
Epitranscriptomic Approach: To Improve the Efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
1.1. Connotation of Immune Checkpoint Markers
1.2. ICB Drug-Resistance and Toxicities
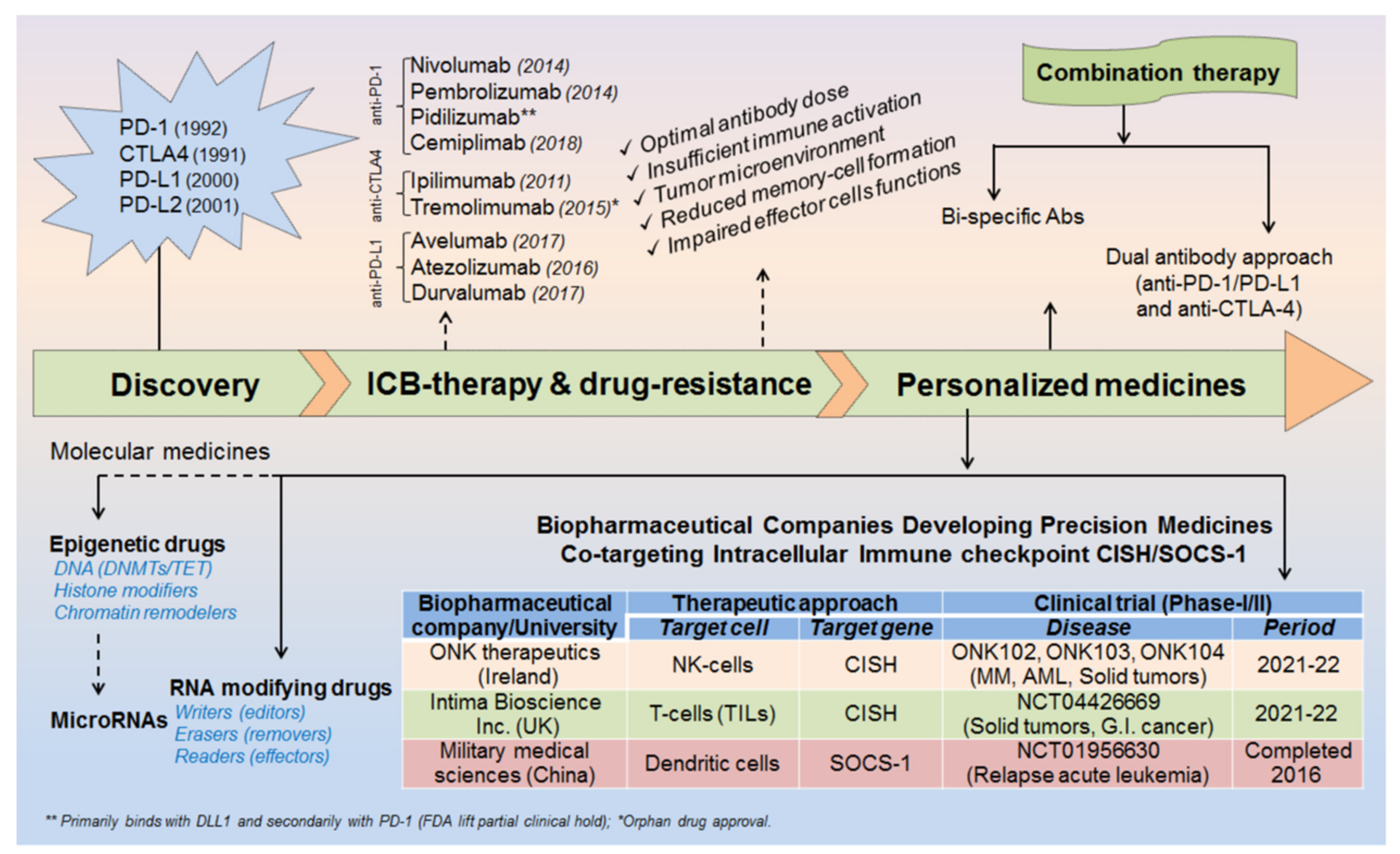
2. Milestones in ICB therapeutics
2.1. Discovery of ICB Therapy
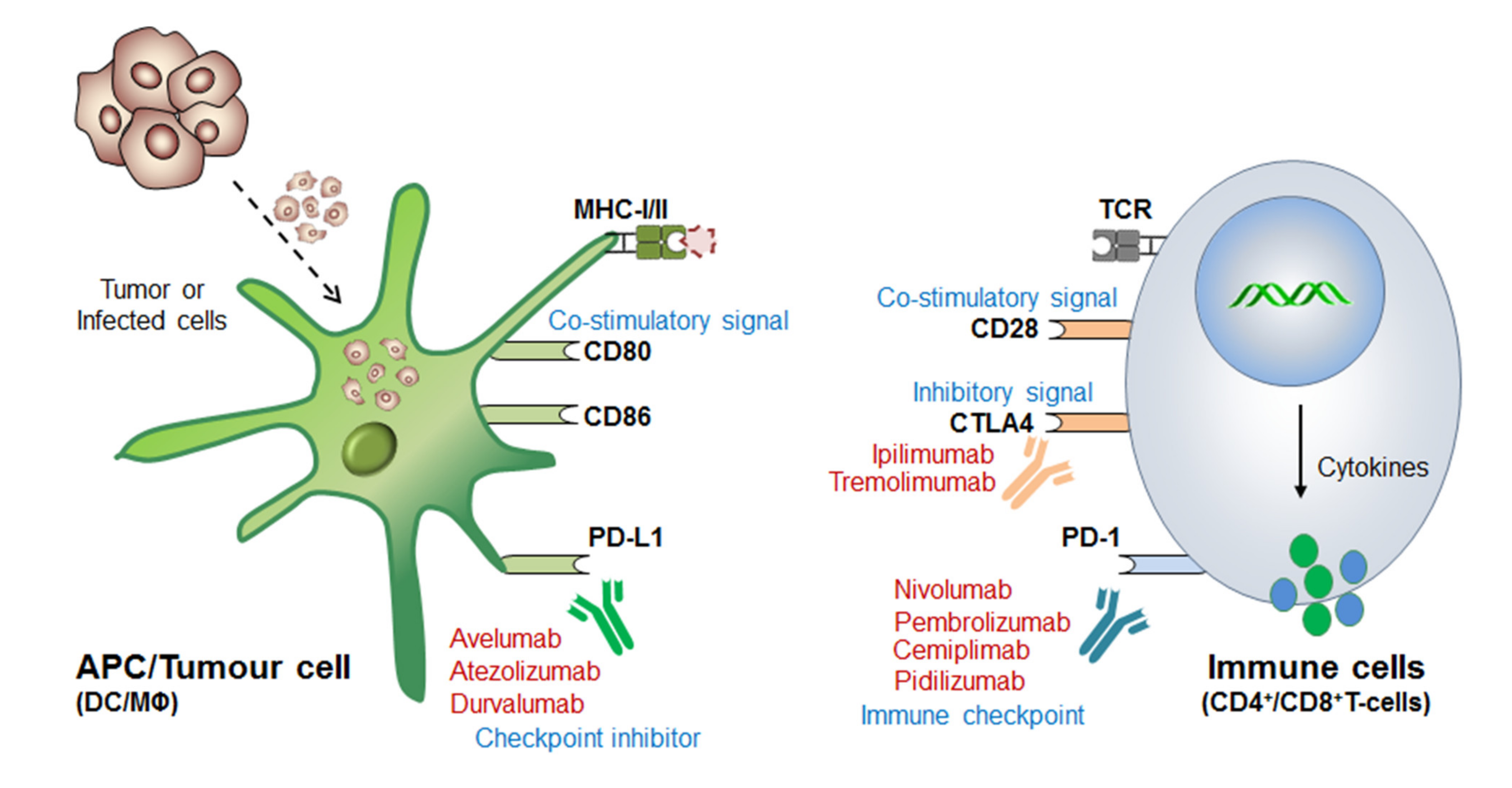
2.2. Mechanism of ICB/ICI-Therapeutics
2.3. Strategies to Overcome ICB Drug-Resistance
3. Epitranscriptomics in ICB-Therapeutics
3.1. Editors (Writers):
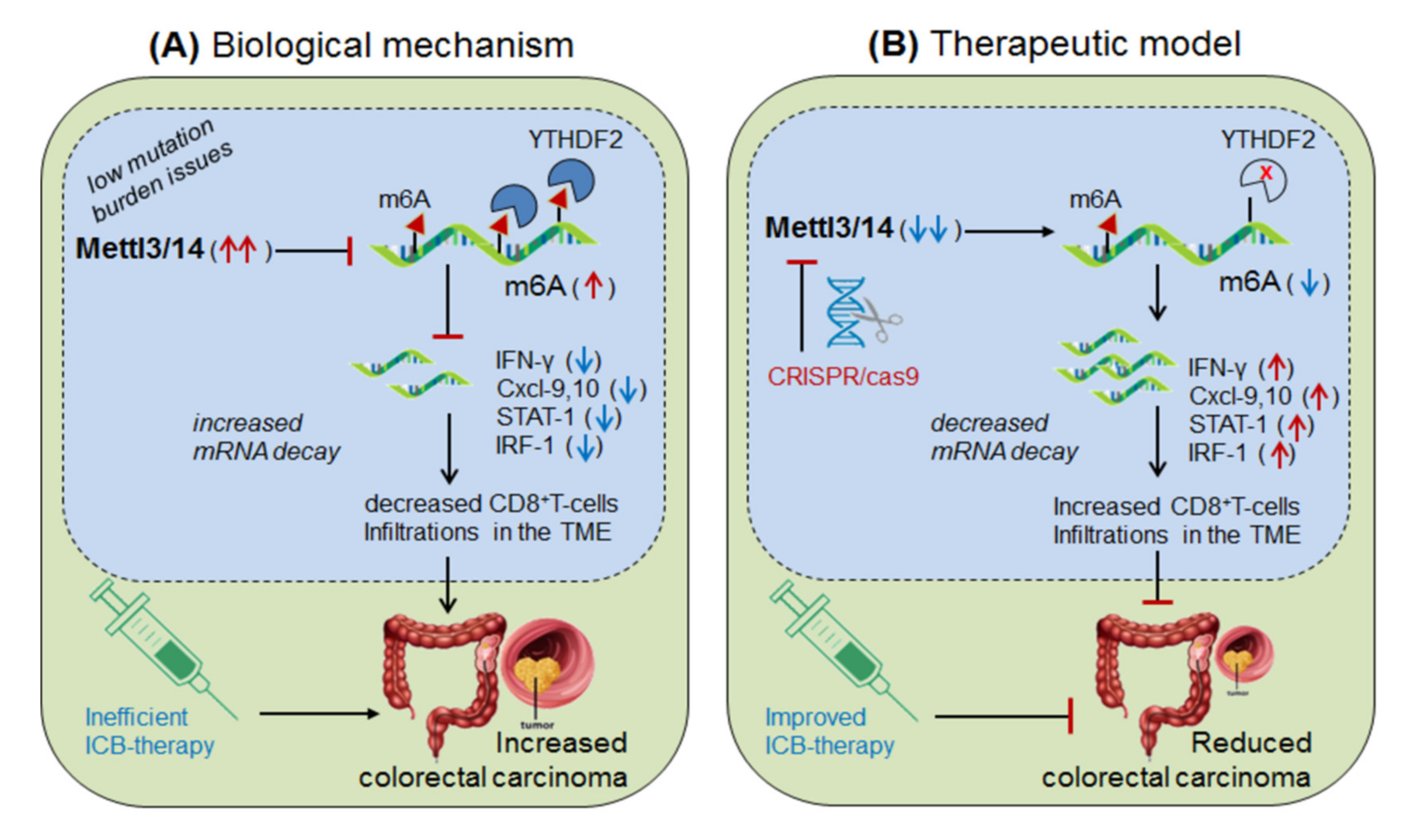
3.1.1. Mettl-3/14 in Anti-PD-1 Resistance (Colorectal Cancer)
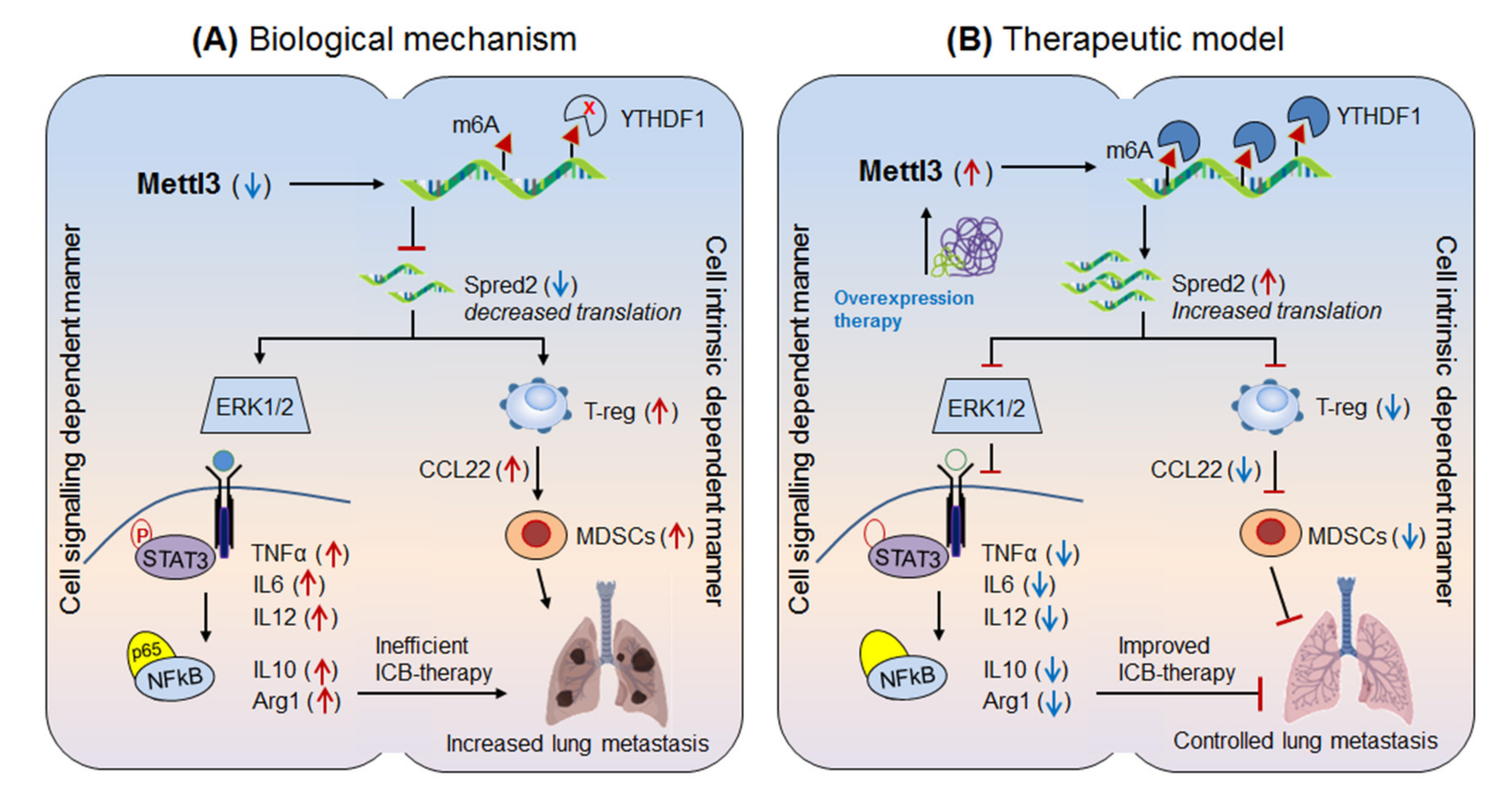
3.1.2. Mettl-3 in Anti-PD-1 Resistance (Lung Metastasis)
3.2. Erasers (Removers):
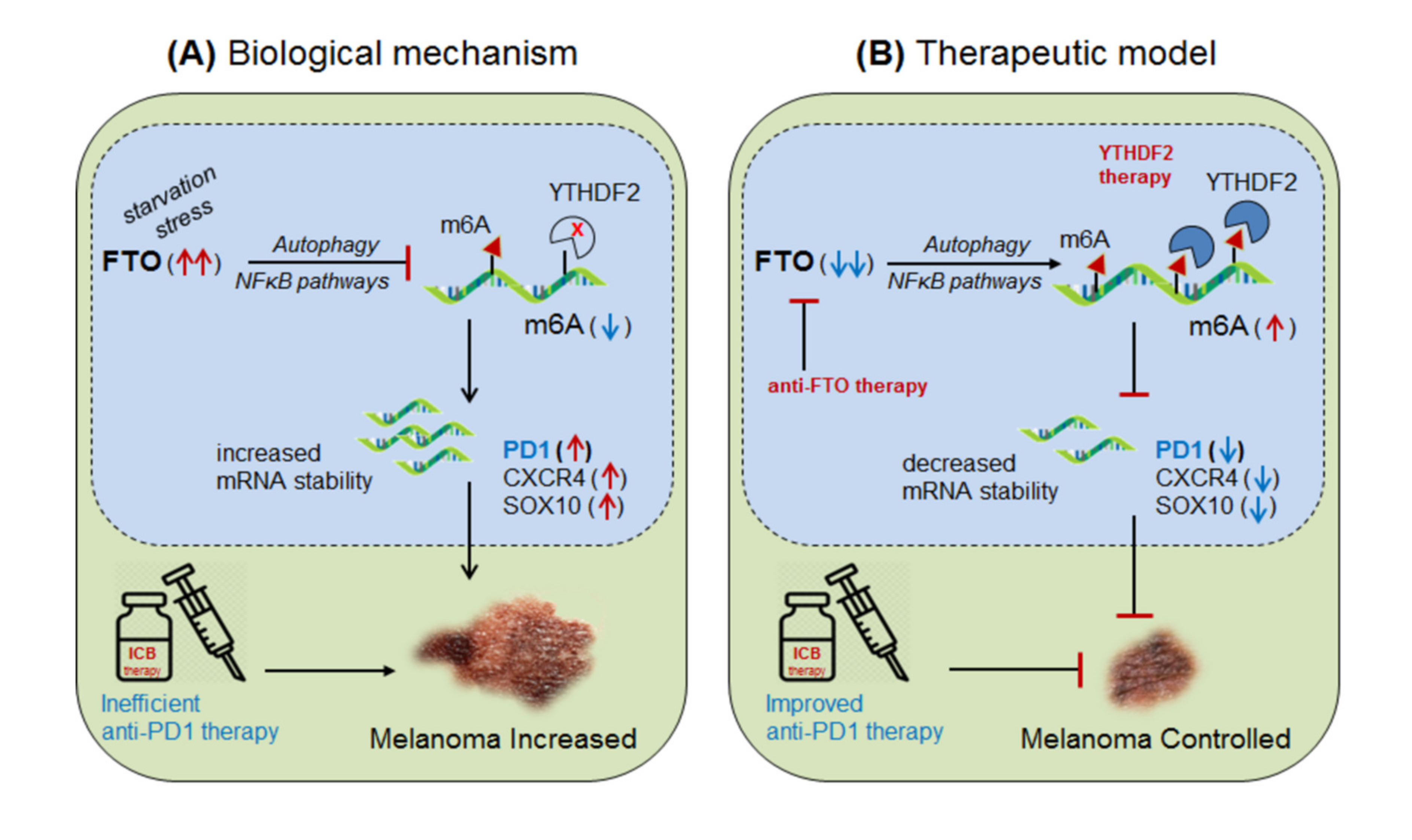
3.2.1. FTO in Anti-PD1 Resistance (Melanoma):
3.2.2. FTO in Anti-PD-1 Resistance (Colon Cancer)
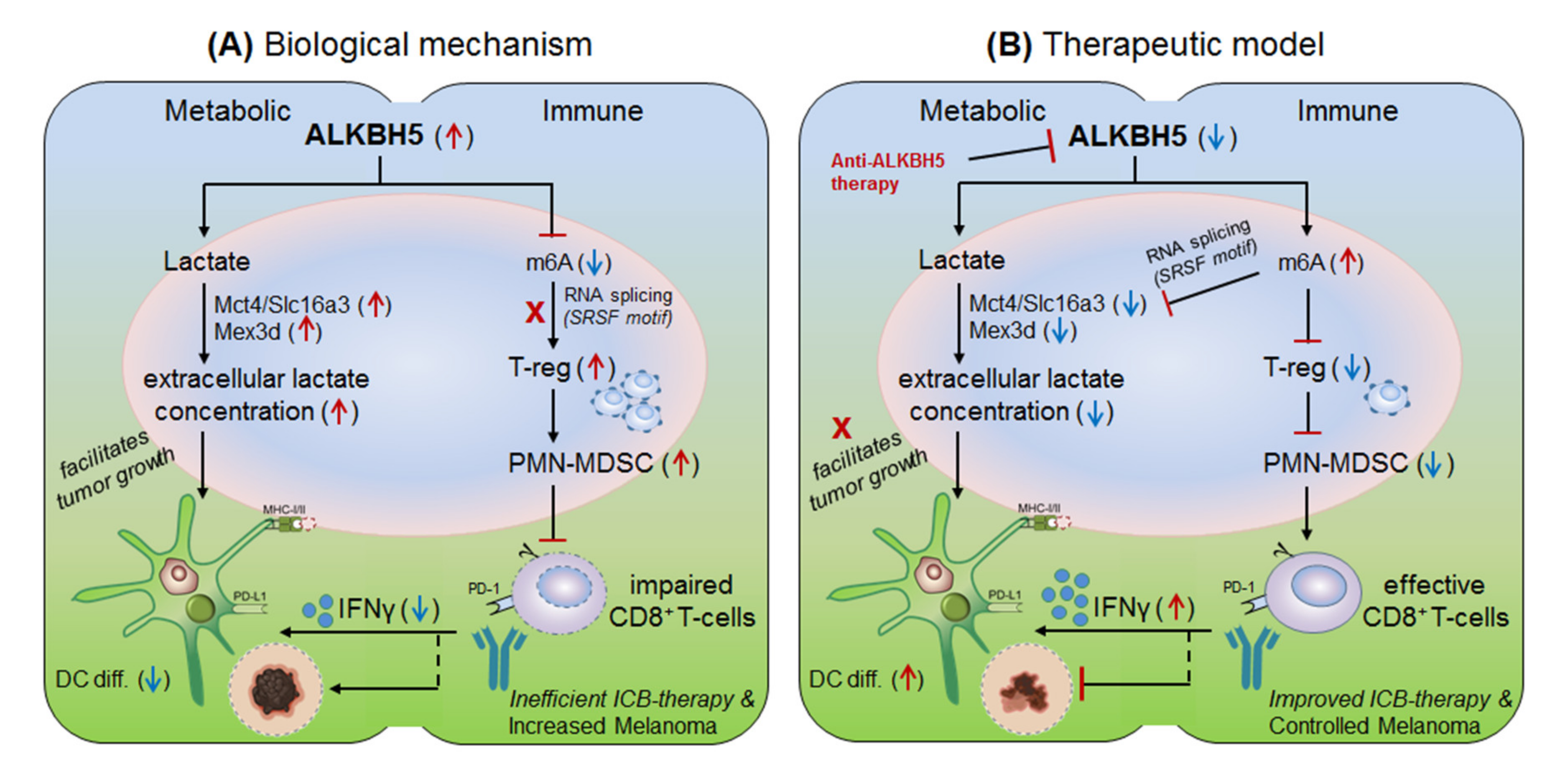
3.2.3. ALKBH5 in Anti-PD-1 Resistance (Melanoma)
3.3. Effectors (Readers):
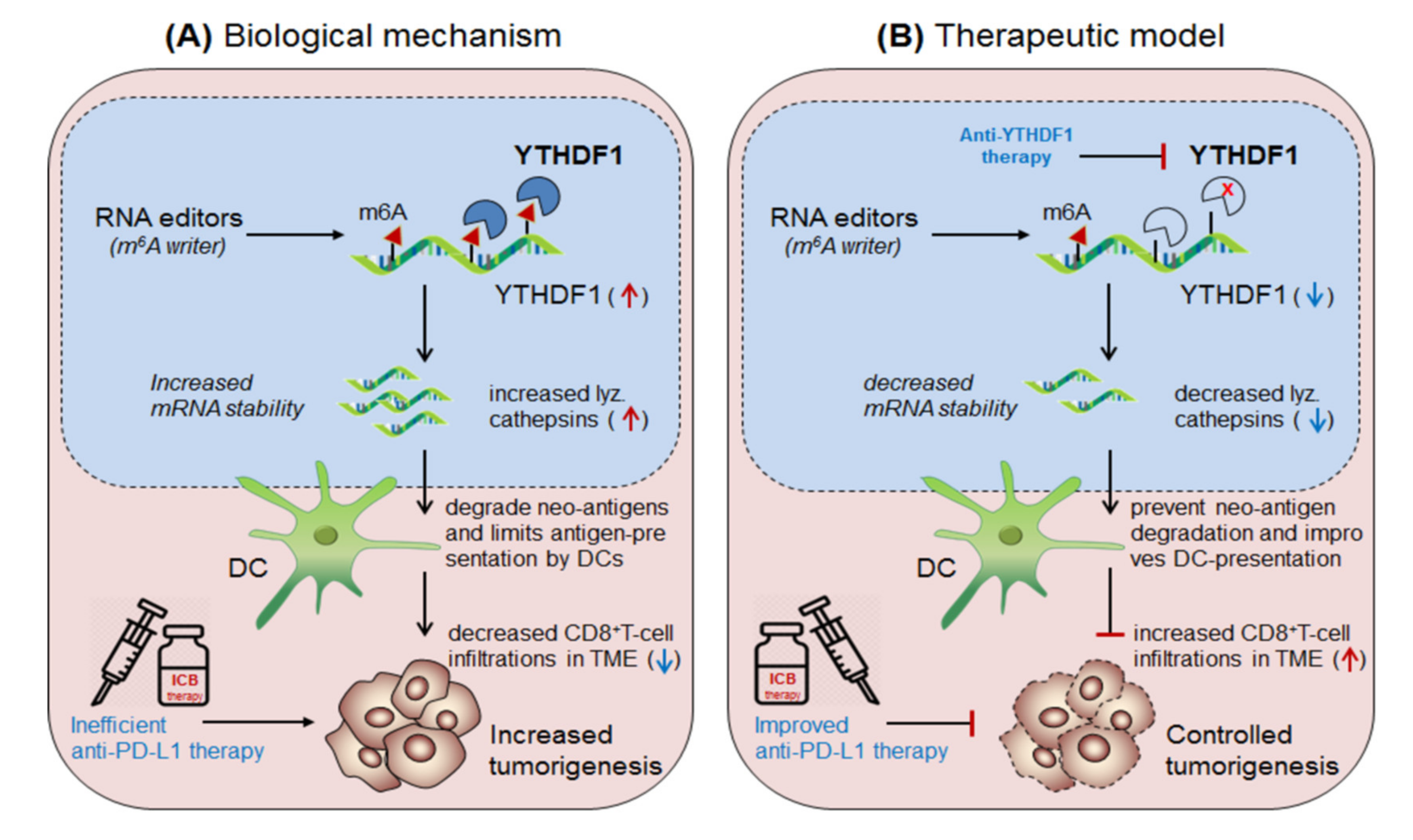
3.3.1. YTHDF1 in Anti-PD1 Resistance (Solid Tumors)
3.3.2. YTHDF2 in Anti-PD1 Resistance (Brain Tumors)
4. Immune Cells: Targeting Intracellular Checkpoint ‘CISH’ in Combination with ICB-Therapeutics and Recent Clinical Trials
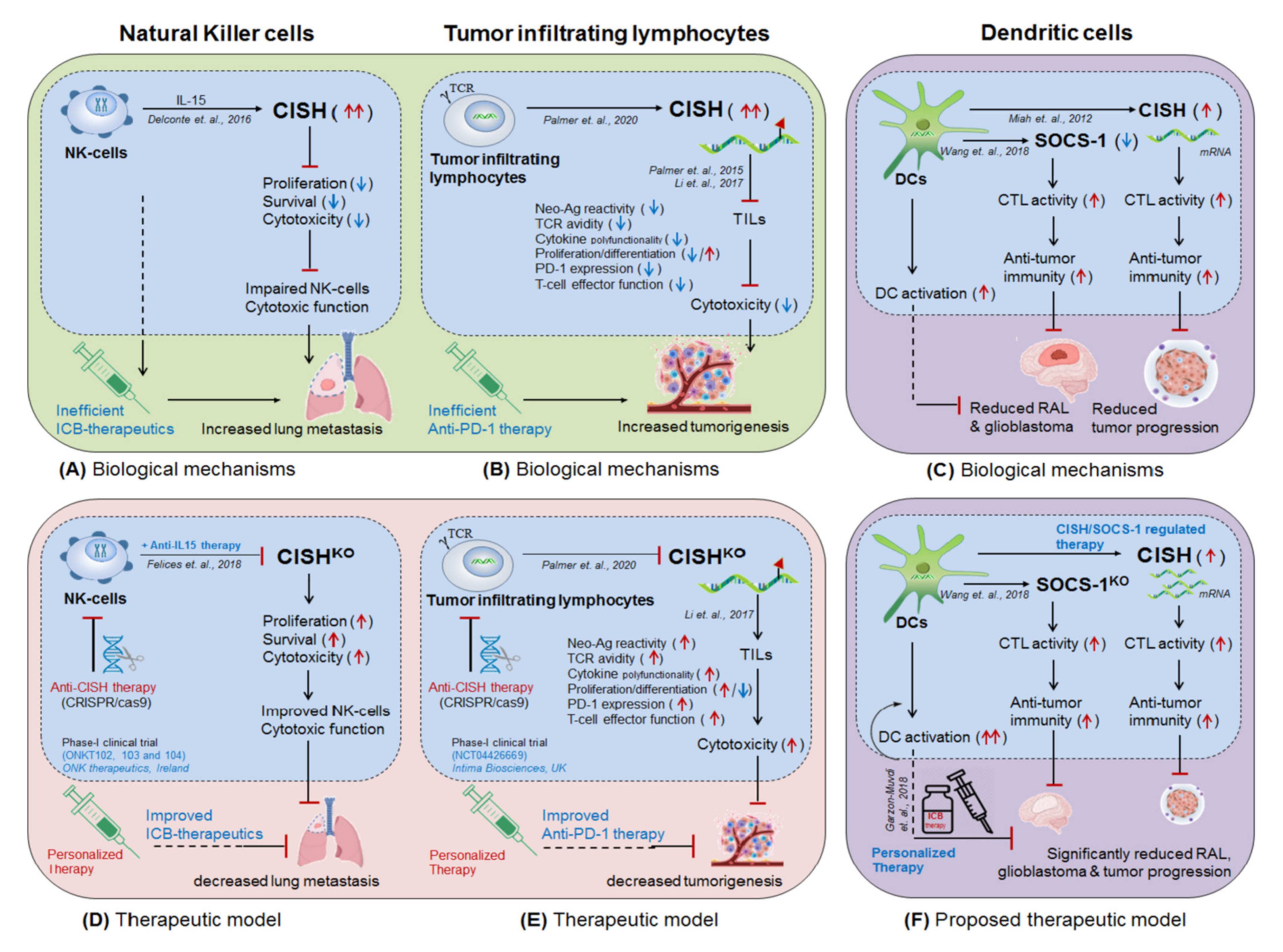
4.1. NK-Cells Targeting CISH in ICB Therapeutics
4.2. T-cells Targeting CISH in ICB Therapeutics
4.3. Dendritic Cells Targeting SOCS-1/CISH in ICB-Therapeutics
5. MicroRNAs and Epigenetic Modifiers (DNA and Histone Proteins) in ICB-Therapy
5.1. MicroRNAs in ICB-Therapeutics
5.2. Epigenetic Modifiers (DNA and Histone Proteins) in ICB-Therapeutics
6. Biopharmaceutical Companies Developing Personalized Medicines: Targeting Intracellular Checkpoint ‘CISH’ in Combination with ICB-Therapeutics and Recent Clinical Trials
6.1. ONK Therapeutics Limited
6.2. Intima Bioscience, Inc.
- ■
- NK-cells targeting intracellular checkpoint CISH: ONK therapeutics, Ireland, estimated to conduct Phase-I clinical trial (ONKT102, ONKT103 and ONKT104) by 2021𠄲2022 for the treatment of haematological malignancies (multiple myeloma and AML) and solid tumors (ovarian, NSCLC and breast cancers) [135].
- ■
- ■
7. Conclusions
8. Future Prospective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AIA | Ag-induced arthritis |
| ALKBH5 | alpha-ketoglutarate-dependent hydroxylase |
| CAND1 | Cullin-associated NEDD8-dissociated protein 1 |
| EZH2 | Enhancer of zeste 2 polycomb repressive complex-2 subunit |
| EBF-1 | Early B-cell factor-1 |
| FTO | Fat Mass and Obesity Associated Protein |
| MPM | Malignant pleural mesothelioma |
| Spred2 | Sprouty related EVH1 domain containing protein-2 |
References
- Palmer, D.; Webber, B.; Patel, Y.; Johnson, M.; Kariya, C.; Lahr, W.; Parkhurst, M.; Gartner, J.; Prickett, T.; Lowery, F.; et al. 333 Targeting the apical intracellular checkpoint CISH unleashes T cell neoantigen reactivity and effector program. J. Immunol. Ther. Cancer 2020, 8, A204. [Google Scholar] [CrossRef]
- Plieth, J. Crispr: Nice Valuation, but Where’s the Clinical Trial? Available online: https://www.evaluate.com/node/13152/pdf (accessed on 20 July 2021).
- Delconte, R.B.; Kolesnik, T.B.; Dagley, L.F.; Rautela, J.; Shi, W.; Putz, E.M.; Stannard, K.; Zhang, J.G.; Teh, C.; Firth, M.; et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat. Immunol. 2016, 17, 816–824. [Google Scholar] [CrossRef]
- Putz, E.M.; Guillerey, C.; Kos, K.; Stannard, K.; Miles, K.; Delconte, R.B.; Takeda, K.; Nicholson, S.E.; Huntington, N.D.; Smyth, M.J. Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 2017, 6, e1267892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, D.C.; Webber, B.R.; Patel, Y.; Johnson, M.J.; Kariya, C.M.; Lahr, W.S.; Parkhurst, M.R.; Gartner, J.J.; Prickett, T.D.; Lowery, F.J.; et al. Internal checkpoint regulates T cellneoantigen reactivity and susceptibility to PD1 blockade. bioRxiv 2020. [Google Scholar] [CrossRef]
- Palmer, D.C.; Guittard, G.C.; Franco, Z.; Crompton, J.G.; Eil, R.L.; Patel, S.J.; Ji, Y.; Van Panhuys, N.; Klebanoff, C.A.; Sukumar, M.; et al. Cish actively silences TCR signaling in CD8+ T cells to maintain tumor tolerance. J. Exp. Med. 2015, 212, 2095–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Huang, X.F.; Hong, B.; Song, X.T.; Hu, L.; Jiang, M.; Zhang, B.; Ning, H.; Li, Y.; Xu, C.; et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Hui, H.; Agrawal, K.; Kang, Y.; Li, N.; Tang, R.; Yuan, J.; Rana, T.M. m(6) A RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020, 39, e104514. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, X.; Yang, P.; Zhang, X.; Peng, Y.; Li, D.; Yu, Y.; Wu, Y.; Wang, Y.; Zhang, J.; et al. RNA m6A methylation orchestrates cancer growth and metastasis via macrophage reprogramming. Nat. Commun. 2021, 12, 1394. [Google Scholar] [CrossRef]
- Yi, L.; Wu, G.; Guo, L.; Zou, X.; Huang, P. Comprehensive Analysis of the PD-L1 and Immune Infiltrates of m(6)A RNA Methylation Regulators in Head and Neck Squamous Cell Carcinoma. Mol. Ther. Nucleic Acids 2020, 21, 299–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wei, J.; Cui, Y.H.; Park, G.; Shah, P.; Deng, Y.; Aplin, A.E.; Lu, Z.; Hwang, S.; He, C.; et al. m(6)A mRNA demethylase FTO regulates melanoma tumorigenicity and response to anti-PD-1 blockade. Nat. Commun. 2019, 10, 2782. [Google Scholar] [CrossRef]
- Tsuruta, N.; Tsuchihashi, K.; Ohmura, H.; Yamaguchi, K.; Ito, M.; Ariyama, H.; Kusaba, H.; Akashi, K.; Baba, E. RNA N6-methyladenosine demethylase FTO regulates PD-L1 expression in colon cancer cells. Biochem. Biophys. Res. Commun. 2020, 530, 235–239. [Google Scholar] [CrossRef]
- Li, N.; Kang, Y.; Wang, L.; Huff, S.; Tang, R.; Hui, H.; Agrawal, K.; Gonzalez, G.M.; Wang, Y.; Patel, S.P.; et al. ALKBH5 regulates anti-PD-1 therapy response by modulating lactate and suppressive immune cell accumulation in tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 20159–20170. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, Z.; Yang, G.; Wen, G.; Zhang, H. YTHDF2 correlates with tumor immune infiltrates in lower-grade glioma. Aging (Albany NY) 2020, 12, 18476–18500. [Google Scholar] [CrossRef] [PubMed]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vos, L.; Grunwald, I.; Bawden, E.G.; Dietrich, J.; Scheckenbach, K.; Wiek, C.; Zarbl, R.; Bootz, F.; Landsberg, J.; Dietrich, D. The landscape of CD28, CD80, CD86, CTLA4, and ICOS DNA methylation in head and neck squamous cell carcinomas. Epigenetics 2020, 15, 1195–1212. [Google Scholar] [CrossRef]
