes, and Their Proteomic Profile Differs from the Control iPSC
3.4. Selection of Mutation Sites Impairing the Function of TRIM28 Protein Domains

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation | Domain | Predicted Influence on TRIM28 Activity | Reference |
|---|---|---|---|
| C91A (structural mutant) | RING | Destabilization of RING domain structure | [28,45,46,47,48] |
| Inhibition of interaction with KRAB domain of KRAB-ZNFs | |||
| Impairment of E3 ubiquitin ligase function | |||
| Restriction of TRIM28 transcription co-repressor function | |||
| S473A (phospho-mutant) | HP1BD | Inhibition of TRIM28 Ser473 phosphorylation | [31,43] |
| HP1BD interaction with HP1 | |||
| TRIM28 functions as transcription co-repressor | |||
| S473D (phospho-mimetic) | HP1BD | Imitation of permanent TRIM28 Ser473 phosphorylation | [31,43] |
| Inhibition of HP1 binding by HP1BD | |||
| Restriction of TRIM28 transcription co-repressor function | |||
| 3YF (phospho-mutant) | HP1BD | Inhibition of triple tyrosine phosphorylation | [19] |
| HP1BD interaction with HP1 | |||
| TRIM28 functions as a transcription repressor | |||
| C628R (structural mutant) | PHD | Destabilization of PHD domain structure | [30,49] |
| Impairment of E3 SUMO ligase function | |||
| Inhibition of NuRD complex and SETDB1 methyltransferase binding | |||
| Restriction of TRIM28 transcription repressor function | |||
| N773G (structural mutant) | BROMO | Destabilization of bromodomain structure | - |
| Low influence on TRIM28 activity | |||
| S824A (phospho-mutant) | BROMO | Inhibition of TRIM28 Ser824 phosphorylation | [18,44] |
| Increased TRIM28 BROMO sumoylation | |||
| Induction of differentiation | |||
| S824D (phospho-mimetic) | BROMO | Imitation of permanent TRIM28 Ser824 phosphorylation | [18,44] |
| Decreased TRIM28 BROMO sumoylation | |||
| Inhibition of spontaneous differentiation | |||
| Induction of SOX2 and NANOG expression |
3.5. Mutations in RING and PHD Domains Are Classified as Mutations with a High Impact on TRIM28 Function
3.6. Constructed Lentiviral System Enables Efficient Transgene Expression and Silencing of Endogenous TRIM28
3.7. Mutations of RING and PHD Domains Influence Human iPSC Morphology and Pluripotency Markers Expression
3.8. Dysfunction of RING and PHD Domains Results in Decreased Proliferation and Inhibition of Embryoid Bodies Formation
3.9. Impairment of RING and PHD Functions Results in Dysregulation of Stem Cell-Associated Signaling Pathways
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BROMO | Bromodomain |
| CC | Coiled-Coil Motif |
| EB | Embryoid Bodies |
| ESC | Embryonic Stem Cells |
| FGFR | Fibroblast Growth Factor Receptor |
| GSEA | Gene Set Enrichment Analysis |
| HP1BD | Heterochromatin Protein 1-Binding Domain |
| iPSC | induced Pluripotent Stem Cells |
| KAP1 | Krüppel-Associated Box-Associated Protein 1 |
| KRAB-ZFPs | KRAB Zinc-Finger Proteins |
| LV | Lentivirus |
| MEF | Mouse Embryonic Fibroblasts |
| NuRD | Nucleosome Remodeling and Deacetylation Complex |
| ORA | Over-Representation Enrichment Analysis |
| OXPHOS | Oxidative Phosphorylation |
| PCA | Principal Component Analysis |
| PHD | Plant Homeodomain |
| PHDF | Primary Human Dermal Fibroblasts |
| RBCC | RING-B-box-Coiled-Coil domain |
| RING | Really Interesting New Gene |
| RPPA | Reverse Phase Protein Array |
| SETDB1 | SET Domain Bifurcated 1 Histone Lysine Methyltransferase |
| shRNA | small hairpin RNA |
| siRNA | small interfering RNA |
| TIF1β | Transcription Intermediary Factor 1-β |
