
Substrate Reduction Therapy Reverses Mitochondrial, mTOR, and Autophagy Alterations in a Cell Model of Gaucher Disease

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Brain Tissues
2.2. Western Blot (WB) Analyses
2.3. GCase Activity Assay
2.4. Glycosphingolipid Analyses
2.5. Oxygen Consumption Rate Assay
2.6. LysoTracker and MitoTracker Staining
2.7. CYTO-ID® Autophagy Dye Staining
2.8. Cell Proliferation and Viability Assessment
2.9. Mitochondrial Membrane Potential Assay
2.10. Mitochondrial Isolation
2.11. Electron Microscope (EM)
2.12. Statistical Analyses
3. Results
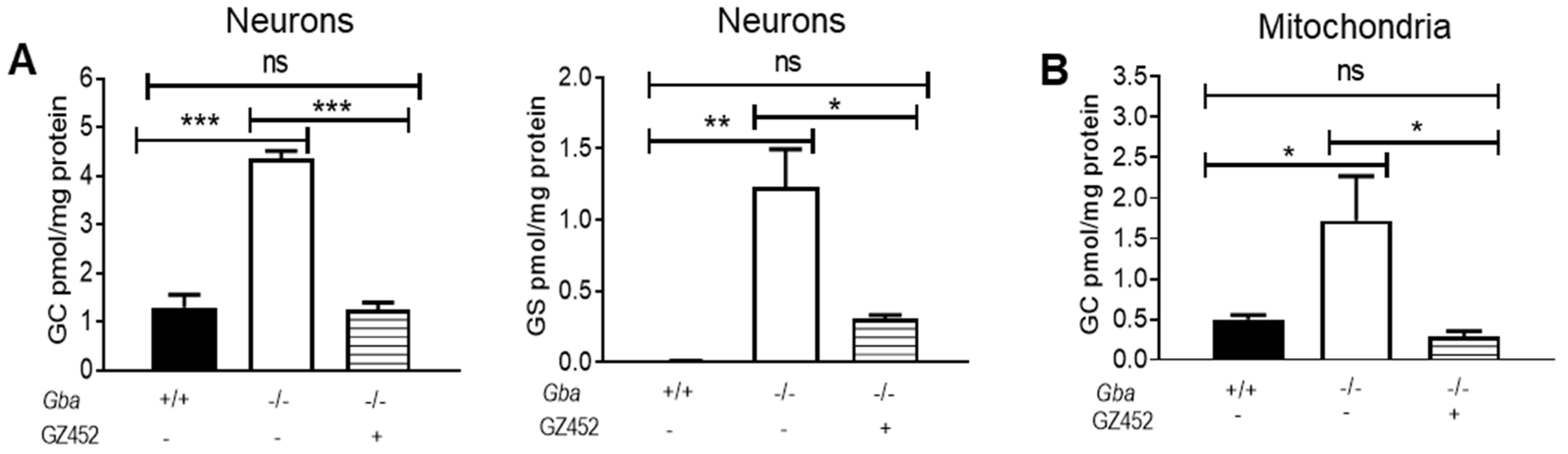
3.1. SRT Corrected Substrate Accumulation in Gba-/- Neurons
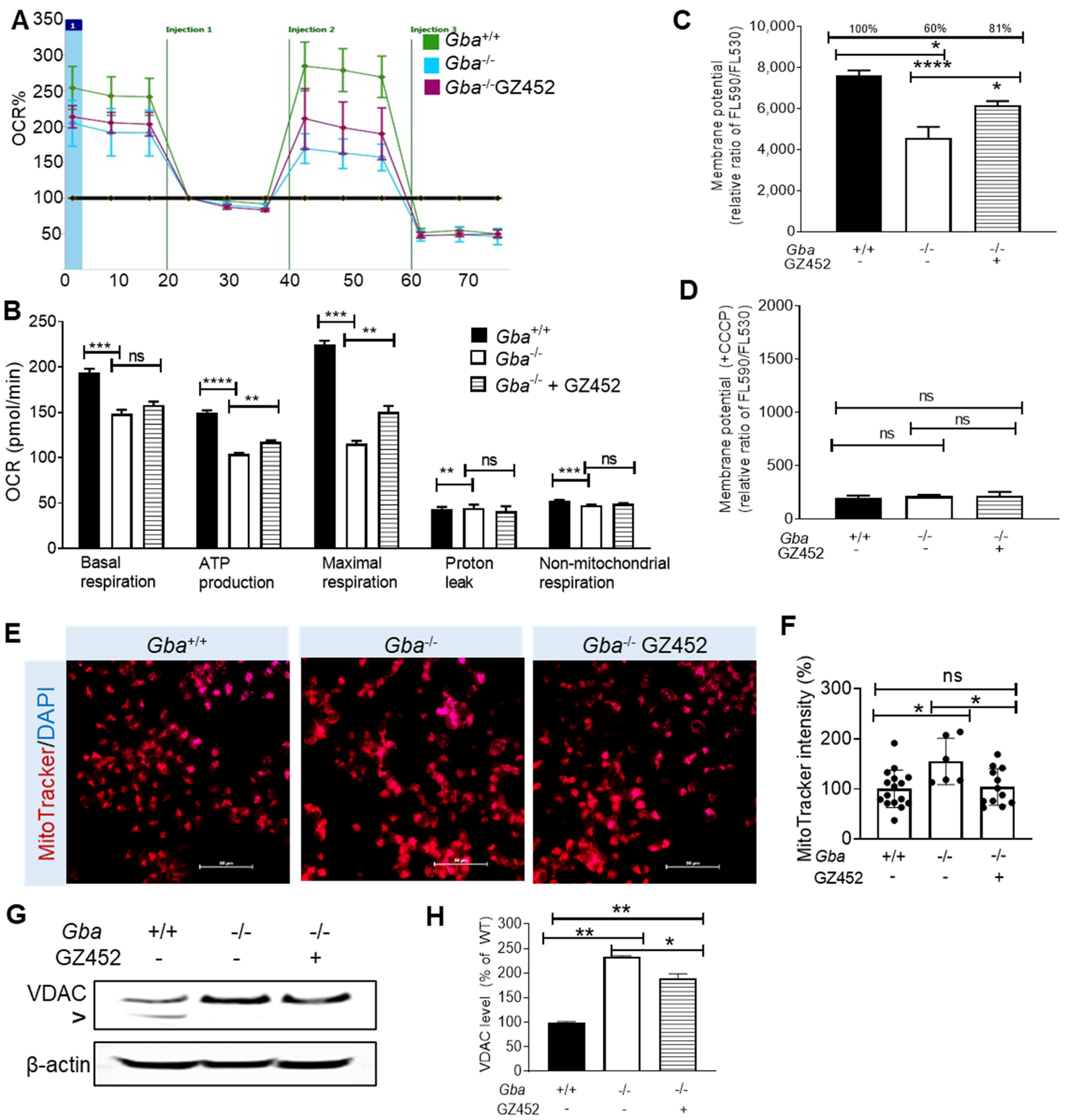
3.2. SRT Improved Oxygen Consumption Rate and Mitochondrial Membrane Potential
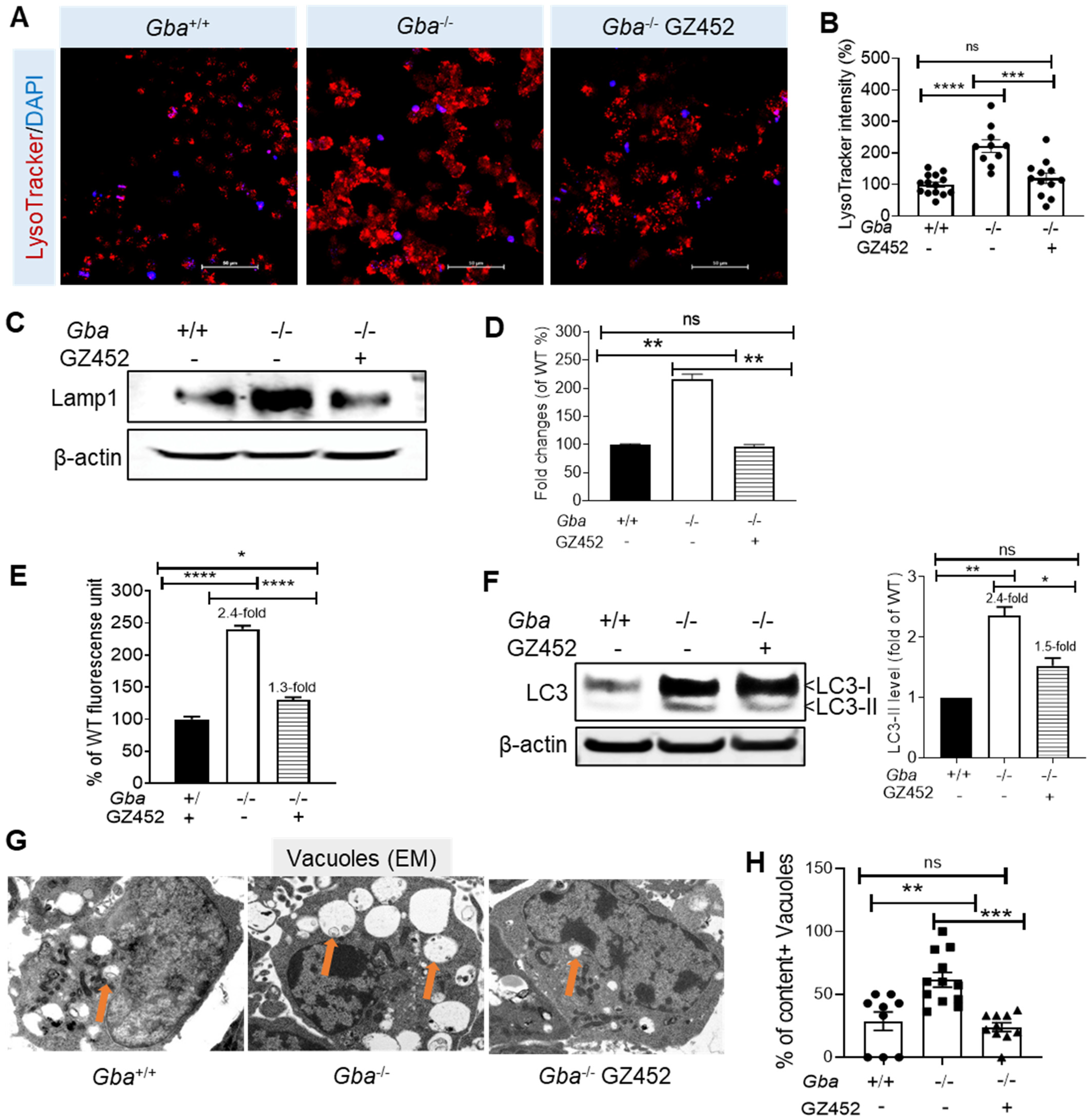
3.3. SRT Protects Lysosomes and Autophagy
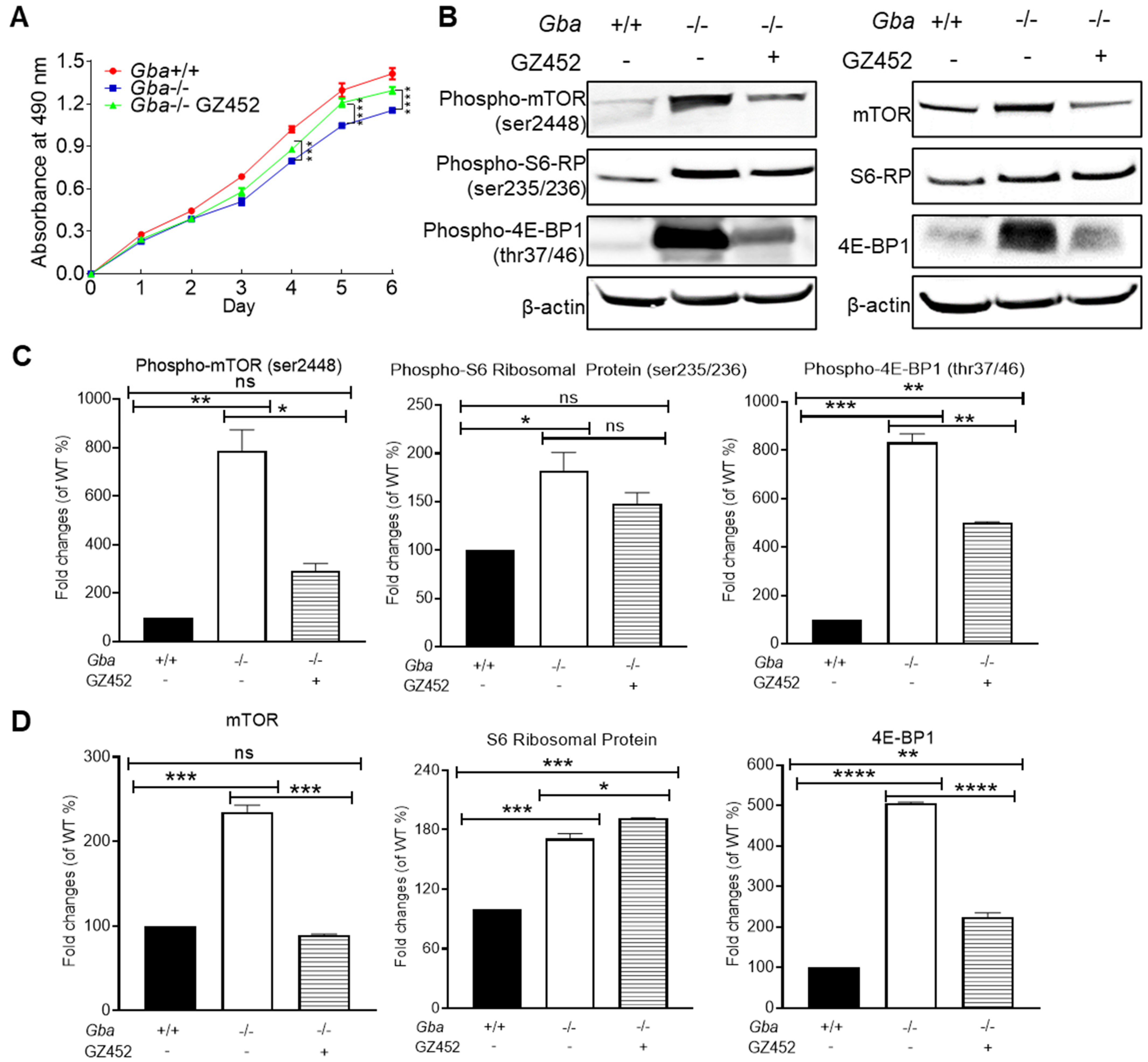
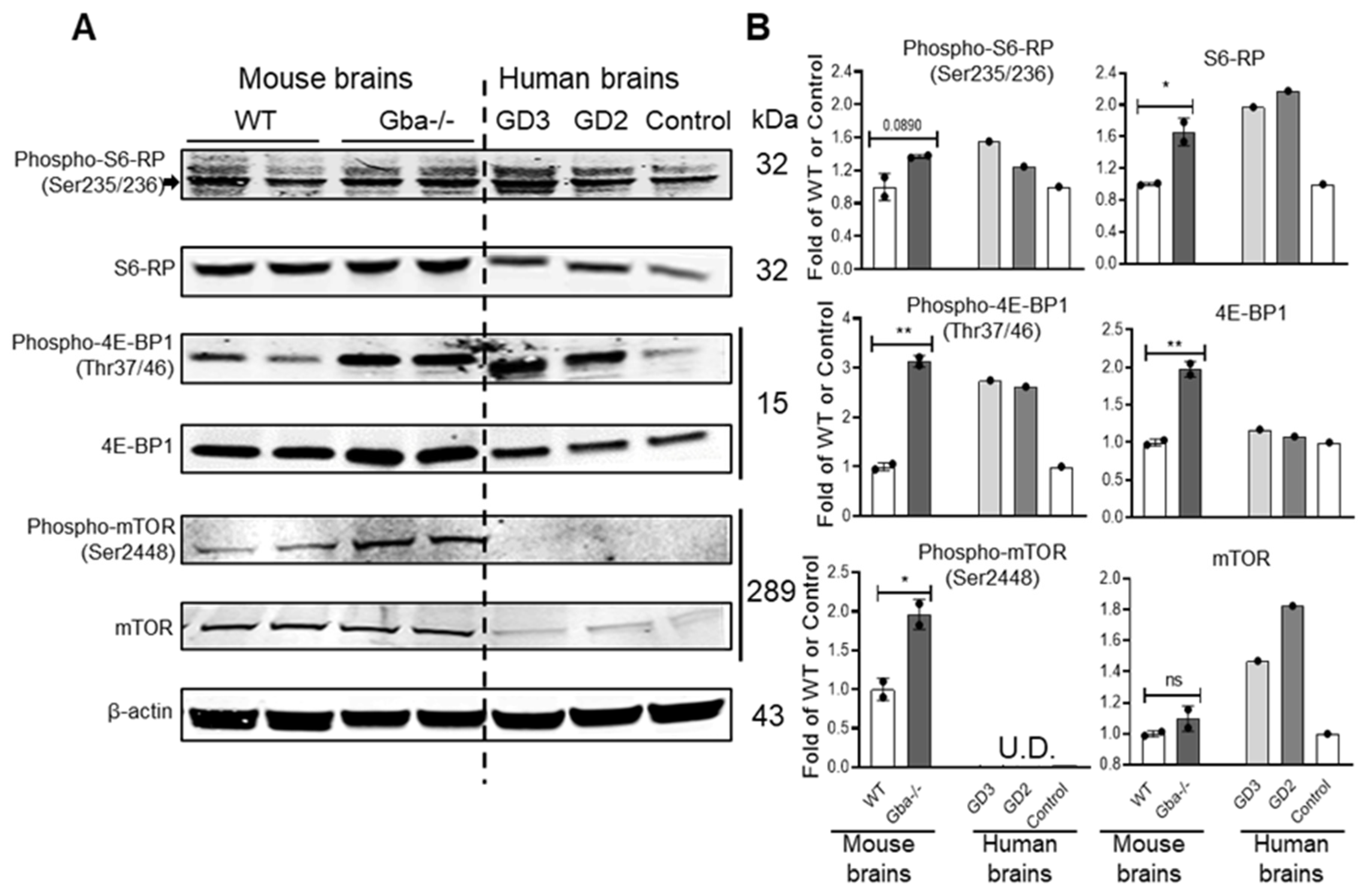
3.4. SRT Lessened Hyperactivity of mTOR Pathway
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Grabowski, G.A.; Petsko, G.A.; Kolodny, E.H. Gaucher disease. In The Online Metabolic and Molecular Bases of Inherited Diseases; Valle, D., Beaudet, A., Vogelstein, B., Kinzler, K.W., Antonarakis, S.E., Ballabio, A., Eds.; The McGraw-Hill Companies, Inc.: New York, NY, USA, 2010. [Google Scholar]