- Micevic, G.; Thakral, D.; McGeary, M.; Bosenberg, M.W. PD-L1 methylation regulates PD-L1 expression and is associated with melanoma survival. Pigment Cell Melanoma Res. 2019, 32, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Marwitz, S.; Scheufele, S.; Perner, S.; Reck, M.; Ammerpohl, O.; Goldmann, T. Epigenetic modifications of the immune-checkpoint genes CTLA4 and PDCD1 in non-small cell lung cancer results in increased expression. Clin. Epigenet. 2017, 9, 51. [Google Scholar] [CrossRef]
- Franzen, A.; Vogt, T.J.; Muller, T.; Dietrich, J.; Schrock, A.; Golletz, C.; Brossart, P.; Bootz, F.; Landsberg, J.; Kristiansen, G.; et al. PD-L1 (CD274) and PD-L2 (PDCD1LG2) promoter methylation is associated with HPV infection and transcriptional repression in head and neck squamous cell carcinomas. Oncotarget 2018, 9, 641–650. [Google Scholar] [CrossRef] [Green Version]
- Goltz, D.; Gevensleben, H.; Dietrich, J.; Dietrich, D. PD-L1 (CD274) promoter methylation predicts survival in colorectal cancer patients. Oncoimmunology 2017, 6, e1257454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Zhao, X.; Zhang, Y.; Shao, P.; Ma, X.; Paradee, W.J.; Liu, C.; Wang, J.; Xue, H.H. TFH cells depend on Tcf1-intrinsic HDAC activity to suppress CTLA4 and guard B-cell help function. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Lienlaf, M.; Perez-Villarroel, P.; Knox, T.; Pabon, M.; Sahakian, E.; Powers, J.; Woan, K.V.; Lee, C.; Cheng, F.; Deng, S.; et al. Essential role of HDAC6 in the regulation of PD-L1 in melanoma. Mol. Oncol. 2016, 10, 735–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darvin, P.; Sasidharan Nair, V.; Elkord, E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J. Oncol. 2019, 2019, 3958908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, C.; Paschall, A.V.; Shi, H.; Savage, N.; Waller, J.L.; Sabbatini, M.E.; Oberlies, N.H.; Pearce, C.; Liu, K. The MLL1-H3K4me3 Axis-Mediated PD-L1 Expression and Pancreatic Cancer Immune Evasion. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, G.; Jin, L.L.; Liu, C.Q.; Wang, Y.C.; Meng, Y.M.; Zhou, Z.G.; Chen, J.; Yu, X.J.; Zhang, Y.J.; Xu, J.; et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J. Immunother. Cancer 2019, 7, 300. [Google Scholar] [CrossRef]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor Belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Han, S.; Kang, Y.; Guo, M.; Hong, S.; Liu, F.; Fu, S.; Wang, L.; Wang, Q.X. SAHA, an HDAC inhibitor, synergizes with tacrolimus to prevent murine cardiac allograft rejection. Cell. Mol. Immunol. 2012, 9, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Fu, C.; Du, J.; Wang, H.; He, R.; Yin, X.; Li, H.; Li, X.; Wang, H.; Li, K.; et al. Enhanced histone H3 acetylation of the PD-L1 promoter via the COP1/c-Jun/HDAC3 axis is required for PD-L1 expression in drug-resistant cancer cells. J. Exp. Clin. Cancer Res. 2020, 39, 29. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.C.; Cheng, Y.Y.; Williams, M.; Kirschner, M.B.; Madore, J.; Lum, T.; Sarun, K.H.; Linton, A.; McCaughan, B.; Klebe, S.; et al. Tumor Suppressor microRNAs Contribute to the Regulation of PD-L1 Expression in Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2017, 12, 1421–1433. [Google Scholar] [CrossRef] [Green Version]
- Audrito, V.; Serra, S.; Stingi, A.; Orso, F.; Gaudino, F.; Bologna, C.; Neri, F.; Garaffo, G.; Nassini, R.; Baroni, G.; et al. PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17-5p. Oncotarget 2017, 8, 15894–15911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, P.; Xiong, Y.; Yu, J.; Chen, L.; Tao, T.; Yi, S.; Hanley, S.J.B.; Yue, J.; Watari, H.; Sakuragi, N. Correction: Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene 2019, 38, 3972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Chen, L.; Zou, L.; Yang, P.; Wu, R.; Mao, Y.; Zhou, H.; Li, R.; Wang, K.; Wang, W.; et al. MiR-20b, -21, and -130b inhibit PTEN expression resulting in B7-H1 over-expression in advanced colorectal cancer. Hum. Immunol. 2014, 75, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Iliopoulos, D.; Kavousanaki, M.; Ioannou, M.; Boumpas, D.; Verginis, P. The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. Eur. J. Immunol. 2011, 41, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Dong, L.; Wang, K.; Zou, H.; Zhao, S.; Wang, Y.; Wang, G. MiR-21 Participates in the PD-1/PD-L1 Pathway-Mediated Imbalance of Th17/Treg Cells in Patients After Gastric Cancer Resection. Ann. Surg. Oncol. 2019, 26, 884–893. [Google Scholar] [CrossRef]
- Liu, J.; Fan, L.; Yu, H.; Zhang, J.; He, Y.; Feng, D.; Wang, F.; Li, X.; Liu, Q.; Li, Y.; et al. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology 2019, 70, 241–258. [Google Scholar] [CrossRef]
- Cioffi, M.; Trabulo, S.M.; Vallespinos, M.; Raj, D.; Kheir, T.B.; Lin, M.L.; Begum, J.; Baker, A.M.; Amgheib, A.; Saif, J.; et al. The miR-25-93-106b cluster regulates tumor metastasis and immune evasion via modulation of CXCL12 and PD-L1. Oncotarget 2017, 8, 21609–21625. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Johnston, N.; Zheng, X.; Wang, H.; Zhang, X.; Gao, D.; Min, W. miR-28 modulates exhaustive differentiation of T cells through silencing programmed cell death-1 and regulating cytokine secretion. Oncotarget 2016, 7, 53735–53750. [Google Scholar] [CrossRef] [Green Version]
- Boldrini, L.; Giordano, M.; Niccoli, C.; Melfi, F.; Lucchi, M.; Mussi, A.; Fontanini, G. Role of microRNA-33a in regulating the expression of PD-1 in lung adenocarcinoma. Cancer Cell. Int. 2017, 17, 105. [Google Scholar] [CrossRef] [Green Version]
- Anastasiadou, E.; Stroopinsky, D.; Alimperti, S.; Jiao, A.L.; Pyzer, A.R.; Cippitelli, C.; Pepe, G.; Severa, M.; Rosenblatt, J.; Etna, M.P.; et al. Epstein-Barr virus-encoded EBNA2 alters immune checkpoint PD-L1 expression by downregulating miR-34a in B-cell lymphomas. Leukemia 2019, 33, 132–147. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Ashraf, M.U.; Kumar, A.; Bae, Y.S. Therapeutic Potential of microRNA Against Th2-associated Immune Disorders. Curr. Top. Med. Chem. 2021, 21, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell. Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef] [Green Version]
- Cortez, M.A.; Ivan, C.; Valdecanas, D.; Wang, X.; Peltier, H.J.; Ye, Y.; Araujo, L.; Carbone, D.P.; Shilo, K.; Giri, D.K.; et al. PDL1 Regulation by p53 via miR-34. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef] [Green Version]
- Xie, W.B.; Liang, L.H.; Wu, K.G.; Wang, L.X.; He, X.; Song, C.; Wang, Y.Q.; Li, Y.H. MiR-140 Expression Regulates Cell Proliferation and Targets PD-L1 in NSCLC. Cell. Physiol. Biochem. 2018, 46, 654–663. [Google Scholar] [CrossRef]
