
Prolyl Hydroxylase Domain-Containing Protein 3 Gene Expression in Chondrocytes Is Not Essential for Bone Development in Mice

Abstract
:1. Introduction
2. Materials and Methods
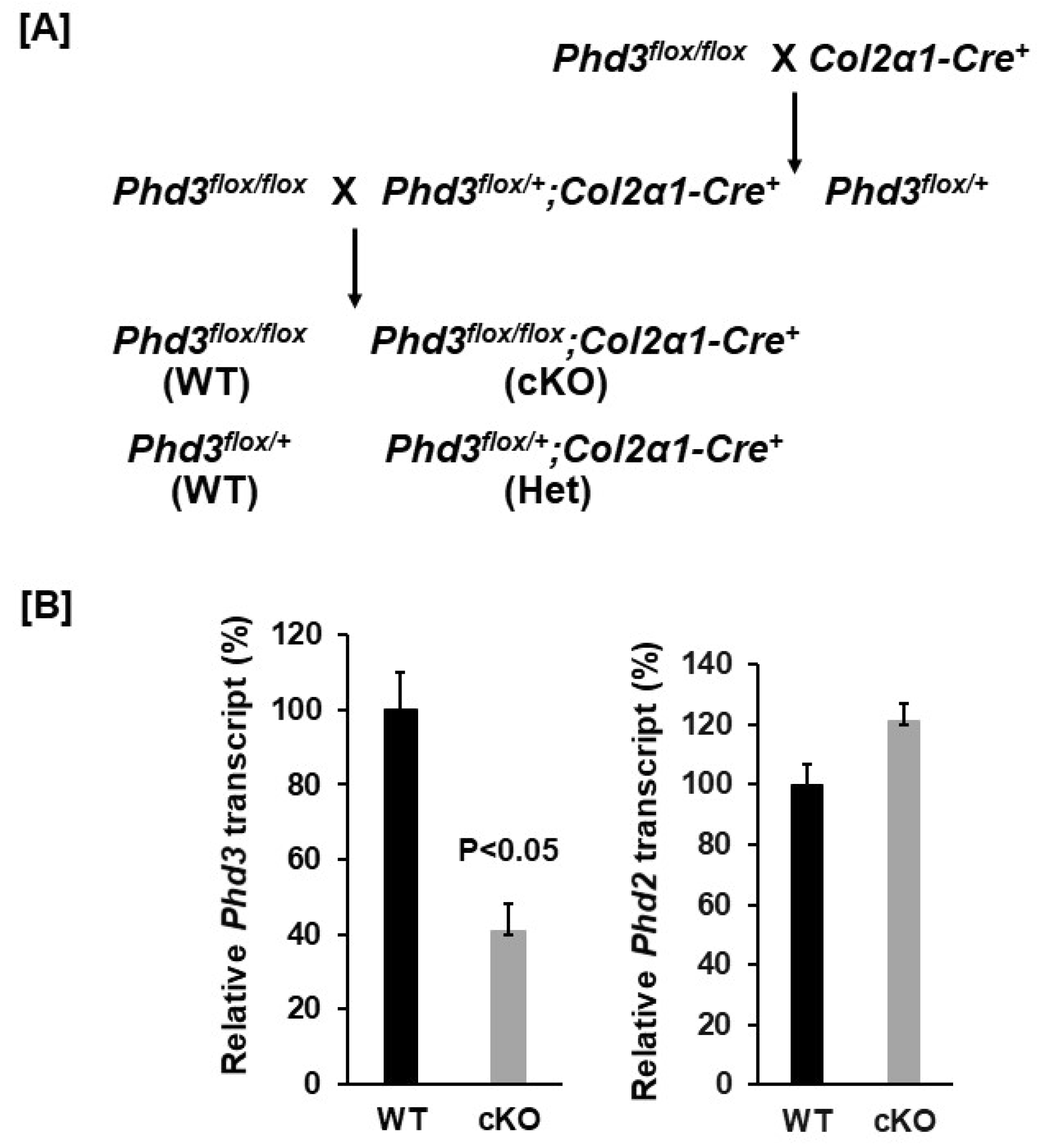
2.1. Generation of Conditional Knockout Mice
2.2. Antibodies and Biological Reagents
- Mouse Phd3-TRCN0000009753:
- CCGGCGGCTTCTGCTACCTGGACAActcgagTTGTCCAGGTAGCAGAAGCCGTTTTT
- Non-target:
- CCGGCAACAAGATGAAGAGCACCAActcgagTTGGTGCTCTTCATCTTGTTGTTTTT
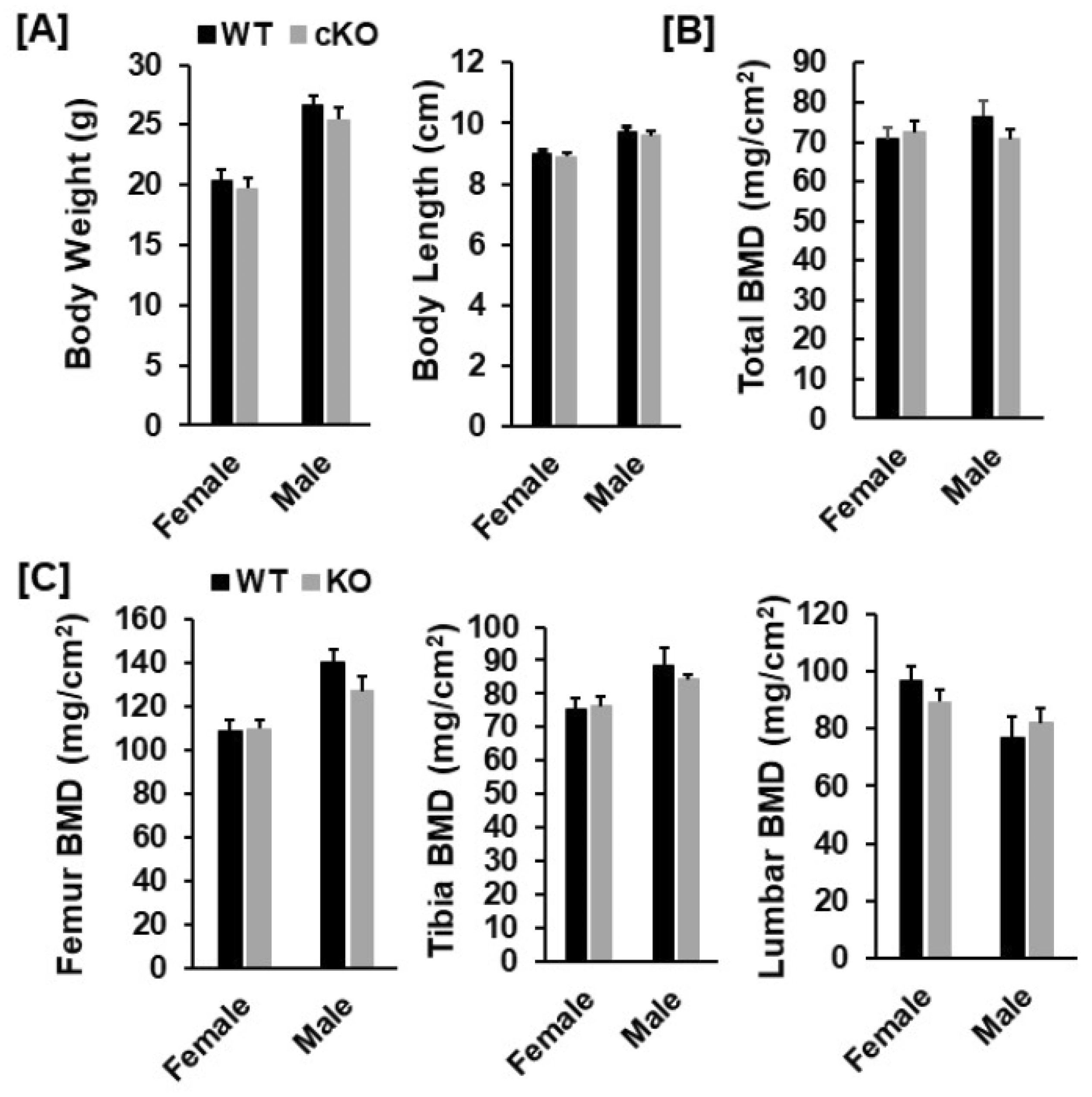
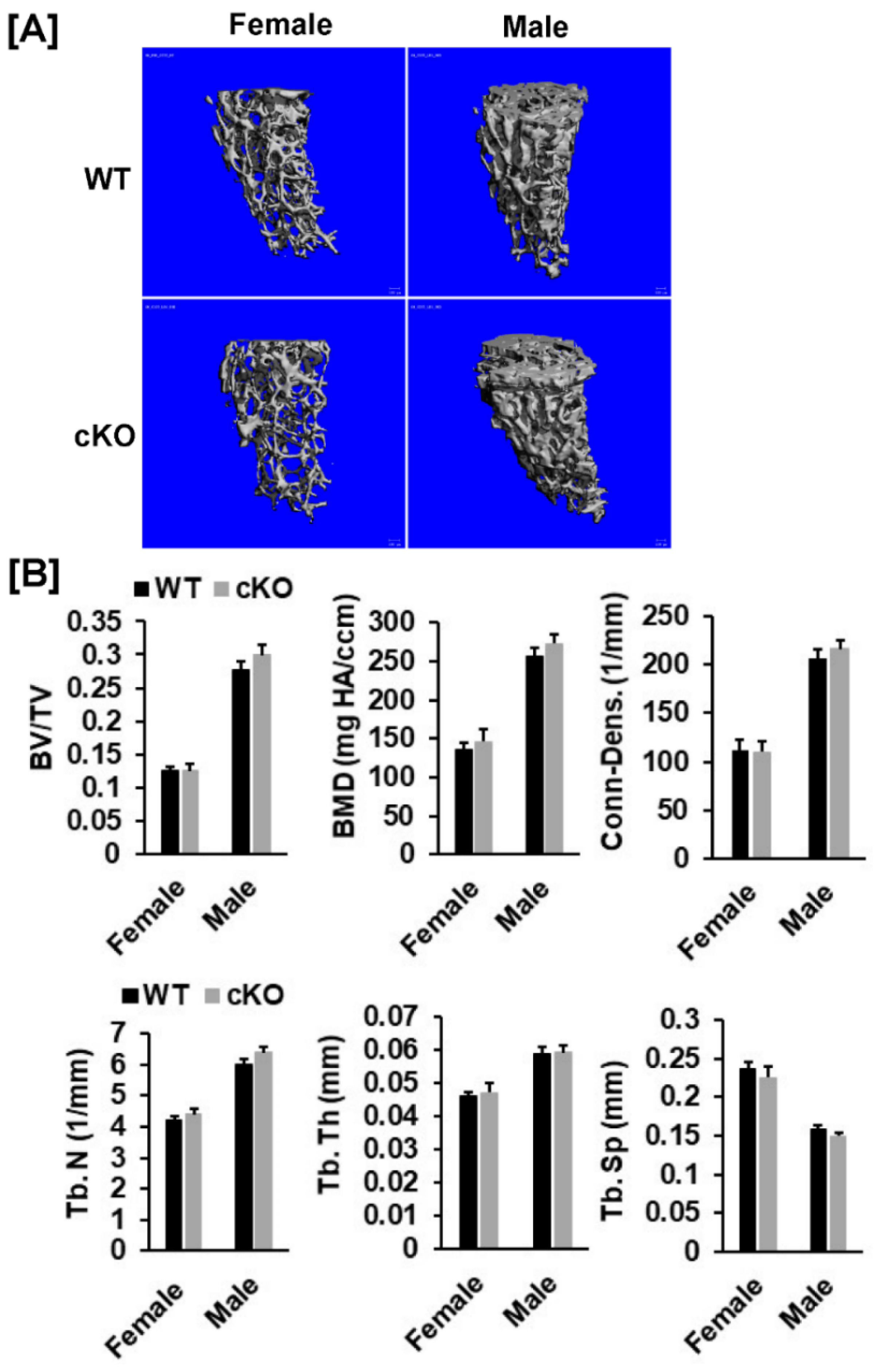
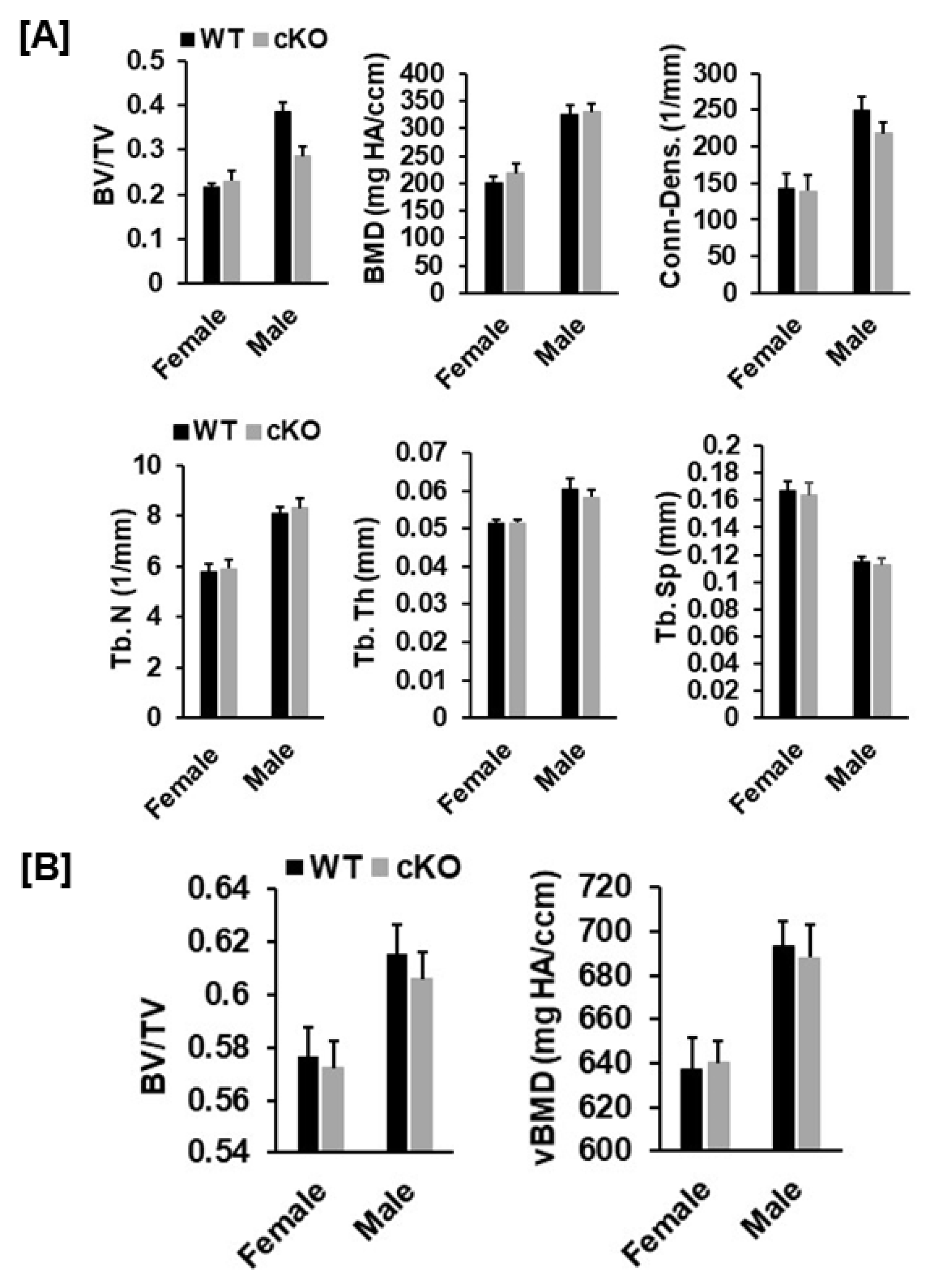
2.3. Evaluation of Bone Phenotypes
2.4. Histology and Immunohistochemistry
2.5. Cell Culture
2.6. RNA Extraction and Real-Time Quantitative Polymerase Chain Reaction
2.7. Statistical Analysis
3. Results
3.1. Conditional Knockout of Phd3 in Col2α1-Expressing Cells Does Not Impair Skeletal Development in Mice
3.2. Disruption of Phd3 Expression in Col2α1-Expressing Cells Does Not Affect Chondrocyte Differentiation and Epiphysis Development in Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R. C elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Berra, E.; Ginouves, A.; Pouyssegur, J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 2006, 7, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Villar, D.; Vara-Vega, A.; Landazuri, M.O.; Del Peso, L. Identification of a region on hypoxia-inducible-factor prolyl 4-hydroxylases that determines their