| TRIM | Tripartite Motif |
| TRIM28 | Tripartite Motif-Containing Protein 28 |
References
- Takahashi, K.; Yamanaka, S. Induced pluripotent stem cells in medicine and biology. Development 2013, 140, 2457–2461. [Google Scholar] [CrossRef] [Green Version]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional Addiction in Cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef] [Green Version]
- Malta, T.M.; Sokolov, A.; Gentles, A.J.; Burzykowski, T.; Poisson, L.; Weinstein, J.N.; Kamińska, B.; Huelsken, J.; Omberg, L.; Gevaert, O.; et al. Machine Learning Identifies Stemness Features Associated with Oncogenic Dedifferentiation. Cell 2018, 173, 338–354.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting Wnt, Notch, and Hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef]
- Singh, A.M.; Reynolds, D.; Cliff, T.; Ohtsuka, S.; Mattheyses, A.L.; Sun, Y.; Menendez, L.; Kulik, M.; Dalton, S. Signaling network crosstalk in human pluripotent cells: A Smad2/3-regulated switch that controls the balance between self-renewal and differentiation. Cell Stem Cell 2012, 10, 312–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585. [Google Scholar] [CrossRef]
- Miles, D.C.; de Vries, N.A.; Gisler, S.; Lieftink, C.; Akhtar, W.; Gogola, E.; Pawlitzky, I.; Hulsman, D.; Tanger, E.; Koppens, M.; et al. TRIM28 is an Epigenetic Barrier to Induced Pluripotent Stem Cell Reprogramming. Stem Cells 2017, 35, 147–157. [Google Scholar] [CrossRef] [Green Version]
- Oleksiewicz, U.; Gładych, M.; Raman, A.T.; Heyn, H.; Mereu, E.; Chlebanowska, P.; Andrzejewska, A.; Sozańska, B.; Samant, N.; Fąk, K.; et al. TRIM28 and Interacting KRAB-ZNFs Control Self-Renewal of Human Pluripotent Stem Cells through Epigenetic Repression of Pro-differentiation Genes. Stem Cell Rep. 2017, 9, 2065–2080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czerwińska, P.; Shah, P.K.; Tomczak, K.; Klimczak, M.; Mazurek, S.; Sozańska, B.; Biecek, P.; Korski, K.; Filas, V.; Mackiewicz, A.; et al. TRIM28 multi-domain protein regulates cancer stem cell population in breast tumor development. Oncotarget 2017, 8, 863–882. [Google Scholar] [CrossRef] [Green Version]
- Czerwińska, P.; Mazurek, S.; Wiznerowicz, M. The complexity of TRIM28 contribution to cancer. J. Biomed. Sci. 2017, 24, 63. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Fredericks, W.J.; Jensen, D.E.; Speicher, D.W.; Huang, X.P.; Neilson, E.G.; Rauscher, F.J. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996, 10, 2067–2078. [Google Scholar] [CrossRef] [Green Version]
- Ryan, R.F.; Schultz, D.C.; Ayyanathan, K.; Singh, P.B.; Friedman, J.R.; Fredericks, W.J.; Rauscher, F.J., III. KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: A potential role for Krüppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol. Cell. Biol. 1999, 19, 4366–4378. [Google Scholar] [CrossRef]
- Cammas, F.; Mark, M.; Dollé, P.; Dierich, A.; Chambon, P.; Losson, R. Mice lacking the transcriptional corepressor TIF1beta are defective in early postimplantation development. Development 2000, 127, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; de Vries, W.; Ito, M.; Solter, D.; Ferguson-Smith, A.; Knowles, B.B. Trim28 Is Required for Epigenetic Stability During Mouse Oocyte to Embryo Transition. Science 2012, 335, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Klimczak, M.; Czerwińska, P.; Mazurek, S.; Sozańska, B.; Biecek, P.; Mackiewicz, A.; Wiznerowicz, M. TRIM28 epigenetic corepressor is indispensable for stable induced pluripotent stem cell formation. Stem Cell Res. 2017, 23, 163–172. [Google Scholar] [CrossRef]