- Jia, L.; Xi, Q.; Wang, H.; Zhang, Z.; Liu, H.; Cheng, Y.; Guo, X.; Zhang, J.; Zhang, Q.; Zhang, L.; et al. miR-142-5p regulates tumor cell PD-L1 expression and enhances anti-tumor immunity. Biochem. Biophys. Res. Commun. 2017, 488, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Q.; Zhang, Y.; Wang, Z.; Ding, J.; Song, Y.; Zhao, W. Cisplatin-mediated down-regulation of miR-145 contributes to up-regulation of PD-L1 via the c-Myc transcription factor in cisplatin-resistant ovarian carcinoma cells. Clin. Exp. Immunol. 2020, 200, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Mastroianni, J.; Stickel, N.; Andrlova, H.; Hanke, K.; Melchinger, W.; Duquesne, S.; Schmidt, D.; Falk, M.; Andrieux, G.; Pfeifer, D.; et al. miR-146a Controls Immune Response in the Melanoma Microenvironment. Cancer Res. 2019, 79, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, M.; Okayama, H.; Ishigame, T.; Thar Min, A.K.; Saito, K.; Ujiie, D.; Murakami, Y.; Kikuchi, T.; Nakayama, Y.; Noda, M.; et al. miRNA-148a-3p Regulates Immunosuppression in DNA Mismatch Repair-Deficient Colorectal Cancer by Targeting PD-L1. Mol. Cancer Res. 2019, 17, 1403–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Sun, R.; Zhao, H.J.; Fu, D.; Zhong, H.J.; Weng, X.Q.; Qu, B.; Zhao, Y.; Wang, L.; Zhao, W.L. MiR155 sensitized B-lymphoma cells to anti-PD-L1 antibody via PD-1/PD-L1-mediated lymphoma cell interaction with CD8+T cells. Mol. Cancer 2019, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Y.; Zhang, J.; Hou, L.D.; Zhang, R.; Chen, W.; Fan, H.N.; Huang, Y.X.; Liu, H.; Zhu, J.S. Upregulation of PD-L1 predicts poor prognosis and is associated with miR-191-5p dysregulation in colon adenocarcinoma. Int. J. Immunopathol. Pharmacol. 2018, 32, 2058738418790318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Z.; Xu, S.; Ruan, H.; Wang, T.; Song, W.; Qian, L.; Chen, K. MiR-195/-16 Family Enhances Radiotherapy via T Cell Activation in the Tumor Microenvironment by Blocking the PD-L1 Immune Checkpoint. Cell. Physiol. Biochem. 2018, 48, 801–814. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Yan, F.; Wu, C. Overexpressed miR-195 attenuated immune escape of diffuse large B-cell lymphoma by targeting PD-L1. Biomed. Pharmacother. 2018, 98, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Yagishita, S.; Hagiwara, K.; Yoshioka, Y.; Kosaka, N.; Takeshita, F.; Fujiwara, T.; Tsuta, K.; Nokihara, H.; Tamura, T.; et al. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol. Ther. 2015, 23, 717–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Gibbons, D.L.; Goswami, S.; Cortez, M.A.; Ahn, Y.H.; Byers, L.A.; Zhang, X.; Yi, X.; Dwyer, D.; Lin, W.; et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun. 2014, 5, 5241. [Google Scholar] [CrossRef]
- Xie, G.; Li, W.; Li, R.; Wu, K.; Zhao, E.; Zhang, Y.; Zhang, P.; Shi, L.; Wang, D.; Yin, Y.; et al. Helicobacter Pylori Promote B7-H1 Expression by Suppressing miR-152 and miR-200b in Gastric Cancer Cells. PLoS ONE 2017, 12, e0168822. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.R.; Zhang, X.; Zhang, Y. MiR-214 prevents the progression of diffuse large B-cell lymphoma by targeting PD-L1. Cell Mol Biol Lett 2019, 24, 68. [Google Scholar] [CrossRef]
- Miao, S.; Mao, X.; Zhao, S.; Song, K.; Xiang, C.; Lv, Y.; Jiang, H.; Wang, L.; Li, B.; Yang, X.; et al. miR-217 inhibits laryngeal cancer metastasis by repressing AEG-1 and PD-L1 expression. Oncotarget 2017, 8, 62143–62153. [Google Scholar] [CrossRef]
- Holla, S.; Stephen-Victor, E.; Prakhar, P.; Sharma, M.; Saha, C.; Udupa, V.; Kaveri, S.V.; Bayry, J.; Balaji, K.N. Mycobacteria-responsive sonic hedgehog signaling mediates programmed death-ligand 1- and prostaglandin E2-induced regulatory T cell expansion. Sci. Rep. 2016, 6, 24193. [Google Scholar] [CrossRef] [Green Version]
- Dong, P.; Xiong, Y.; Yu, J.; Chen, L.; Tao, T.; Yi, S.; Hanley, S.J.B.; Yue, J.; Watari, H.; Sakuragi, N. Control of PD-L1 expression by miR-140/142/340/383 and oncogenic activation of the OCT4-miR-18a pathway in cervical cancer. Oncogene 2018, 37, 5257–5268. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Zhao, Y.; Sun, Y.; Yan, X.; Wang, P. miR-375 inhibits IFN-gamma-induced programmed death 1 ligand 1 surface expression in head and neck squamous cell carcinoma cells by blocking JAK2/STAT1 signaling. Oncol. Rep. 2018, 39, 1461–1468. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Tao, Z.; Hai, B.; Liang, H.; Shi, Y.; Wang, T.; Song, W.; Chen, Y.; OuYang, J.; Chen, J.; et al. miR-424(322) reverses chemoresistance via T-cell immune response activation by blocking the PD-L1 immune checkpoint. Nat. Commun. 2016, 7, 11406. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yuan, C.; Wangmo, D.; Subramanian, S. Tumor-Secreted Extracellular Vesicles Regulate T-Cell Costimulation and Can Be Manipulated To Induce Tumor-Specific T-Cell Responses. Gastroenterology 2021, 161, 560–574. [Google Scholar] [CrossRef] [PubMed]
- Qu, F.; Ye, J.; Pan, X.; Wang, J.; Gan, S.; Chu, C.; Chu, J.; Zhang, X.; Liu, M.; He, H.; et al. MicroRNA-497-5p down-regulation increases PD-L1 expression in clear cell renal cell carcinoma. J. Drug Target. 2019, 27, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, A.Y.; Zhou, R.; Hu, G.; Liu, J.; Sosnowska, D.; Drescher, K.M.; Dong, H.; Chen, X.M. Cryptosporidium parvum induces B7-H1 expression in cholangiocytes by down-regulating microRNA-513. J. Infect. Dis. 2010, 201, 160–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Li, F.; Mao, Y.; Zhou, H.; Sun, J.; Li, R.; Liu, C.; Chen, W.; Hua, D.; Zhang, X. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum. Genet. 2013, 132, 641–648. [Google Scholar] [CrossRef]
- Gao, L.; Guo, Q.; Li, X.; Yang, X.; Ni, H.; Wang, T.; Zhao, Q.; Liu, H.; Xing, Y.; Xi, T.; et al. MiR-873/PD-L1 axis regulates the stemness of breast cancer cells. EBioMedicine 2019, 41, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, D.; Zhao, D.; Wu, Y.; Yao, R.; Zhou, L.; Lu, L.; Gao, W.; Sun, Y. The miR-3127-5p/p-STAT3 axis up-regulates PD-L1 inducing chemoresistance in non-small-cell lung cancer. J. Cell. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, X.; Yang, M.; Kan, Q.; Duan, Z. miR3609 sensitizes breast cancer cells to adriamycin by blocking the programmed death-ligand 1 immune checkpoint. Exp. Cell. Res. 2019, 380, 20–28. [Google Scholar] [CrossRef]