- Grabowski, G.A.; Zimran, A.; Ida, H. Gaucher disease types 1 and 3: Phenotypic characterization of large populations from the ICGG Gaucher Registry. Am. J. Hematol. 2015, 90, S12. [Google Scholar] [CrossRef]
- Grabowski, G.A.; Antommaria, A.H.M.; Kolodny, E.H.; Mistry, P.K. Gaucher disease: Basic and translational science needs for more complete therapy and management. Mol. Genet. Metab. 2021, 132, 59–75. [Google Scholar] [CrossRef]
- Choy, F.Y.; Zhang, W.; Shi, H.P.; Zay, A.; Campbell, T.; Tang, N.; Ferreira, P. Gaucher disease among Chinese patients: Review on genotype/phenotype correlation from 29 patients and identification of novel and rare alleles. Blood Cells Mol. Dis. 2007, 38, 287–293. [Google Scholar] [CrossRef]
- Koprivica, V.; Stone, D.L.; Park, J.K.; Callahan, M.; Frisch, A.; Cohen, I.J.; Tayebi, N.; Sidransky, E. Analysis and classification of 304 mutant alleles in patients with type 1 and type 3 Gaucher disease. Am. J. Hum. Genet. 2000, 66, 1777–1786. [Google Scholar] [CrossRef] [Green Version]
- Ankleshwaria, C.; Mistri, M.; Bavdekar, A.; Muranjan, M.; Dave, U.; Tamhankar, P.; Khanna, V.; Jasinge, E.; Nampoothiri, S.; Edayankara Kadangot, S.; et al. Novel mutations in the glucocerebrosidase gene of Indian patients with Gaucher disease. J. Hum. Genet. 2015, 60, 285. [Google Scholar] [CrossRef] [Green Version]
- Mignot, C.; Gelot, A.; Bessieres, B.; Daffos, F.; Voyer, M.; Menez, F.; Fallet Bianco, C.; Odent, S.; Le Duff, D.; Loget, P.; et al. Perinatal-lethal Gaucher disease. Am. J. Med. Genet. A. 2003, 120A, 338–344. [Google Scholar] [CrossRef]