- Fazzio, T.G.; Huff, J.T.; Panning, B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell 2008, 134, 162–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Leung, D.; Miyashita, H.; Maksakova, I.A.; Miyachi, H.; Kimura, H.; Tachibana, M.; Lorincz, M.C.; Shinkai, Y. Proviral silencing in embryonic stem cells requires the histone methyltransferase ESET. Nature 2010, 464, 927–931. [Google Scholar] [CrossRef] [Green Version]
- Seki, Y.; Kurisaki, A.; Watanabe-Susaki, K.; Nakajima, Y.; Nakanishi, M.; Arai, Y.; Shiota, K.; Sugino, H.; Asashima, M. TIF1beta regulates the pluripotency of embryonic stem cells in a phosphorylation-dependent manner. Proc. Natl. Acad. Sci. USA 2010, 107, 10926–10931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, S.; Fukumoto, Y.; Aoyama, K.; Ishibashi, K.; Yuki, R.; Morinaga, T.; Honda, T.; Yamaguchi, N.; Kuga, T.; Tomonaga, T.; et al. Phosphorylation of KRAB-associated protein 1 (KAP1) at Tyr-449, Tyr-458, and Tyr-517 by nuclear tyrosine kinases inhibits the association of KAP1 and heterochromatin protein 1α (HP1α) with heterochromatin. J. Biol. Chem. 2013, 288, 17871–17883. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.F.; Li, C.F.; Wang, W.J.; Yang, W.M.; Wang, D.D.H.; Chang, W.C.; Lee, W.H.; Wang, J.M. Loss of ZBRK1 Contributes to the Increase of KAP1 and Promotes KAP1-Mediated Metastasis and Invasion in Cervical Cancer. PLoS ONE 2013, 8, e73033. [Google Scholar] [CrossRef] [Green Version]
- Yokoe, T.; Toiyama, Y.; Okugawa, Y.; Tanaka, K.; Ohi, M.; Inoue, Y.; Mohri, Y.; Miki, C.; Kusunoki, M. KAP1 is associated with peritoneal carcinomatosis in gastric cancer. Ann. Surg. Oncol. 2010, 17, 821–828. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Li, L.; Zhao, Z.S.; Wang, H.J. Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J. Surg. Oncol. 2013, 11, 81. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Fu, X.; Cui, Y.; Xu, S.; Xu, Y.; Dong, Q.; Sun, L. Expression of KAP1 in epithelial ovarian cancer and its correlation with drug-resistance. Int. J. Clin. Exp. Med. 2015, 8, 17308–17320. [Google Scholar] [PubMed]
- Cui, Y.; Yang, S.; Fu, X.; Feng, J.; Xu, S.; Ying, G. High levels of KAP1 expression are associated with aggressive clinical features in ovarian cancer. Int. J. Mol. Sci. 2015, 16, 363–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovčevska, I.; Zupanec, N.; Urlep, Ž.; Vranic, A.; Matos, B.; Stokin, C.L.; Muyldermans, S.; Myers, M.P.; Buzdin, A.A.; Petrov, I.; et al. Differentially expressed proteins in glioblastoma multiforme identified with a nanobody-based anti-proteome approach and confirmed by OncoFinder as possible tumor-class predictive biomarker candidates. Oncotarget 2017, 8, 44141–44158. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.S.; Begg, G.E.; Speicher, D.W.; Rauscher, F.J., III. Molecular determinants for targeting heterochromatin protein 1-mediated gene silencing: Direct chromoshadow domain-KAP-1 corepressor interaction is essential. Mol. Cell. Biol. 2000, 20, 6449–6465. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011, 286, 26267–26276. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.V.; Peng, H.; Yurchenko, V.; Yap, K.L.; Negorev, D.G.; Schultz, D.C.; Psulkowski, E.; Fredericks, W.J.; White, D.E.; Maul, G.G.; et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol. Cell 2007, 28, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher, F.J.; Zhou, M.M. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Schultz, D.C.; Ayyanathan, K.; Negorev, D.; Maul, G.G.; Rauscher, F.J., III. SETDB1: A novel KAP-1-associated histone H3, lysine 9-specific methyltransferase that contributes to HP1-mediated silencing of euchromatic genes by KRAB zinc-finger proteins. Genes Dev. 2002, 16, 919–932. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-W.; Chou, H.-Y.; Lin, Y.-S.; Huang, K.-H.; Chang, C.-J.; Hsu, T.-C.; Lee, S.-C. Phosphorylation at Ser473 regulates heterochromatin protein 1 binding and corepressor function of TIF1beta/KAP1. BMC Mol. Biol. 2008, 9, 61. [Google Scholar] [CrossRef] [Green Version]
- Scherr, M.; Battmer, K.; Blömer, U.; Ganser, A.; Grez, M. Quantitative Determination of Lentiviral Vector Particle Numbers by Real-Time PCR. Biotechniques 2001, 31, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Rohde, C.; Zhang, Y.; Reinhardt, R.; Jeltsch, A. BISMA—Fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinform. 2010, 11, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Starruß, J.; De Back, W.; Brusch, L.; Deutsch, A. Morpheus: A user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Boyle, E.I.; Weng, S.; Gollub, J.; Jin, H.; Botstein, D.; Cherry, J.M.; Sherlock, G. GO::TermFinder—Open source software for accessing Gene Ontology information and finding significantly enriched Gene Ontology terms associated with a list of genes. Bioinformatics 2004, 20, 3710–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleefeldt, J.M.; Pozarska, A.; Nardiello, C.; Pfeffer, T.; Vadász, I.; Herold, S.; Seeger, W.; Morty, R.E. Commercially available transfection reagents and negative control siRNA are not inert. Anal. Biochem. 2020, 606, 113828. [Google Scholar] [CrossRef]
- Prigione, A.; Fauler, B.; Lurz, R.; Lehrach, H.; Adjaye, J. The Senescence-Related Mitochondrial/Oxidative Stress Pathway is Repressed in Human Induced Pluripotent Stem Cells. Stem Cells 2010, 28, 721–733. [Google Scholar] [CrossRef]
- Choi, H.W.; Kim, J.H.; Chung, M.K.; Hong, Y.J.; Jang, H.S.; Seo, B.J.; Jung, T.H.; Kim, J.S.; Chung, H.M.; Byun, S.J.; et al. Mitochondrial and Metabolic Remodeling During Reprogramming and Differentiation of the Reprogrammed Cells. Stem Cells Dev. 2015, 24, 1366–1373. [Google Scholar] [CrossRef]
- Zhu, S.; Li, W.; Zhou, H.; Wei, W.; Ambasudhan, R.; Lin, T.; Kim, J.; Zhang, K.; Ding, S. Reprogramming of human primary somatic cells by OCT4 and chemical compounds. Cell Stem Cell 2010, 7, 651–655. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Zhang, S.; Gao, X.; Gao, X.; Xu, X.; Lv, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Li, Q.; et al. Roles of Kruppel-associated Box (KRAB)-associated Co-repressor KAP1 Ser-473 Phosphorylation in DNA Damage Response. J. Biol. Chem. 2012, 287, 18937–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Lee, Y.-K.; Jeng, J.-C.; Yen, Y.; Schultz, D.C.; Shih, H.-M.; Ann, D.K. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J. Biol. Chem. 2007, 282, 36177–36189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, H.; Begg, G.E.; Schultz, D.C.; Friedman, J.R.; Jensen, D.E.; Speicher, D.W.; Rauscher, F.J. Reconstitution of the KRAB-KAP-1 repressor complex: A model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J. Mol. Biol. 2000, 295, 1139–1162. [Google Scholar] [CrossRef]