- Zhang, G.; Li, N.; Li, Z.; Zhu, Q.; Li, F.; Yang, C.; Han, Q.; Lv, Y.; Zhou, Z.; Liu, Z. microRNA-4717 differentially interacts with its polymorphic target in the PD1 3′ untranslated region: A mechanism for regulating PD-1 expression and function in HBV-associated liver diseases. Oncotarget 2015, 6, 18933–18944. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.Y.; Johnson, D.B.; Davis, E.J. Toxicities Associated With PD-1/PD-L1 Blockade. Cancer J. 2018, 24, 36–40. [Google Scholar] [CrossRef]
- Jenkins, R.W.; Barbie, D.A.; Flaherty, K.T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 2018, 118, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef]
- Kalbasi, A.; Ribas, A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat. Rev. Immunol. 2020, 20, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Bashyam, H. CTLA-4: From conflict to clinic. J. Exp. Med. 2007, 204, 1243. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef]
- Tivol, E.A.; Borriello, F.; Schweitzer, A.N.; Lynch, W.P.; Bluestone, J.A.; Sharpe, A.H. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 1995, 3, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- De Sousa Linhares, A.; Battin, C.; Jutz, S.; Leitner, J.; Hafner, C.; Tobias, J.; Wiedermann, U.; Kundi, M.; Zlabinger, G.J.; Grabmeier-Pfistershammer, K.; et al. Therapeutic PD-L1 antibodies are more effective than PD-1 antibodies in blocking PD-1/PD-L1 signaling. Sci. Rep. 2019, 9, 11472. [Google Scholar] [CrossRef]
- Lucibello, G.; Mograbi, B.; Milano, G.; Hofman, P.; Brest, P. PD-L1 regulation revisited: Impact on immunotherapeutic strategies. Trends. Mol. Med. 2021. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef]
- Wu, Z.; Man, S.; Sun, R.; Li, Z.; Wu, Y.; Zuo, D. Recent advances and challenges of immune checkpoint inhibitors in immunotherapy of non-small cell lung cancer. Int. Immunopharmacol. 2020, 85, 106613. [Google Scholar] [CrossRef] [PubMed]
- Naing, A.; Infante, J.; Goel, S.; Burris, H.; Black, C.; Marshall, S.; Achour, I.; Barbee, S.; May, R.; Morehouse, C.; et al. Anti-PD-1 monoclonal antibody MEDI0680 in a phase I study of patients with advanced solid malignancies. J. Immunother. Cancer 2019, 7, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naing, A.; Gainor, J.F.; Gelderblom, H.; Forde, P.M.; Butler, M.O.; Lin, C.C.; Sharma, S.; Ochoa de Olza, M.; Varga, A.; Taylor, M.; et al. A first-in-human phase 1 dose escalation study of spartalizumab (PDR001), an anti-PD-1 antibody, in patients with advanced solid tumors. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Wang, H.; Hahn, N.M.; Twardowski, P.; Pal, S.K.; Albany, C.; Fleming, M.T.; Starodub, A.; Hauke, R.J.; Yu, M.; et al. Phase 2 Trial of Gemcitabine, Cisplatin, plus Ipilimumab in Patients with Metastatic Urothelial Cancer and Impact of DNA Damage Response Gene Mutations on Outcomes. Eur. Urol. 2018, 73, 751–759. [Google Scholar] [CrossRef]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar] [CrossRef]
- Iwama, S.; De Remigis, A.; Callahan, M.K.; Slovin, S.F.; Wolchok, J.D.; Caturegli, P. Pituitary expression of CTLA-4 mediates hypophysitis secondary to administration of CTLA-4 blocking antibody. Sci. Transl. Med. 2014, 6, 230ra245. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Tomillero, A.; Moral, M.A. Gateways to clinical trials. Methods Find Exp. Clin. Pharmacol. 2008, 30, 643–672. [Google Scholar] [CrossRef]
- Poust, J. Targeting metastatic melanoma. Am. J. Health Syst. Pharm. 2008, 65, S9–S15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reuben, J.M.; Lee, B.N.; Li, C.; Gomez-Navarro, J.; Bozon, V.A.; Parker, C.A.; Hernandez, I.M.; Gutierrez, C.; Lopez-Berestein, G.; Camacho, L.H. Biologic and immunomodulatory events after CTLA-4 blockade with ticilimumab in patients with advanced malignant melanoma. Cancer 2006, 106, 2437–2444. [Google Scholar] [CrossRef] [PubMed]
- Senan, S.; Okamoto, I.; Lee, G.W.; Chen, Y.; Niho, S.; Mak, G.; Yao, W.; Shire, N.; Jiang, H.; Cho, B.C. Design and Rationale for a Phase III, Randomized, Placebo-controlled Trial of Durvalumab With or Without Tremelimumab After Concurrent Chemoradiotherapy for Patients With Limited-stage Small-cell Lung Cancer: The ADRIATIC Study. Clin. Lung. Cancer 2020, 21, e84–e88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Kefford, R.; Marshall, M.A.; Punt, C.J.; Haanen, J.B.; Marmol, M.; Garbe, C.; Gogas, H.; Schachter, J.; Linette, G.; et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J. Clin. Oncol. 2013, 31, 616–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulovic, S.; Demey, W.; Ullen, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2020, 383, 1218–1230. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Ozguroglu, M.; Ji, J.H.; et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): A randomised, controlled, open-label, phase 3 trial. Lancet 2019, 394, 1929–1939. [Google Scholar] [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Yokoi, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Durvalumab after Chemoradiotherapy in Stage III Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 377, 1919–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N. Engl. J. Med. 2018, 379, 2342–2350. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.L.; Bosch, R.J.; Ritz, J.; Hataye, J.M.; Aga, E.; Tressler, R.L.; Mason, S.W.; Hwang, C.K.; Grasela, D.M.; Ray, N.; et al. Clinical Trial of the Anti-PD-L1 Antibody BMS-936559 in HIV-1 Infected Participants on Suppressive Antiretroviral Therapy. J. Infect. Dis. 2017, 215, 1725–1733. [Google Scholar] [CrossRef] [Green Version]
- Hotchkiss, R.S.; Colston, E.; Yende, S.; Angus, D.C.; Moldawer, L.L.; Crouser, E.D.; Martin, G.S.; Coopersmith, C.M.; Brakenridge, S.; Mayr, F.B.; et al. Immune Checkpoint Inhibition in Sepsis: A Phase 1b Randomized, Placebo-Controlled, Single Ascending Dose Study of Antiprogrammed Cell Death-Ligand 1 Antibody (BMS-936559). Crit. Care Med. 2019, 47, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Corrales, L.; Scilla, K.; Caglevic, C.; Miller, K.; Oliveira, J.; Rolfo, C. Immunotherapy in Lung Cancer: A New Age in Cancer Treatment. Adv. Exp. Med. Biol. 2018, 995, 65–95. [Google Scholar] [CrossRef]
- Floudas, C.S.; Brar, G.; Mabry-Hrones, D.; Duffy, A.G.; Wood, B.; Levy, E.; Krishnasamy, V.; Fioravanti, S.; Bonilla, C.M.; Walker, M.; et al. A Pilot Study of the PD-1 Targeting Agent AMP-224 Used With Low-Dose Cyclophosphamide and Stereotactic Body Radiation Therapy in Patients With Metastatic Colorectal Cancer. Clin. Colorectal. Cancer 2019, 18, e349–e360. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, H.; Okazaki, T.; Tanaka, Y.; Nakatani, K.; Hara, M.; Matsumori, A.; Sasayama, S.; Mizoguchi, A.; Hiai, H.; Minato, N.; et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 2001, 291, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [Green Version]