specificity for the oxygen degradation domains. Biochem. J. 2007, 408, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.E.; Bunn, H.F. Hypoxia-inducible factor and its biomedical relevance. J. Biol. Chem. 2003, 278, 19575–19578. [Google Scholar] [CrossRef] [Green Version]
- Wan, C.; Shao, J.; Gilbert, S.R.; Riddle, R.C.; Long, F.; Johnson, R.S. Role of HIF-1alpha in skeletal development. Ann. N. Y. Acad. Sci. 2010, 1192, 322–326. [Google Scholar] [CrossRef] [Green Version]
- Cummins, E.P.; Berra, E.; Comerford, K.M.; Ginouves, A.; Fitzgerald, K.T.; Seeballuck, F. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc. Natl. Acad. Sci. USA 2006, 103, 18154–18159. [Google Scholar] [CrossRef] [Green Version]
- Xie, L.; Xiao, K.; Whalen, E.J.; Forrester, M.T.; Freeman, R.S.; Fong, G. Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci. Signal. 2009, 2, ra33. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, J.E., III; Wu, Y.; Smith, K.; Charles, P.; Powers, K.; Wang, H. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol. Cell. Biol. 2007, 27, 6407–6419. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.H.; Ongusaha, P.P.; Myllyharju, J.; Cheng, D.; Pakkanen, O.; Shi, Y. Prolyl 4-hydroxylation regulates Argonaute 2 stability. Nature 2008, 455, 421–424. [Google Scholar] [CrossRef]