- Huntley, S.; Baggott, D.M.; Hamilton, A.T.; Tran-Gyamfi, M.; Yang, S.; Kim, J.; Gordon, L.; Branscomb, E.; Stubbs, L. A comprehensive catalog of human KRAB-associated zinc finger genes: Insights into the evolutionary history of a large family of transcriptional repressors. Genome Res. 2006, 16, 669–677. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Feldman, I.; Rauscher, F.J. Hetero-oligomerization Among the TIF Family of RBCC/TRIM Domain-containing Nuclear Cofactors: A Potential Mechanism for Regulating the Switch Between Coactivation and Corepression. J. Mol. Biol. 2002, 320, 629–644. [Google Scholar] [CrossRef]
- Uchida, C.; Kitagawa, M. RING-, HECT-, and RBR-type E3 Ubiquitin Ligases: Involvement in Human Cancer. Curr. Cancer Drug Targets 2016, 16, 157–174. [Google Scholar] [CrossRef]
- Schultz, D.C.; Friedman, J.R.; Rauscher, F.J. Targeting histone deacetylase complexes via KRAB-zinc finger proteins: The PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2α subunit of NuRD. Genes Dev. 2001, 15, 428–443. [Google Scholar] [CrossRef] [Green Version]
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sevilla, A.; Forero, E.; Zimmer, M.; Martinez, H.; Reggio, K.; Paull, D.; Egli, D.; Noggle, S. Derivation and characterization of the NYSCFe003-A human embryonic stem cell line. Stem Cell Res. 2017, 25, 217–220. [Google Scholar] [CrossRef]
- Lachner, M.; O’Carroll, D.; Rea, S.; Mechtler, K.; Jenuwein, T. Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 2001, 410, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Bryan, L.C.; Weilandt, D.R.; Bachmann, A.L.; Kilic, S.; Lechner, C.C.; Odermatt, P.D.; Fantner, G.E.; Georgeon, S.; Hantschel, O.; Hatzimanikatis, V.; et al. Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 2017, 45, 10504–10517. [Google Scholar] [CrossRef]
- Draper, J.S.; Pigott, C.; Thomson, J.A.; Andrews, P.W. Surface antigens of human embryonic stem cells: Changes upon differentiation in culture. J. Anat. 2002, 200, 249–258. [Google Scholar] [CrossRef]
- Fouse, S.D.; Shen, Y.; Pellegrini, M.; Cole, S.; Meissner, A.; Van Neste, L.; Jaenisch, R.; Fan, G. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell 2008, 2, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhao, Y.; Hu, C.; Zhu, J. Differential repression of human and mouse TERT genes during cell differentiation. Nucleic Acids Res. 2009, 37, 2618–2629. [Google Scholar] [CrossRef]
- Saretzki, G.; Walter, T.; Atkinson, S.; Passos, J.F.; Bareth, B.; Keith, W.N.; Stewart, R.; Hoare, S.; Stojkovic, M.; Armstrong, L.; et al. Downregulation of Multiple Stress Defense Mechanisms During Differentiation of Human Embryonic Stem Cells. Stem Cells 2008, 26, 455–464. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Liu, Y.; Hsu, Y.J.; Fujiwara, Y.; Kim, J.; Mao, X.; Yuan, G.C.; Orkin, S.H. EZH1 Mediates Methylation on Histone H3 Lysine 27 and Complements EZH2 in Maintaining Stem Cell Identity and Executing Pluripotency. Mol. Cell 2008, 32, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar] [CrossRef]
- McIntyre, B.A.S.; Ramos-Mejia, V.; Rampalli, S.; Mechael, R.; Lee, J.-H.; Alev, C.; Sheng, G.; Bhatia, M. Gli3-mediated hedgehog inhibition in human pluripotent stem cells initiates and augments developmental programming of adult hematopoiesis. Blood 2013, 121, 1543–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ornitz, D.M.; Itoh, N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 215–266. [Google Scholar] [CrossRef] [Green Version]
- Choo, A.B.; Tan, H.L.; Ang, S.N.; Fong, W.J.; Chin, A.; Lo, J.; Zheng, L.; Hentze, H.; Philp, R.J.; Oh, S.K.W.; et al. Selection Against Undifferentiated Human Embryonic Stem Cells by a Cytotoxic Antibody Recognizing Podocalyxin-Like Protein-1. Stem Cells 2008, 26, 1454–1463. [Google Scholar] [CrossRef] [PubMed]