- Okazaki, T.; Tanaka, Y.; Nishio, R.; Mitsuiye, T.; Mizoguchi, A.; Wang, J.; Ishida, M.; Hiai, H.; Matsumori, A.; Minato, N.; et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nat. Med. 2003, 9, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Okazaki, I.M.; Yoshida, T.; Chikuma, S.; Kato, Y.; Nakaki, F.; Hiai, H.; Honjo, T.; Okazaki, T. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int. Immunol. 2010, 22, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yoshida, T.; Nakaki, F.; Hiai, H.; Okazaki, T.; Honjo, T. Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc. Natl. Acad. Sci. USA 2005, 102, 11823–11828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Jiang, F.; Honjo, T.; Okazaki, T. PD-1 deficiency reveals various tissue-specific autoimmunity by H-2b and dose-dependent requirement of H-2g7 for diabetes in NOD mice. Proc. Natl. Acad. Sci. USA 2008, 105, 3533–3538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, T.; Otaka, Y.; Wang, J.; Hiai, H.; Takai, T.; Ravetch, J.V.; Honjo, T. Hydronephrosis associated with antiurothelial and antinuclear autoantibodies in BALB/c-Fcgr2b-/-Pdcd1-/- mice. J. Exp. Med. 2005, 202, 1643–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosso, J.F.; Jure-Kunkel, M.N. CTLA-4 blockade in tumor models: An overview of preclinical and translational research. Cancer Immunol. 2013, 13, 5. [Google Scholar] [PubMed]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734–1736. [Google Scholar] [CrossRef] [Green Version]
- Hodi, F.S.; Mihm, M.C.; Soiffer, R.J.; Haluska, F.G.; Butler, M.; Seiden, M.V.; Davis, T.; Henry-Spires, R.; MacRae, S.; Willman, A.; et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc. Natl. Acad. Sci. USA 2003, 100, 4712–4717. [Google Scholar] [CrossRef] [Green Version]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, Y.; Hamanishi, J.; Chamoto, K.; Honjo, T. Cancer immunotherapies targeting the PD-1 signaling pathway. J. Biomed. Sci. 2017, 24, 26. [Google Scholar] [CrossRef] [Green Version]
- Chiossone, L.; Dumas, P.Y.; Vienne, M.; Vivier, E. Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 2018, 18, 671–688. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrueto, L.; Caminero, F.; Cash, L.; Makris, C.; Lamichhane, P.; Deshmukh, R.R. Resistance to Checkpoint Inhibition in Cancer Immunotherapy. Transl. Oncol. 2020, 13, 100738. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Nagpal, R.; Kumar, A.; Ashraf, M.U.; Bae, Y.S. Immunotherapeutic Potential of m6A-Modifiers and MicroRNAs in Controlling Acute Myeloid Leukaemia. Biomedicines 2021, 9, 690. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.S.; Roundtree, I.A.; He, C. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell. Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Cheong, K.H. Novel Immunotherapies to Combine with PD-1/PD-L1 Treatment. Available online: https://media.nature.com/original/magazine-assets/d43747-020-00338-3/d43747-020-00338-3.pdf (accessed on 25 July 2021).
- Dovedi, S.J.; Elder, M.J.; Yang, C.; Sitnikova, S.I.; Irving, L.; Hansen, A.; Hair, J.; Jones, D.C.; Hasani, S.; Wang, B.; et al. Design and Efficacy of a Monovalent Bispecific PD-1/CTLA4 Antibody That Enhances CTLA4 Blockade on PD-1(+) Activated T Cells. Cancer Discov. 2021, 11, 1100–1117. [Google Scholar] [CrossRef] [PubMed]
- Kakimi, K.; Karasaki, T.; Matsushita, H.; Sugie, T. Advances in personalized cancer immunotherapy. Breast Cancer 2017, 24, 16–24. [Google Scholar] [CrossRef]
- Sahin, U.; Tureci, O. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.J.; Liu, Z.; Cheng, Q. Regulatory mechanisms of immune checkpoints PD-L1 and CTLA-4 in cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Catela Ivkovic, T.; Voss, G.; Cornella, H.; Ceder, Y. microRNAs as cancer therapeutics: A step closer to clinical application. Cancer Lett. 2017, 407, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Nowers, C. Maximizing Synergy and Mitigating Resistance: Novel Dual-Targeted Natural Killer Cell Therapies for Cancer. Available online: https://www.onktherapeutics.com/wp/wp-content/uploads/2021/03/Nature-Biotech-Dealmakers-ONK-Therapeutics-March-2021.pdf (accessed on 20 July 2021).
- Ryan, B.S.M.; Gaidarova, S.; Abujarour, R.; Clarke, R.; Stokely, L.; Rogers, P.; Ge, M.; Robinson, M.; Rezner, B.; Lee, T.T.; et al. Abstract 3576: FT500, an off-the-shelf NK cell cancer immunotherapy derived from a master pluripotent cell line, enhances T-cell activation and recruitment to overcome checkpoint blockade resistance. Cancer Res. Immunol. 2018. [Google Scholar] [CrossRef]
- Guo, H.; He, Y.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Zhou, C. Combinational immunotherapy based on immune checkpoints inhibitors in small cell lung cancer: Is this the beginning to reverse the refractory situation? J. Thorac. Dis. 2020, 12, 6070–6089. [Google Scholar] [CrossRef]
- Wang, F.; Lau, J.K.C.; Yu, J. The role of natural killer cell in gastrointestinal cancer: Killer or helper. Oncogene 2021, 40, 717–730. [Google Scholar] [CrossRef]
- Shin, M.H.; Kim, J.; Lim, S.A.; Kim, J.; Kim, S.J.; Lee, K.M. NK Cell-Based Immunotherapies in Cancer. Immune Netw. 2020, 20, e14. [Google Scholar] [CrossRef]
- Goodridge, J.P.; Mahmood, S.; Zhu, H.; Gaidarova, S.; Blum, R.; Bjordahl, R.; Cichocki, F.; Chu, H.-y.; Bonello, G.; Lee, T.; et al. FT596: Translation of First-of-Kind Multi-Antigen Targeted Off-the-Shelf CAR-NK Cell with Engineered Persistence for the Treatment of B Cell Malignancies. Blood 2019, 134, 301. [Google Scholar] [CrossRef]
- Bjordahl, R.; Gaidarova, S.; Woan, K.; Cichocki, F.; Bonello, G.; Robinson, M.; Ruller, C.; Pribadi, M.; Dinella, J.; Fong, L.; et al. FT538: Preclinical Development of an Off-the-Shelf Adoptive NK Cell Immunotherapy with Targeted Disruption of CD38 to Prevent Anti-CD38 Antibody-Mediated Fratricide and Enhance ADCC in Multiple Myeloma When Combined with Daratumumab. Blood 2019, 134, 133. [Google Scholar] [CrossRef]
- Veluchamy, J.P.; Kok, N.; van der Vliet, H.J.; Verheul, H.M.W.; de Gruijl, T.D.; Spanholtz, J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front. Immunol. 2017, 8, 631. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, Y.; Shi, C. Targeting Natural Killer Cells for Tumor Immunotherapy. Front. Immunol. 2020, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- U.S. National Library of Medicine. Available online: https://clinicaltrials.gov/ (accessed on 7 July 2021).