- Burrow, T.A.; Sun, Y.; Prada, C.E.; Bailey, L.; Zhang, W.; Brewer, A.; Wu, S.W.; Setchell, K.D.R.; Witte, D.; Cohen, M.B.; et al. CNS, lung, and lymph node involvement in Gaucher disease type 3 after 11 years of therapy: Clinical, histopathologic, and biochemical findings. Mol. Genet. Metab. 2015, 114, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Conradi, N.G.; Sourander, P.; Nilsson, O.; Svennerholm, L.; Erikson, A. Neuropathology of the Norrbottnian type of Gaucher disease. Morphological and biochemical studies. Acta. Neuropathol. 1984, 65, 99–109. [Google Scholar] [CrossRef]
- Xu, Y.H.; Barnes, S.; Sun, Y.; Grabowski, G.A. Multi-system disorders of glycosphingolipid and ganglioside metabolism. J. Lipid Res. 2010, 51, 1643–1675. [Google Scholar] [CrossRef] [Green Version]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Grabowski, G.A. Impaired autophagosomes and lysosomes in neuronopathic Gaucher disease. Autophagy 2010, 6, 648–649. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Duchen, M.R. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy 2013, 9, 1633–1635. [Google Scholar] [CrossRef] [Green Version]
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, N.; Xu, Y.H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic Gaucher disease: Dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef]