- Takeda, K.; Ho, V.C.; Takeda, H.; Duan, L.J.; Nagy, A.; Fong, G.H. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell. Biol. 2006, 26, 8336–8346. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Mohan, S. Conditional disruption of the prolyl hydroxylase domain-containing protein 2 (Phd2) gene defines its key role in skeletal development. J. Bone Miner. Res. 2014, 29, 2276–2286. [Google Scholar] [CrossRef]
- Cheng, S.; Xing, W.; Pourteymoor, S.; Schulte, J.; Mohan, S. Conditional deletion of prolyl hydroxylase domain-containing protein 2 (Phd2) gene reveals its essential role in chondrocyte function and endochondral bone formation. Endocrinology 2015, 1, en20151473. [Google Scholar] [CrossRef] [Green Version]
- Duan, L.J.; Takeda, K.; Fong, G.H. Hematological, hepatic, and retinal phenotypes in mice deficient for prolyl hydroxylase domain proteins in the liver. Am. J. Pathol. 2014, 184, 1240–1250. [Google Scholar] [CrossRef] [Green Version]
- Minamishima, Y.A.; Moslehi, J.; Bardeesy, N.; Cullen, D.; Bronson, R.T.; Kaelin, W.G., Jr. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 2008, 111, 3236–3244. [Google Scholar] [CrossRef] [Green Version]
- Ovchinnikov, D.A.; Deng, J.M.; Ogunrinu, G.; Behringer, R.R. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis 2000, 26, 145–146. [Google Scholar] [CrossRef]
- Xing, W.; Liu, J.; Cheng, S.; Vogel, P.; Mohan, S.; Brommage, R. Targeted disruption of leucine-rich repeat kinase 1 but not leucine-rich repeat kinase 2 in mice causes severe osteopetrosis. J. Bone Miner. Res. 2013, 28, 1962–1974. [Google Scholar] [CrossRef]