- Barta, S.K.; Zain, J.; MacFarlane, A.W.t.; Smith, S.M.; Ruan, J.; Fung, H.C.; Tan, C.R.; Yang, Y.; Alpaugh, R.K.; Dulaimi, E.; et al. Phase II Study of the PD-1 Inhibitor Pembrolizumab for the Treatment of Relapsed or Refractory Mature T-cell Lymphoma. Clin. Lymphoma Myeloma Leuk 2019, 19, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Barta, S.K.; Fowler, N.H.; Zain, J.; Ruan, J.; Smith, S.M.; Schuster, S.J.; Nasta, S.D.; Svoboda, J.; Gerson, J.N.; Landsburg, D.J.; et al. Pembrolizumab and Copanlisib for the Treatment of Relapsed or Refractory Mature T-Cell Lymphomas. Blood 2019, 134, 4031. [Google Scholar] [CrossRef]
- Periasamy, S.; Dhiman, R.; Barnes, P.F.; Paidipally, P.; Tvinnereim, A.; Bandaru, A.; Valluri, V.L.; Vankayalapati, R. Programmed death 1 and cytokine inducible SH2-containing protein dependent expansion of regulatory T cells upon stimulation With Mycobacterium tuberculosis. J. Infect. Dis. 2011, 203, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Xue, J.; Deng, T.; Zhou, X.; Yu, K.; Deng, L.; Huang, M.; Yi, X.; Liang, M.; Wang, Y.; et al. Safety and feasibility of CRISPR-edited T cells in patients with refractory non-small-cell lung cancer. Nat. Med. 2020, 26, 732–740. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367. [Google Scholar] [CrossRef]
- Versteven, M.; Van den Bergh, J.M.J.; Marcq, E.; Smits, E.L.J.; Van Tendeloo, V.F.I.; Hobo, W.; Lion, E. Dendritic Cells and Programmed Death-1 Blockade: A Joint Venture to Combat Cancer. Front. Immunol. 2018, 9, 394. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Kong, S.; Tao, M.; Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol. Cancer 2020, 19, 88. [Google Scholar] [CrossRef]
- Li, Y.; Gu, J.; Xu, F.; Zhu, Q.; Chen, Y.; Ge, D.; Lu, C. Molecular characterization, biological function, tumor microenvironment association and clinical significance of m6A regulators in lung adenocarcinoma. Brief. Bioinform. 2020. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Han, S.H.; Choe, J. Diverse molecular functions of m(6)A mRNA modification in cancer. Exp. Mol. Med. 2020, 52, 738–749. [Google Scholar] [CrossRef]
- Dai, X.Y.; Shi, L.; Li, Z.; Yang, H.Y.; Wei, J.F.; Ding, Q. Main N6-Methyladenosine Readers: YTH Family Proteins in Cancers. Front. Oncol. 2021, 11, 635329. [Google Scholar] [CrossRef]
- Elcheva, I.A.; Spiegelman, V.S. Targeting RNA-binding proteins in acute and chronic leukemia. Leukemia 2021, 35, 360–376. [Google Scholar] [CrossRef] [PubMed]
- Hou, G.; Zhao, X.; Li, L.; Yang, Q.; Liu, X.; Huang, C.; Lu, R.; Chen, R.; Wang, Y.; Jiang, B.; et al. SUMOylation of YTHDF2 promotes mRNA degradation and cancer progression by increasing its binding affinity with m6A-modified mRNAs. Nucleic Acids Res. 2021, 49, 2859–2877. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Iwasaki, A. YTHDF1 Control of Dendritic Cell Cross-Priming as a Possible Target of Cancer Immunotherapy. Biochemistry 2019, 58, 1945–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kachroo, N.; Valencia, T.; Warren, A.Y.; Gnanapragasam, V.J. Evidence for downregulation of the negative regulator SPRED2 in clinical prostate cancer. Br. J. Cancer 2013, 108, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, I.; Tzelepis, K.; Pandolfini, L.; Shi, J.; Millan-Zambrano, G.; Robson, S.C.; Aspris, D.; Migliori, V.; Bannister, A.J.; Han, N.; et al. Promoter-bound METTL3 maintains myeloid leukaemia by m(6)A-dependent translation control. Nature 2017, 552, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Gulati, P.; Cheung, M.K.; Antrobus, R.; Church, C.D.; Harding, H.P.; Tung, Y.C.; Rimmington, D.; Ma, M.; Ron, D.; Lehner, P.J.; et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc. Natl. Acad. Sci. USA 2013, 110, 2557–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, B.; Kinne, H.E.; Milligan, R.D.; Washburn, L.J.; Olsen, M.; Lucci, A. Important Role of FTO in the Survival of Rare Panresistant Triple-Negative Inflammatory Breast Cancer Cells Facing a Severe Metabolic Challenge. PLoS ONE 2016, 11, e0159072. [Google Scholar] [CrossRef]
- Su, R.; Dong, L.; Li, Y.; Gao, M.; Han, L.; Wunderlich, M.; Deng, X.; Li, H.; Huang, Y.; Gao, L.; et al. Targeting FTO Suppresses Cancer Stem Cell Maintenance and Immune Evasion. Cancer Cell 2020, 38, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, T.; Kambayashi, Y.; Aiba, S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology 2012, 1, 1433–1434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, S.; Xiao, W.; Zhao, Y.L.; Yang, Y.G. m(6)A: Signaling for mRNA splicing. RNA Biol. 2016, 13, 756–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Yang, Y.; Sun, B.F.; Shi, Y.; Yang, X.; Xiao, W.; Hao, Y.J.; Ping, X.L.; Chen, Y.S.; Wang, W.J.; et al. FTO-dependent demethylation of N6-methyladenosine regulates mRNA splicing and is required for adipogenesis. Cell Res. 2014, 24, 1403–1419. [Google Scholar] [CrossRef]
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA m6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320. [Google Scholar] [CrossRef]
- Ding, Z.; Li, Q.; Zhang, R.; Xie, L.; Shu, Y.; Gao, S.; Wang, P.; Su, X.; Qin, Y.; Wang, Y.; et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal. Transduct. Target. Ther. 2021, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Ito, M.; Chikuma, S.; Akanuma, T.; Nakatsukasa, H. Negative Regulation of Cytokine Signaling in Immunity. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Nishinakamura, H.; Matsumura, Y.; Hanada, T. Negative regulation of cytokine signaling and immune responses by SOCS proteins. Arthritis Res. Ther. 2005, 7, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Shouda, T.; Yoshida, T.; Hanada, T.; Wakioka, T.; Oishi, M.; Miyoshi, K.; Komiya, S.; Kosai, K.; Hanakawa, Y.; Hashimoto, K.; et al. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J. Clin. Investig. 2001, 108, 1781–1788. [Google Scholar] [CrossRef]
- Hunter, M.G.; Jacob, A.; O’Donnell, L.C.; Agler, A.; Druhan, L.J.; Coggeshall, K.M.; Avalos, B.R. Loss of SHIP and CIS recruitment to the granulocyte colony-stimulating factor receptor contribute to hyperproliferative responses in severe congenital neutropenia/acute myelogenous leukemia. J. Immunol. 2004, 173, 5036–5045. [Google Scholar] [CrossRef]
- Ochoa, D.; Hercules, A.; Carmona, M.; Suveges, D.; Gonzalez-Uriarte, A.; Malangone, C.; Miranda, A.; Fumis, L.; Carvalho-Silva, D.; Spitzer, M.; et al. Open Targets Platform: Supporting systematic drug-target identification and prioritisation. Nucleic Acids Res. 2021, 49, D1302–D1310. [Google Scholar] [CrossRef]
- Martz, L. Innate harmony. Available online: https://www.innate-pharma.com/sites/default/files/072816in_coverstory_innateharmony.pdf (accessed on 21 July 2021).
- Trengove, M.C.; Ward, A.C. SOCS proteins in development and disease. Am. J. Clin. Exp. Immunol. 2013, 2, 1–29. [Google Scholar]
- Hernandez, C.; Bogdanov, P.; Gomez-Guerrero, C.; Sampedro, J.; Sola-Adell, C.; Espejo, C.; Garcia-Ramirez, M.; Prieto, I.; Egido, J.; Simo, R. SOCS1-Derived Peptide Administered by Eye Drops Prevents Retinal Neuroinflammation and Vascular Leakage in Experimental Diabetes. Int. J. Mol. Sci. 2019, 20, 3615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chikuma, S.; Kanamori, M.; Mise-Omata, S.; Yoshimura, A. Suppressors of cytokine signaling: Potential immune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017, 108, 574–580. [Google Scholar] [CrossRef] [Green Version]
- Bernard, P.-L.; Delconte, R.B.; Pastor, S.; Laletin, V.; Goubard, A.; Josselin, E.; Castellano, R.; Vernerey, J.; Vivier, E.; Huntington, N.D.; et al. CISH targeting in NK cells activates natural cytotoxicity receptor signaling and reduce cell exhaustion to unsilence primary anti-tumor response. bioRxiv 2021. [Google Scholar] [CrossRef]
- Felices, M.; Lenvik, A.J.; McElmurry, R.; Chu, S.; Hinderlie, P.; Bendzick, L.; Geller, M.A.; Tolar, J.; Blazar, B.R.; Miller, J.S. Continuous treatment with IL-15 exhausts human NK cells via a metabolic defect. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Andre, P.; Denis, C.; Soulas, C.; Bourbon-Caillet, C.; Lopez, J.; Arnoux, T.; Blery, M.; Bonnafous, C.; Gauthier, L.; Morel, A.; et al. Anti-NKG2A mAb Is a Checkpoint Inhibitor that Promotes Anti-tumor Immunity by Unleashing Both T and NK Cells. Cell 2018, 175, 1731–1743. [Google Scholar] [CrossRef] [Green Version]
- Gilboa, E. Knocking the SOCS1 off dendritic cells. Nat. Biotechnol. 2004, 22, 1521–1522. [Google Scholar] [CrossRef]
- Shen, L.; Evel-Kabler, K.; Strube, R.; Chen, S.Y. Silencing of SOCS1 enhances antigen presentation by dendritic cells and antigen-specific anti-tumor immunity. Nat. Biotechnol. 2004, 22, 1546–1553. [Google Scholar] [CrossRef]
- Miah, M.A.; Yoon, C.H.; Kim, J.; Jang, J.; Seong, Y.R.; Bae, Y.S. CISH is induced during DC development and regulates DC-mediated CTL activation. Eur. J. Immunol. 2012, 42, 58–68. [Google Scholar] [CrossRef]
- Kobayashi, T.; Yoshimura, A. Keeping DCs awake by putting SOCS1 to sleep. Trends Immunol. 2005, 26, 177–179. [Google Scholar] [CrossRef]
- Zhang, W.; Song, Z.; Xiao, J.; Liu, X.; Luo, Y.; Yang, Z.; Luo, R.; Li, A. Blocking the PD-1/PD-L1 axis in dendritic cell-stimulated Cytokine-Induced Killer Cells with pembrolizumab enhances their therapeutic effects against hepatocellular carcinoma. J. Cancer 2019, 10, 2578–2587. [Google Scholar] [CrossRef] [Green Version]
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology 2016, 5, e1085146. [Google Scholar] [CrossRef] [Green Version]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef]
- Go, D.M.; Lee, S.H.; Lee, S.H.; Woo, S.H.; Kim, K.; Kim, K.; Park, K.S.; Park, J.H.; Ha, S.J.; Kim, W.H.; et al. Programmed Death Ligand 1-Expressing Classical Dendritic Cells MitigateHelicobacter-Induced Gastritis. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 715–739. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Blum, R.H.; Bernareggi, D.; Ask, E.H.; Wu, Z.; Hoel, H.J.; Meng, Z.; Wu, C.; Guan, K.L.; Malmberg, K.J.; et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell 2020, 27, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Li, H.B.; Tong, J.; Zhu, S.; Batista, P.J.; Duffy, E.E.; Zhao, J.; Bailis, W.; Cao, G.; Kroehling, L.; Chen, Y.; et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature 2017, 548, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Jeong, Y.; Ashraf, M.U.; Bae, Y.S. Dendritic Cell-Mediated Th2 Immunity and Immune Disorders. Int. J. Mol. Sci. 2019, 20, 2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huemer, F.; Leisch, M.; Geisberger, R.; Zaborsky, N.; Greil, R. miRNA-Based Therapeutics in the Era of Immune-Checkpoint Inhibitors. Pharmaceuticals (Basel) 2021, 14, 89. [Google Scholar] [CrossRef] [PubMed]