- Cleeter, M.W.; Chau, K.Y.; Gluck, C.; Mehta, A.; Hughes, D.A.; Duchen, M.; Wood, N.W.; Hardy, J.; Mark Cooper, J.; Schapira, A.H. Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem. Int. 2013, 62, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Xu, K.; Sun, Y.; Liou, B.; Quinn, B.; Li, R.H.; Xue, L.; Zhang, W.; Setchell, K.D.; Witte, D.; et al. Multiple pathogenic proteins implicated in neuronopathic Gaucher disease mice. Hum. Mol. Genet. 2014, 23, 3943–3957. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. mTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. Dis. Models Mech. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Srikanth, M.P.; Jones, J.W.; Kane, M.; Awad, O.; Park, T.S.; Zambidis, E.T.; Feldman, R.A. Elevated glucosylsphingosine in Gaucher disease induced pluripotent stem cell neurons deregulates lysosomal compartment through mammalian target of rapamycin complex 1. Stem Cells Transl. Med. 2021, 10, 1081–1094. [Google Scholar] [CrossRef]
- Radin, N.S. Treatment of Gaucher disease with an enzyme inhibitor. Glycoconj. J. 1996, 13, 153–157. [Google Scholar] [CrossRef] [Green Version]
- Sam, R.; Ryan, E.; Daykin, E.; Sidransky, E. Current and emerging pharmacotherapy for Gaucher disease in pediatric populations. Expert Opin. Pharm. 2021, 10, 1489–1503. [Google Scholar] [CrossRef]