- Gires, O.; Klein, C.A.; Baeuerle, P.A. On the abundance of EpCAM on cancer stem cells. Nat. Rev. Cancer 2009, 9, 143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, V.Y.; Ang, S.N.; Chan, J.X.; Choo, A.B.H. Characterization of Epithelial Cell Adhesion Molecule as a Surface Marker on Undifferentiated Human Embryonic Stem Cells. Stem Cells 2010, 28, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Niida, A.; Hiroko, T.; Kasai, M.; Furukawa, Y.; Nakamura, Y.; Suzuki, Y.; Sugano, S.; Akiyama, T. DKK1, a negative regulator of Wnt signaling, is a target of the β-catenin/TCF pathway. Oncogene 2004, 23, 8520–8526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila Mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Wu, L.; Sun, T.; Kobayashi, K.; Gao, P.; Griffin, J.D. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol. Cell. Biol. 2002, 22, 7688–7700. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Johkura, K.; Yue, F.; Ogiwara, N.; Okouchi, Y.; Asanuma, K.; Sasaki, K. Spatial Distribution and Initial Changes of SSEA-1 and Other Cell Adhesion-related Molecules on Mouse Embryonic Stem Cells Before and During Differentiation. J. Histochem. Cytochem. 2004, 52, 1447–1457. [Google Scholar] [CrossRef]
- Nagano, K.; Yoshida, Y.; Isobe, T. Cell surface biomarkers of embryonic stem cells. Proteomics 2008, 8, 4025–4035. [Google Scholar] [CrossRef]
- Hu, G.; Kim, J.; Xu, Q.; Leng, Y.; Orkin, S.H.; Elledge, S.J. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009, 23, 837–848. [Google Scholar] [CrossRef] [Green Version]
- Cheng, B.; Ren, X.; Kerppola, T.K. KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Mol. Cell. Biol. 2014, 34, 2075–2091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.B.; Zhang, Y. Mi2, an auto-antigen for dermatomyositis, is an ATP-dependent nucleosome remodeling factor. Nucleic Acids Res. 2001, 29, 2517–2521. [Google Scholar] [CrossRef]
- Tanaka, S.; Pfleger, C.; Lai, J.F.; Roan, F.; Sun, S.C.; Ziegler, S.F. KAP1 Regulates Regulatory T Cell Function and Proliferation in Both Foxp3-Dependent and -Independent Manners. Cell Rep. 2018, 23, 796–807. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, L.; Wang, J.; Zhao, X.; Xu, Q.; Lu, Y.; Zuo, Y.; Chen, L.; Du, J.; Lian, Y.; et al. Downregulation of TRIM28 inhibits growth and increases apoptosis of nude mice with non-small cell lung cancer xenografts. Mol. Med. Rep. 2018, 17, 835–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell–like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Pinto, J.P.; Kalathur, R.K.; Oliveira, D.V.; Barata, T.; Machado, R.S.R.; Machado, S.; Pacheco-Leyva, I.; Duarte, I.; Futschik, M.E. StemChecker: A web-based tool to discover and explore stemness signatures in gene sets. Nucleic Acids Res. 2015, 43, W72–W77. [Google Scholar] [CrossRef] [PubMed]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Su, P.; Wang, L.; Chen, J.; Zimmermann, M.; Genbacev, O.; Afonja, O.; Horne, M.C.; Tanaka, T.; Duan, E.; et al. mTOR supports long-term self-renewal and suppresses mesoderm and endoderm activities of human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2009, 106, 7840–7845. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.S.L.; Cui, W. Proliferation, survival and metabolism: The role of PI3K/AKT/ mTOR signalling in pluripotency and cell fate determination. Development 2016, 143, 3050–3060. [Google Scholar] [CrossRef] [Green Version]