- Skafi, N.; Fayyad-Kazan, M.; Badran, B. Immunomodulatory role for MicroRNAs: Regulation of PD-1/PD-L1 and CTLA-4 immune checkpoints expression. Gene 2020, 754, 144888. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, Y. An endoparasitoid wasp influences host DNA methylation. Sci. Rep. 2017, 7, 43287. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Venkata, P.; Kim, Y. Suppressive activity of a viral histone H4 against two host chromatin remodelling factors: Lysine demethylase and SWI/SNF. J. Gen. Virol. 2016, 97, 2780–2796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Lan, F. RNA m(6)A meets transposable elements and chromatin. Protein Cell 2021. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Allison, J.P. Immune checkpoint targeting in cancer therapy: Toward combination strategies with curative potential. Cell 2015, 161, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Blass, E.; Ott, P.A. Advances in the development of personalized neoantigen-based therapeutic cancer vaccines. Nat. Rev. Clin. Oncol. 2021, 18, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Perrier, A.; Didelot, A.; Laurent-Puig, P.; Blons, H.; Garinet, S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules 2020, 10, 1061. [Google Scholar] [CrossRef]
- Wang, H.; Hu, X.; Huang, M.; Liu, J.; Gu, Y.; Ma, L.; Zhou, Q.; Cao, X. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nat. Commun. 2019, 10, 1898. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Xu, Z.; Wang, Z.; Ren, Z.; Li, L.; Ruan, Y. Exosomes from dendritic cells with Mettl3 gene knockdown prevent immune rejection in a mouse cardiac allograft model. Immunogenetics 2020, 72, 423–430. [Google Scholar] [CrossRef]
- Feng, Y.; Dong, H.; Sun, B.; Hu, Y.; Yang, Y.; Jia, Y.; Jia, L.; Zhong, X.; Zhao, R. METTL3/METTL14 Transactivation and m(6)A-Dependent TGF-beta1 Translation in Activated Kupffer Cells. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 839–856. [Google Scholar] [CrossRef]
- Yao, Y.; Yang, Y.; Guo, W.; Xu, L.; You, M.; Zhang, Y.C.; Sun, Z.; Cui, X.; Yu, G.; Qi, Z.; et al. METTL3-dependent m(6)A modification programs T follicular helper cell differentiation. Nat. Commun. 2021, 12, 1333. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RNA (m6A)-Modifiers (Editors/Erasers/Effectors) | |||||
|---|---|---|---|---|---|
| RNA Modifiers | Disease Condition | Target | Disease Mechanism | Therapeutic Strategies | Ref. |
| Writers Mettl3/14 | up-regulated in colorectal cancer and melanoma | IFNγ, STAT1, IRF1, Cxcl-9 and Cxcl-10 | By reducing CD8+T-cells infiltrations in TME | CRISPR/cas9 silencing of Mettl3/14 via YTHDF2 | [8] |
| Mettl-3 | down-regulated in M1/M2-med. lung metastasis | Spred-2 | By recruiting immunosuppresive T-reg and MDSCs | Overexpressing Mettl3 via polarizing M1/M2-macrophages | [9] |
| m6A | m6A-mediated regulation of PD-L1 in HNSCC | G2M checkpoint and PI3K/AKT/ mTOR signaling | Analysed via cancer genome atlas TCGA and GSE65858 cohort | By targeting m6A regulatated signature genes | [10] |
| Erasers FTO | up-regulated in melanoma | PD-1, CXCR4 and SOX10 | Impairs anti-PD1 effect by reducing target gene expressions | Selective inhibition of FTO to enhance anti-PD1 effects | [11] |
| FTO | up-regulated in colon cancer | PD-L1 | Up-regulates PD-L1 expression in IFNγ signaling-independent manner | Selective inhibition of FTO inhibits PD-L1 to control colon cancer | [12] |
| ALKBH5 | up-regulated in melanoma | Mct4/Slc16a3 | By recruiting immunosuppresive T-reg and MDSCs | Anti-ALKBH5 enhances the effect of anti-PD1 therapy. | [13] |
| Readers YTHDF1 | up-regulated in solid tumors | Lysosomal cathepsins | Degrade neo-antigen and impair dendritic cell presentation | Anti-YTHDF1 suppress cathepsins and enhance DC cross-presentation | [14] |
| YTHDF2 | up-regulated in LGG (brain tumor) and several other immune cells | PD-1, CTLA4, TIM3 | Impair immune checkpoint signalling | Anti-YTHDF2 in combination with immunecheckpoint immunotherapy | [15,16] |
| DNA and Histone Modifiers in ICB-Therapeutics | |||||
| Epigenetic Regulators | Disease Condition | Target | Mechanism | Therapeutic Strategies | Ref. |
| DNA methylation | down-ragulates CTLA4 in HNSCC | CTLA4, CD28, CD80/86, ICOS | DNA methylation affects HNSCC | Selective DNA (DNMTs) inhibitors | [17] |
| DNA methylation | down-regulates PD-L1 in melanoma | Interfron signalling | cpG DNA methylation regulate melanoma | [18] | |
| DNA methylation | up-regulates PD-1 & CTLA4 in NSCLC | PD-1 (PDCD-1) CTLA4 | Hypo-methylation increases PD-1, CTLA4 expression in NSCLC | Selective DNA (5hmC) inhibitors | [19] |
| DNA methylation | up-regulates PD-L1 & PD-L2 in HNSCC | PD-L1 (CD274) PD-L2 (PDCD1LG2) | Hypo-methylation increases PD-L1 & PD-L2 expression | Combining DNA inh. with Nivolumab and Pembrolizumab | [20] |
| DNA methylation | up-regulates PD-L1 in CRC | PD-L1 (CD274) | DNA-methylation control PD-L1 exp. | Selective DNA (TETs) inhibitors | [21] |
| HDAC | up-ragulates CTLA4 in B-cell associated function | CTLA4 and LAG3 | Tcf1 regulate CTLA4 expression in TFH-cells | HDACi control CTLA4-mediated B-cell help | [22] |
| HDAC6 | up-regulates PD-L1 in melanoma | PD-L1 (CD274) STAT3 | HDAC6 increase PD-L1 expression by recruiting STAT3 | HDAC6-inhibitor decreases PD-L1 by de-activating STAT3 | [23] |
| Active H3K4me3 | up-regulates PD-L1 in breast cancer | EMT-induced PD-L1 expression | Active H3K4me3 modifications in Breast cancer | Selective histone inhi. enhance the efficacy of ICB-Abs | [24] |
| Active H3K4me3 | up-regulates PD-L1 in pancreatic cancer | PD-L1 (CD274) | MLL1 catalyzed H3K4me3 to bind with PD-L1 promoter and increase its expression | MLL1 inhibitor in combination with anti-PD-L1,anti-PD-1 improves efficacy | [25] |
| Repressive H3K27me3 | down-regulates PD-L1 in HCC | PD-L1, IRF1 | EZH2 negatively regulate PD-L1 exp. by recruiting repressive H3K27me3 in HCC | Selective H3K27me3 inhibitor could enhance ICB efficacy | [26] |
| HDACi (Belinostat) | up-regulates PD-L1 & CTLA4 in HCC | Increase IFN-γ & reduce T-reg populations | Belinostat treatment increase anti-tumor immunity against HCC | Combining belinostat enhances the efficacy of ICB therapy | [27] |
| SAHA | Increases CTLA4 and Foxp3 exp. cardiac transplant | Foxp3 CTLA4 | SAHA increases suppressive function of T-reg to prolong allograft survival | SAHA (HDACi) couls be a promissing immunosuppressive agent with CNI drug | [28] |
| H3Ac | up-regulates PD-L1 in drug resistant cancer cell | H3Ac enhance PD-L1 exp. | drug resistant issues in cancer cells | HDACi in combination with anti-PD-L1 | [29] |
| MicroRNAs in ICB-Therapeutics | |||||
| miRNAs | Disease Condition | Target | Mechanism | Therapeutic Strategies | Ref. |
| miR-15a,b miR-16, miR-193a-3p | down-regulated in MPM | Direct target of PD-L1 | miR-15a, miR-16 and miR-193a-3p (−)vely regulates PD-L1 | Respective miRNA mimics combined ICB-therapeutics | [30] |
| miR-17-5p | down-regulated in melanoma | Directly binds 3′-UTR PD-L1 | miR-17-5p (−)vely regulates PD-L1 | miR-17-5p mimics with anti-PD-L1 Abs | [31] |
| miR-18a (miR-140, 142, 340, 383) | up-regulated in cervical cancer | PI3K/AKT, WNK2, SOX6, p53 PTEN, MEK | miR-18a (+)vely and miR-140, 142, 340, 383 (−)ly regulates PD-L1 | Respective miRNA antagomiR & mimics with ICB-therapy | [32] |
| miR-20b-21-130b | up-regulated in colorectal cancer | PTEN, B7-H1 (PD-1) | miRs (+)vely regulates B7-H1 (PD-1) exp. | Respective miRNAs AntagomiRs in combination with ICB-therapeutics | [33] |
| miR-21 (CD4+T-cells) | up-regulated in arthritis and GC | PDCD4, Th17, STAT5, T-reg | miR-21 (−)vely regulates PDCD4, PD-1 | [34,35] | |
| miR-23a-3p | up-regulated in (MΦ) liver cancer | PTEN, AKT pathways | miR-23a-3p (+)vely regulates PD-L1 exp. | Anti-miR-23a-3p (antagomiR therapy) with anti-PD-L1 Abs | [36] |
| miR-25-93- 106b cluster | down-regulated in pancreatic cancer | CXCL12, PD-L1 | miR-25-93- 106b−/− mice increases PD-L1 | miR-93, miR-106b mimics with BET inh. | [37] |
| miR-28 | melanoma | PD-1 | miR-28 (−)vely regulates PD-1 | miR-28 mimics | [38] |
| miR-33a | down-regulated in Lung A. carcinoma | PD-L1,CTLA4, PD-1, CAND1 | miR-33a (−)vely regulates PD-1/PD-L1 | miR-33a mimics with combined ICB-Abs | [39] |
| miR-34a | down-regulated in AML, lymphoma | EBF-1 and 3′-UTR PD-L1 | miR-34a (−)vely regulates PD-L1 exp. | ICB therapy combined miRNA | [40,41,42,43,44] |
| miR-138-5p | down-regulated in CRC | Target 3′-UTR PD-L1 | miR-138-5p (−)vely regulatesPD-L1 exp. | miR-138-5p mimics combined ICB-Abs | [45] |
| miR-140 | down-regulated in NSCLC | miR-140/ PD-L1/cyclinE pathways | miR-140 target 3′-UTR PD-L1 (−)vely regulates its exp. | miR-140 mimics with anti-PD-L1 therapy | [46] |
| miR-142-5p | down-regulated in pancreatic cancer | miR-142-5p target 3′-UTR PD-L1 | miR-142-5p (−)vely regulates PD-L1 exp. | miR-142-5p mimics + anti-PD-L1 therapy | [47] |
| miR-145 | down-regulated in ovarian carcinoma | Cisplatin cMYc (TcF) | miR-145 (−)vely regulates PD-L1 exp. | miR-145 mimic (restoration therapy) with anti-PD-L1 Abs | [48] |
| miR-146a | up-regulated in melanoma | STAT1-IFNγ axis | miR-146a (+)vely regulates PD-L1 exp. | miR-146a antagomiR with anti-PD-L1 Abs | [49] |
| miR-148a -3p | down-regulated in dMMR/MSI-H CRC | miR-148a-3p binds to 3′-UTR PD-L1 | miR-148a-3p (−)vely regulates PD-L1 exp. | Respective miRNA mimics with anti-PD-L1 therapy | [50] |
| miR-155 | up-regulated in B-cell lymphoma | AKT and ERK | miR-155 (+)vely regulates PD-L1 exp. | miR-155 antagomiR + PD-L1 antagonists | [51] |
| miR-191-5p | down-regulated in colon-adenocarcinoma | PD-L1 | miR-191-5p (−)vely regulates PD-L1 exp. | miR-191-5p mimics | [52] |
| miR-195 | down-regulated in PC and DLBCL | PD-L1 | miR-191-5p (−)vely regulates PD-L1 exp. | miR-191 mimics | [53,54] |
| miR-197 | down-regulated in NSCLC | CKS1B/STAT3 (Bcl-2, c-Myc, CyclinD1) | miR-197 (−)vely regulates PD-L1 exp. | miR-193 mimics (replacement therapy) + ICB-therapeutics | [55] |
| miR-200b, miR-152 | down-regulated in gastric cancer (GC) | B7-H1 (PD-1) | miR-200b and miR-152 (−)vely regulates B7-H1 | Respective miRNA mimics combined PD-L1 antagonists | [43,56,57] |
| miR-214 | down-regulated in B-cell lymphoma (DLBCL) | miR-214 atrget 3′-UTR PD-L1 | miR-214 (−)vely regulates PD-L1 exp. | miR-214 mimic in combination with anti-PD-L1 Abs | [58] |
| miR-217 | down-regulated in laryngeal cancer | AEG-1 and PD-L1 | miR-217 (−)vely regulates PD-L1 exp | miR-217 mimics with anti-PD-L1 therapy | [59] |
| miR-324-5p miR-338-5p | downregulated in Mycobateria-responsive hedgehog sign | PD-L1, SHH signaling | (−)vely regulate PD-L1 | miRNA mimics | [60] |
| miR-340 | down-regulated in Cervical cancer | PD-L1 | miR-340 (−)vely regulates PD-L1 exp. | miR-340 mimics | [61] |
| miR-375 | down-regulated in HNSCC | JAK2 | Inhibits JAK2-STAT1 axis suppressing PD-L1 exp. | miR-375 mimics | [62] |
| miR-424 (322) | down-regulated in ovarian cancer | PD-1/PD-L1, CD80/CTLA4 | miR-424 (322) (−)vely regulates PD-1/PD-L1, CD80/CTLA4 exp. | miR-424 (322) mimics (restoration therapy) + ICB-therapeutics | [63] |
| up-regulated in Colon cancer | CD28, CD80 and CD86 | up regulated miR-424 impairs anti-tumor immunity | modified tumor-secreted EVs with miR-424 knocked down | [64] | |
| miR-497-5p | down-regulated in RCC (ccRCC) | Cell proliferation | miR-497-5p (−)vely regulates PD-L1 exp. | miR-497-5p mimic with anti-PD-L1 Abs | [65] |
| miR-513 | cholangiocytes in response to C. parvum infection | B7-H1 (PD-1) | miR-513 (−)vely regulates PD-1 exp. | miR-513 mimics | [66] |
| miR-570 | down-regulated in gastric cancer | B7-H1 (PD-1) | SNP (polymorphism) disrupts miR-570- B7-H1 interactions | Restoration therapy combined ICB-Abs | [43,67] |