- Marshall, J.; Sun, Y.; Bangari, D.S.; Budman, E.; Park, H.; Nietupski, J.B.; Allaire, A.; Cromwell, M.A.; Wang, B.; Grabowski, G.A.; et al. CNS-accessible inhibitor of glucosylceramide synthase for substrate reduction therapy of neuronopathic Gaucher disease. Molecular 2016, 24, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Cabrera-Salazar, M.A.; Deriso, M.; Bercury, S.D.; Li, L.; Lydon, J.T.; Weber, W.; Pande, N.; Cromwell, M.A.; Copeland, D.; Leonard, J.; et al. Systemic delivery of a glucosylceramide synthase inhibitor reduces CNS substrates and increases lifespan in a mouse model of type 2 Gaucher disease. PLoS ONE 2012, 7, e43310. [Google Scholar] [CrossRef] [Green Version]
- Blumenreich, S.; Yaacobi, C.; Vardi, A.; Barav, O.B.; Vitner, E.B.; Park, H.; Wang, B.; Cheng, S.H.; Sardi, S.P.; Futerman, A.H. Substrate reduction therapy using Genz-667161 reduces levels of pathogenic components in a mouse model of neuronopathic forms of Gaucher disease. J. Neurochem. 2021, 156, 692–701. [Google Scholar] [CrossRef]
- Westbroek, W.; Nguyen, M.; Siebert, M.; Lindstrom, T.; Burnett, R.A.; Aflaki, E.; Jung, O.; Tamargo, R.; Rodriguez-Gil, J.L.; Acosta, W.; et al. A new glucocerebrosidase-deficient neuronal cell model provides a tool to probe pathophysiology and therapeutics for Gaucher disease. Dis. Models Mech. 2016, 9, 769–778. [Google Scholar] [CrossRef] [Green Version]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [Green Version]