- Do, E.K.; Moon, H.J.; Kang, K.T.; Yoon, J.W.; Kim, Y.S.; Seo, J.K.; Kim, J.H. Kap1 regulates the self-renewal of embryonic stem cells and cellular reprogramming by modulating Oct4 protein stability. Cell Death Differ. 2021, 28, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Takebe, N.; Lorusso, P. Targeting the Hedgehog pathway in cancer. Ther. Adv. Med. Oncol. 2010, 2, 237–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2017, 18, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Warneke, V.S.; Behrens, H.-M.; Haag, J.; Krüger, S.; Simon, E.; Mathiak, M.; Ebert, M.P.A.; Röcken, C. Members of the EpCAM signalling pathway are expressed in gastric cancer tissue and are correlated with patient prognosis. Br. J. Cancer 2013, 109, 2217–2227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, F.Q.; Qi, Y.M.; Xu, H.; Wang, Q.Y.; Gao, X.S.; Guo, H.G. Expression of EpCAM and Wnt/ β-catenin in human colon cancer. Genet. Mol. Res. 2015, 14, 4485–4494. [Google Scholar] [CrossRef]
- Teng, Y.; Zeisberg, M.; Kalluri, R. Transcriptional regulation of epithelial-mesenchymal transition. J. Clin. Investig. 2007, 117, 304–306. [Google Scholar] [CrossRef] [PubMed]
- Venkov, C.D.; Link, A.J.; Jennings, J.L.; Plieth, D.; Inoue, T.; Nagai, K.; Xu, C.; Dimitrova, Y.N.; Rauscher, F.J.; Neilson, E.G.; et al. A proximal activator of transcription in epithelial-mesenchymal transition. J. Clin. Investig. 2007, 117, 482–491. [Google Scholar] [CrossRef]
- Nishimine, M.; Nakamura, M.; Mishima, K.; Kishi, M.; Kirita, T.; Sugimura, M.; Konishi, N. Id proteins are overexpressed in human oral squamous cell carcinomas. J. Oral Pathol. Med. 2003, 32, 350–357. [Google Scholar] [CrossRef]
- Cho, Y.; Cho, E.J.; Lee, J.-H.; Yu, S.J.; Kim, Y.J.; Kim, C.Y.; Yoon, J.-H. Fucoidan-induced ID-1 suppression inhibits the in vitro and in vivo invasion of hepatocellular carcinoma cells. Biomed. Pharmacother. 2016, 83, 607–616. [Google Scholar] [CrossRef]
- Sun, B.; Dong, C.; Lei, H.; Gong, Y.; Li, M.; Zhang, Y.; Zhang, H.; Sun, L. Knockdown of inhibitor of differentiation 1 suppresses proliferation and induces apoptosis by inactivating PI3K/Akt/mTOR signaling in hemangioma-derived endothelial cells. Biomed. Pharmacother. 2019, 111, 236–243. [Google Scholar] [CrossRef]
- Crosnier, C.; Stamataki, D.; Lewis, J. Organizing cell renewal in the intestine: Stem cells, signals and combinatorial control. Nat. Rev. Genet. 2006, 7, 349–359. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mazurek, S.; Oleksiewicz, U.; Czerwińska, P.; Wróblewska, J.; Klimczak, M.; Wiznerowicz, M. Disruption of RING and PHD Domains of TRIM28 Evokes Differentiation in Human iPSCs. Cells 2021, 10, 1933. https://doi.org/10.3390/cells10081933
Mazurek S, Oleksiewicz U, Czerwińska P, Wróblewska J, Klimczak M, Wiznerowicz M. Disruption of RING and PHD Domains of TRIM28 Evokes Differentiation in Human iPSCs. Cells. 2021; 10(8):1933. https://doi.org/10.3390/cells10081933
Chicago/Turabian StyleMazurek, Sylwia, Urszula Oleksiewicz, Patrycja Czerwińska, Joanna Wróblewska, Marta Klimczak, and Maciej Wiznerowicz. 2021. "Disruption of RING and PHD Domains of TRIM28 Evokes Differentiation in Human iPSCs" Cells 10, no. 8: 1933. https://doi.org/10.3390/cells10081933
APA StyleMazurek, S., Oleksiewicz, U., Czerwińska, P., Wróblewska, J., Klimczak, M., & Wiznerowicz, M. (2021). Disruption of RING and PHD Domains of TRIM28 Evokes Differentiation in Human iPSCs. Cells, 10(8), 1933. https://doi.org/10.3390/cells10081933