- Bouxsein, M.L.; Boyd, S.K.; Christiansen, B.A.; Guldberg, R.E.; Jepsen, K.J.; Muller, R. Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J. Bone Miner. Res. 2010, 25, 1468–1486. [Google Scholar] [CrossRef]
- Xing, W.; Pourteymoor, S.; Mohan, S. Ascorbic acid regulates osterix expression in osteoblasts by activation of prolyl hydroxylase and ubiquitination-mediated proteosomal degradation pathway. Physiol. Genom. 2011, 43, 749–757. [Google Scholar] [CrossRef] [Green Version]
- Xing, W.; Kim, J.; Wergedal, J.; Chen, S.T.; Mohan, S. Ephrin B1 regulates bone marrow stromal cell differentiation and bone formation by influencing TAZ transactivation via complex formation with NHERF1. Mol. Cell. Biol. 2010, 30, 711–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beamer, W.G.; Donahue, L.R.; Rosen, C.J.; Baylink, D.J. Genetic variability in adult bone density among inbred strains of mice. Bone 1996, 18, 397–403. [Google Scholar] [CrossRef]
- Qin, X.; Wergedal, J.E.; Rehage, M.; Tran, K.; Newton, J.; Lam, P. Pregnancy-associated plasma protein-A increases osteoblast proliferation in vitro and bone formation in vivo. Endocrinology 2006, 147, 5653–5661. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.; Xing, W.; Zhou, X.; Mohan, S. Haploinsufficiency of osterix in chondrocytes impairs skeletal growth in mice. Physiol. Genom. 2013, 45, 917–923. [Google Scholar] [CrossRef] [Green Version]