- Lin-Moshier, Y.; Marchant, J.S. A rapid Western blotting protocol for the Xenopus oocyte. Cold Spring Harb Protoc. Cold Spring Harbor Protoc. 2013, 2013, pdb-prot072793. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.H.; Reboulet, R.; Quinn, B.; Huelsken, J.; Witte, D.; Grabowski, G.A. Dependence of reversibility and progression of mouse neuronopathic Gaucher disease on acid beta-glucosidase residual activity levels. Mol. Genet. Metab. 2008, 94, 190–203. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Zhang, W.; Xu, Y.H.; Quinn, B.; Dasgupta, N.; Liou, B.; Setchell, K.D.; Grabowski, G.A. Substrate compositional variation with tissue/region and Gba1 mutations in mouse models--implications for Gaucher disease. PLoS ONE 2013, 8, e57560. [Google Scholar] [CrossRef] [Green Version]
- Liou, B.; Peng, Y.; Li, R.; Inskeep, V.; Zhang, W.; Quinn, B.; Dasgupta, N.; Blackwood, R.; Setchell, K.D.; Fleming, S.; et al. Modulating ryanodine receptors with dantrolene attenuates neuronopathic phenotype in Gaucher disease mice. Hum. Mol. Genet. 2016, 25, 5126–5141. [Google Scholar] [CrossRef]
- Chu, Z.; Sun, Y.; Kuan, C.Y.; Grabowski, G.A.; Qi, X. Saposin C: Neuronal effect and CNS delivery by liposomes. Ann. N. Y. Acad. Sci. 2005, 1053, 237–246. [Google Scholar] [CrossRef]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
- Cottet-Rousselle, C.; Ronot, X.; Leverve, X.; Mayol, J.F. Cytometric assessment of mitochondria using fluorescent probes. Cytom. A. 2011, 79, 405–425. [Google Scholar] [CrossRef]