| miR-873 | down-regulated in breast cancer | PI3K/Akt, ERK1/2 pathways | miR-873 (−)vely regulates PD-L1 by binding to 3′-UTR | miR-873 mimics with PD-1/PD-L1 inhibitor | [68] |
| miR-3127-5p | up-regulated in NSCLC | pSTAT3 | Upregulates PD-L1 by suppressing p-STAT3 | Anti-miR-3127-5p (antagomiR therapy) | [69] |
| miR-3609 | down-regulated in breast cancer | PD-L1 | miR-3609 (−)vely regulates PD-L1 exp. | miR-3609 mimics | [70] |
| miR-4717 | down-regulated in HBV | PD-1 | miR-4717 (−)vely regulates PD-1 exp. | miR-4717 mimics | [71] |
| Immune Cell Targeted Antibodies (Anti-PD-1 Therapy) | ||||||
|---|---|---|---|---|---|---|
| Company | Antibody FDA Approval | Brand/ Other Name | Combination | Disease | Clinical Trial | Ref. |
| Bristol-Meyers Squibb | Nivolumab (Human IgG4) 2014 | Opdivo®, BMS-936558, MDX-1106 ONO-4538 | LAG3 (BMS-986016), B7-H3 (Enoblituzumab), KIR (Lirilumab), 4-1BB (Urelumab), ICOS (JTX-2011), CD27 (Varlilumab), GM.CD40L (vaccine for lung NSCLC) | Broad range of tumor types and Lymphomas | NCT01968109 NCT02817633 NCT01714739 NCT02253992 NCT02904226 NCT02335918 NCT02466568 NCT01673867 | [83,84] |
| Medimmune | MEDI0680 (AMP-514) | - | NCT02118337 Phase I | [85,86] | ||
| Regeneron/ Sanofi | REGN2810 | - | Phase I/II NCT02383212 NCT02760498 | |||
| Novartis | PDR001 | GITR (GWN323) | NCT02740270 | [87] | ||
| Merck | Pembrolizumab (Humanized IgG4k) 2014 | Keytruda® MK-3475, lambrolizumab | B7-H3 (Enoblituzumab), Multi-kinase inhibitor (Sunitinib) | Melanoma, Lung, NSCLC, HNC, cervical, thyroid cancer | NCT02475213 NCT02599779 NCT01295827 | [83,88] |
| Cure Tech | Pidilizumab (Humanized IgG1k) | CT-011 | Pidilizumab (formerly CT-011), anti-delta like-1 (DLL1), anti-PD-1 | Malignant gliomas | Phase I/ II NCT01952769 | |
| Sanofi | Cemiplimab 2018 | Libtayo® | Cervical cancer CSCC | Phase III | [83] | |
| Immune Cell Targeted Antibodies (Anti-CTLA4 Therapy) | ||||||
| Medarex/ Bristol-Meyers Squibb | Ipilimumab (IgG1 isotype) 2011 | Yervoy® (BMS-734016, MDX-010, MDX-101) | Nivolumab, Gemcitabine, Cisplatin | Melanoma, SCLC, Bladder, prostate cancer | NCT00527735 NCT01524991 NCT00323882 | [83,89,90,91,92] |
| Pfizer/ AstraZeneca | Tremolimumab (IgG2 isotype) 2015 | Orphan drug approval, CP-675, 206 | Metastatic melanoma, Solid Tumor | Phase III NCT02527434 NCT03703297 | [93,94,95,96,97] | |
| Tumor Cell/APC-Targeted Antibodies (Anti-PD-L1/L2 Therapy) | ||||||
| Roche/ Genentech | Anti-PD-L1 Atezolizumab (Humanized IgG1k), 2016 | Tecentriq®, MPDL3280A, RG7446, RO5541267 | CD27 (Varlilumab), VEGF inhibitors (Bevacizumab cediranib) | Ovarian, Urothelial, Lung Cancer, HNCLC | NCT02543645 NCT02659384 | [83] |
| Merck, EMD, Serono/Pfizer | Avelumab 2017 | Bavencio® MSB0010718C | Metastatic MCC | Urothelial, RCC, Merkel | NCT02603432 | [83,98] |
| Medimmune/ AstraZeneca | Anti-PD-L1 Durvalumab (Human IgG1k), 2017 | Imfinzi® MEDI4736 | Osimertinib, Olaparib and Sunitinib | NSCLC, Solid Tumor, urothelial carcinoma | Reference [70] NCT02221960 NCT02484404 | [99,100,101] |
| Bristol-Meyers Squibb | Anti-PD-L1 (Human IgG4) | BMS-936559 (MDX1105) | - | HIV-1, Sepsis, NSCLC | Phase I NCT02028403 | [102,103,104] |
| Amplimmune/ Glaxo Smith Klein | Anti-PD-L2 | AMP-224 | - | MCC | NCT02298946 | [105] |
| Anti-PD-L2 AMP-514 (fusion protein) | MEDI0680 | - | kidney cancer, melanoma | Phase I NCT02013804 | [86] | |
| Biopharmaceutical Company/University | Target | Combined Therapeutic Approach | Clinical Trial | Indication | Ref. |
|---|---|---|---|---|---|
| Natural Killer Cells (NK-cells) Clinicaltrials.gov, accessed on 15 July 2021 | |||||
| ONK therapeutics (Ireland) 2015 www.onktherapeutics.com | CISH−/− NK-cells NK-cells | CISH−/− NK-cells in combination with ICB-antibodies | ONK102 ONK103 ONK104 | M. Myeloma NSCLC AML | [135] |
| Fate Therapeutics San Diego, USA | iPSC-derived NK Cells (FT500) | Nivolumab (anti-PD-1) Pembrolizumab (anti-PD-1) Atezolizumab (anti-PD-L1) Interleukin-2 (IL-2) | NCT03841110 NCT04106167 (Phase-I) | Advanced solid tumors and lymphoma | [136,137,138,139,140] |
| Innate Pharma S. A | NK cell (NKG2A) | Durvalumab (Phase-I/II) Nivolumab (Phase-I) Ipilimumab (Phase-I) Nivolumab + 5-Aza (Ph-I) | NCT02671435 NCT01592370 NCT01750580 NCT02599649 | Metastatic Cancer | [141,142] |
| Altor Biosciences corporation | IL-15 super agonist mediated NK-cells | Nivolumab (anti-PD-1) | NCT02523469 (Phase-I/II) | NSCLC | [142] |
| ImmunityBio, Inc. | High-affinity Natural Killer (haNK) Cell | Avelumab (Bavencio®) (anti-PD-L1) | NCT03387085 (Phase-I/II) | Triple Negative Breast Cancer | - |
| SignalRX Pharmaceuticals, Inc. | SF1126 (dual inhibitor of PI3K and BRD4) | Nivolumab (anti-PD-1) | NCT03059147 | Advanced HCC | [83] |
| Effector Therapeutics | Tomivosertib (eFT-508) | Pembrolizumab (anti-PD-1) | NCT03616834 Phase-II Completed 2021 | Solid tumors and NSCLC | [83] |
| NantKwest Inc., and Chan Soon-Shiong Institute for Medicine, USA | CD16-targeted NK-cell (haNKTM) with N-803 (IL-15 superagonist) | Avelumab (Bavencio®) (anti-PD-L1) | NCT03853317 (Phase-II) | Merkel cell carcinoma | [139,143] |
| National Cancer Institute, Naples | NK-cells (Tregs and NKs) | Nivolumab (anti-PD-1) | NCT03891485 | Renal cell carcinoma | [144] |
| Gachon University & Severance hospital, Republic of Korea | Allogeneic NK-Cells (SMT-NK) | Pembrolizumab (anti-PD-1) Keytruda | NCT03937895 (Phase-I/II) | Biliary tract cancer | [139] |
| Fox Chase Cancer Center, USA | NK-cells and T-cells | Pembrolizumab (anti-PD-1) | NCT02535247 (Phase-I/II) | Lymphoma | [144,145,146] |
| Jilin University Hospital, China | NK-cells | PD-1 Ab | NCT03958097 (Phase-II) | Non-small cell lung cancer | [139] |
| MD Anderson Cancer Center, USA | DF1001 (a new molecule targeting NK-cell activations) | Drug: DF1001 Pembrolizumab (anti-PD-1) | NCT04143711 (Phase-I/II) | Advanced Solid Tumors | [139,144] |
| T-Cells: Tumor-Infiltrating Lymphocytes (TILs) | |||||
| Intima Bioscience, Inc. with University of Minnesota | CISH-deleted Tumor-Infiltrating Lymphocytes (TIL) | CISH checkpoint-deleted TILs combined with Cyclophosphamide, Fludarabine, Aldesleukin and ICB-therapeutics | NCT04426669 (Phase-I/II) | Solid tumors & gastro-intestinal cancers | [1,147] |
| CISH−/− T-cells (TILs) | NCT03538613 (Phase-I/II) | Gastro-intestinal cancers | [2,5] | ||
| Hangzhou Cancer Hospital in collabration with Anhui Kedgene Biotechnology Co.,Ltd | PD-1 Knockout T-Cells | CRISPR/Cas9-deleted PD-1 in T-Cells with hydrocortisone | NCT03081715 (Phase-I) Completed, 2018 | Advanced Esophageal Squamous Cell Carcinoma | [2,144] |
| Sichuan University in collabration with Chengdu MedGenCell | PD-1 Knockout T-Cells | CRISPR/Cas9-deleted PD-1 in T-Cells with Cyclophosphamide | NCT02793856 (Phase-I) Completed, 2020 | Metastatic Non-small Cell Lung Cancer | [2,144,148] |
| Peking University and (Cell Biotech) | PD-1 Knockout Engineered T Cells | PD-1-KO-T-cells with IL-2 and Cyclophosphamide | NCT02863913 NCT02867345 NCT02867332 (Phase-I) | Bladder, Prostate and Renal Cell Carcinoma | [2,5] |
| University of Pennsylvania, with Tmunity Therapeutics | NY-ESO-1 redirected autologous T cells | TCR-deleted and PD-1-deleted T cells | NCT03399448 | Myeloma, melanoma and several cancers | [2,5,149] |
| Nanjing University Medical School | PD-1 Knockout EBV-CTLs | PD-1-KO-EBV-CTL with IL-2, Fludarabine and Cyclophosphamide | NCT03044743 (Phase-I/II) | EBV associated Malignancies | [2,5] |
| Dendritic Cells (DCs) | |||||
| H. Lee Moffitt Cancer Center, BMS and MultiVir, Inc. | DC-based p53 Vaccine | Ipilimumab (anti-CTLA4) Nivolumab (anti-PD-1) | NCT03406715 (Phase-II) | Small Cell Lung Cancer | [137] |
| Allife Medical Sc. and Technology Co., Ltd. | DC-NK YNYY-01 (DC-NK Cells) | Pembrolizumab (anti-PD-1) Keytruda | NCT03815084 (Phase-I) | Solid tumors | [144,150] |
| Bristol-Myers Squibb and Duke Cancer Inst. | DC Vaccines | Nivolumab (anti-PD-1) | NCT02529072 NCT02775292 (Phase-I) | Recurrent Brain Tumors | |
| Northwest Biotherapeutics, BMS and JCCC | Autologous DCs pulsed with tumor lysate | Nivolumab (anti-PD-1) | NCT03014804 (Phase-II) | Recurrent Glioblastoma | |
| University of Pennsylvania | Autologous DC pulsed peptide | Pembrolizumab (anti-PD-1) | NCT03092453 (Phase-I) | Advanced Melanoma | |
| Mayo Clinic in collabration with National Cancer Inst. | Autologous DC pulsed tumor Ags | Pembrolizumab (anti-PD-1) | NCT03035331 (Phase-I/II) | Aggressive Non-Hodgkin Lymphoma | |
| Oslo University Hospital in collabration with NCS and MSDC | Autologous DC | Pembrolizumab (anti-PD-1) Rituximab, GM-CSF and anti-TNF-alpha therapy | NCT02677155 (Phase-II) | Follicular Lymphoma | |
| Capital Medical Univ. in collabration with Duke Univ. | Autologous DC-CIK cell | Pembrolizumab Anti-PD-1 + DC-CIK (Ph-I) Anti-PD-1 alone (Ph-II) | NCT03190811 NCT03360630 | Advanced Solid Tumors and NSCLC | |
| Sun Yat-sen University | DC-CIK cell (Cytokine-induced Killer Cell) | Anti-PD-1 antibody | NCT02886897 (Phase-I) Completed, 2019 | Refractory Solid Tumors | |
| Beth Israel Deaconess Medical Center | Dendritic Cell Fusion Vaccine | Pidilizumab (anti-PD-1) | NCT01067287 (Phase-I) | Multiple Myeloma | |
| Cancer Insight in collabration with Elios Therapeutics, LLC | Autologous DC (TLPLDC Vaccine) | Checkpoint Inhibitor | NCT02678741 (Phase-I/II) | Metastatic Melanoma | |
| Grupo Espanol Multidisciplinario del Cancer Digestivo | Autologous DC Vaccine (AVEVAC) | Avelumab (Bavencio®) (anti-PD-L1) | NCT03152565 (Phase-I/II) Completed, 2020 | Metastatic Colorectal Carcinoma | |
| Dana-Farber Cancer Institute in collabration with Celgene | DC/AML Fusion Vaccine | Durvalumab (Imfinzi®) (anti-PD-L1) | NCT03059485 (Phase-II) | Acute Myelogenous Leukemia | |
| Radboud University in collabration with Dutch Cancer Society | MiHA-loaded PD-L1/L2 silenced DC Vaccination | PD-L1/L2-silenced DC (siRNA silenced) | NCT02528682 (Phase-I/II) Completed, 2021 | Hematological Malignancies | |
| Johns Hopkins University, USA | TLR3 agonist enhace DC activation | Anti-PD-1 in combination with DCs | - | Glioblastoma | [16] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Sarthi, P.; Mani, I.; Ashraf, M.U.; Kang, M.-H.; Kumar, V.; Bae, Y.-S. Epitranscriptomic Approach: To Improve the Efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH. Cells 2021, 10, 2250. https://doi.org/10.3390/cells10092250
Kumar S, Sarthi P, Mani I, Ashraf MU, Kang M-H, Kumar V, Bae Y-S. Epitranscriptomic Approach: To Improve the Efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH. Cells. 2021; 10(9):2250. https://doi.org/10.3390/cells10092250
Chicago/Turabian StyleKumar, Sunil, Parth Sarthi, Indra Mani, Muhammad Umer Ashraf, Myeong-Ho Kang, Vishal Kumar, and Yong-Soo Bae. 2021. "Epitranscriptomic Approach: To Improve the Efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH" Cells 10, no. 9: 2250. https://doi.org/10.3390/cells10092250
APA StyleKumar, S., Sarthi, P., Mani, I., Ashraf, M. U., Kang, M. -H., Kumar, V., & Bae, Y. -S. (2021). Epitranscriptomic Approach: To Improve the Efficacy of ICB Therapy by Co-Targeting Intracellular Checkpoint CISH. Cells, 10(9), 2250. https://doi.org/10.3390/cells10092250