- Xing, W.; Singgih, A.; Kapoor, A.; Alarcon, C.M.; Baylink, D.J.; Mohan, S. Nuclear factor-E2-related factor-1 mediates ascorbic acid induction of osterix expression via interaction with antioxidant-responsive element in bone cells. J. Biol. Chem. 2007, 282, 22052–22061. [Google Scholar] [CrossRef] [Green Version]
- Xing, W.; Baylink, D.; Kesavan, C.; Hu, Y.; Kapoor, S.; Chadwick, R.B. Global gene expression analysis in the bones reveals involvement of several novel genes and pathways in mediating an anabolic response of mechanical loading in mice. J. Cell. Biochem. 2005, 96, 1049–1060. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Lieb, M.E.; Menzies, K.; Moschella, M.C.; Ni, R.; Taubman, M.B. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem. Cell Biol. 2002, 80, 421–426. [Google Scholar] [CrossRef]
- Takeda, K.; Aguila, H.L.; Parikh, N.S.; Li, X.; Lamothe, K.; Duan, L.J. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood 2008, 111, 3229–3235. [Google Scholar] [CrossRef] [Green Version]
- Laperre, K.F.P.; Van Looveren, R.; Bouillon, R.; Carmeliet, P.; Carmeliet, G. Deletion of the oxygen-sensor PHD2 in chondrocytes results in increased cartilage and bone mineralization. J. Bone Miner. Res. 2008, 1, S44. [Google Scholar]
- Fong, G.H.; Takeda, K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008, 15, 635–641. [Google Scholar] [CrossRef] [Green Version]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzen, E.; Berchner-Pfannschmidt, U.; Stengel, P.; Marxsen, J.H.; Stolze, I.; Klinger, M. Intracellular localisation of human HIF-1 alpha hydroxylases: Implications for oxygen sensing. J. Cell Sci. 2003, 116, 1319–1326. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Cowan, A.; Fong, G.H. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 2007, 116, 774–781. [Google Scholar] [CrossRef] [PubMed]
- Provot, S.; Zinyk, D.; Gunes, Y.; Kathri, R.; Le, Q.; Kronenberg, H.M. Hif-1alpha regulates differentiation of limb bud mesenchyme and joint development. J. Cell Biol. 2007, 177, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Gilbert, S.R.; Wang, Y.; Cao, X.; Shen, X.; Ramaswamy, G. Activation of the hypoxia-inducible factor-1alpha pathway accelerates bone regeneration. Proc. Natl. Acad. Sci. USA 2008, 105, 686–691. [Google Scholar] [CrossRef] [Green Version]
- Shomento, S.H.; Wan, C.; Cao, X.; Faugere, M.C.; Bouxsein, M.L.; Clemens, T.L. Hypoxia-inducible factors 1alpha and 2alpha exert both distinct and overlapping functions in long bone development. J. Cell. Biochem. 2010, 109, 196–204. [Google Scholar] [CrossRef]
- Lindsey, R.C.; Cheng, S.; Mohan, S. Vitamin C effects on 5-hydroxymethylcytosine and gene expression in osteoblasts and chondrocytes: Potential involvement of PHD2. PLoS ONE 2019, 14, e0220653. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer | Reverse Primer |
|---|---|---|
| Ppia | 5′-CCATGGCAAATGCTGGACCA | 5′-TCCTGGACCCAAAACGCTCC |
| Phd3 Phd2 | 5′-GGGACGCCAAGTTACACGGA 5′-GAAGCTGGGCAACTACAGGA | 5′-GGGCTCCACGTCTGCTACAA 5′-CATGTCACGCATCTTCCATC |
| Col2 | 5′-TGGCTTCCACTTCAGCTATG | 5′-AGGTAGGCGATGCTGTTCTT |
| Col10 | 5′-ACGGCACGCCTACGATGT | 5′-CCATGATTGCACTCCCTGAA |
| Acan | 5′-GACCAGGAAGGGAGGAGTAG | 5′-CAGCCGAGAAATGACACC |
| Sox9 | 5′-CGGAGGAAGTCGGTGAAGA | 5′-GTCGGTTTTGGGAGTGGTG |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, W.; Pourteymoor, S.; Gomez, G.A.; Chen, Y.; Mohan, S. Prolyl Hydroxylase Domain-Containing Protein 3 Gene Expression in Chondrocytes Is Not Essential for Bone Development in Mice. Cells 2021, 10, 2200. https://doi.org/10.3390/cells10092200
Xing W, Pourteymoor S, Gomez GA, Chen Y, Mohan S. Prolyl Hydroxylase Domain-Containing Protein 3 Gene Expression in Chondrocytes Is Not Essential for Bone Development in Mice. Cells. 2021; 10(9):2200. https://doi.org/10.3390/cells10092200
Chicago/Turabian StyleXing, Weirong, Sheila Pourteymoor, Gustavo A. Gomez, Yian Chen, and Subburaman Mohan. 2021. "Prolyl Hydroxylase Domain-Containing Protein 3 Gene Expression in Chondrocytes Is Not Essential for Bone Development in Mice" Cells 10, no. 9: 2200. https://doi.org/10.3390/cells10092200
APA StyleXing, W., Pourteymoor, S., Gomez, G. A., Chen, Y., & Mohan, S. (2021). Prolyl Hydroxylase Domain-Containing Protein 3 Gene Expression in Chondrocytes Is Not Essential for Bone Development in Mice. Cells, 10(9), 2200. https://doi.org/10.3390/cells10092200