- Lim, J.A.; Li, L.; Kakhlon, O.; Myerowitz, R.; Raben, N. Defects in calcium homeostasis and mitochondria can be reversed in Pompe disease. Autophagy 2015, 11, 385–402. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef]
- Jastrzebski, K.; Hannan, K.M.; Tchoubrieva, E.B.; Hannan, R.D.; Pearson, R.B. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors (Chur Switz.) 2007, 25, 209–226. [Google Scholar] [CrossRef]
- Vega-Rubin-de-Celis, S.; Pena-Llopis, S.; Konda, M.; Brugarolas, J. Multistep regulation of TFEB by MTORC1. Autophagy 2017, 13, 464–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Sasaki, S.; Horie, Y.; Iwata, M. Mitochondrial alterations in dorsal root ganglion cells in sporadic amyotrophic lateral sclerosis. Acta. Neuropathol. 2007, 114, 633–639. [Google Scholar] [CrossRef]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Beal, M.F. Mitochondrial dysfunction in neurodegenerative diseases. J. Pharm. Exp. 2012, 342, 619–630. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Liou, B.; Inskeep, V.; Blackwood, R.; Mayhew, C.N.; Grabowski, G.A.; Sun, Y. Intravenous infusion of iPSC-derived neural precursor cells increases acid beta-glucosidase function in the brain and lessens the neuronopathic phenotype in a mouse model of Gaucher disease. Hum. Mol. Genet. 2019, 28, 3406–3421. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.; Moyle, J. Estimation of membrane potential and pH difference across the cristae membrane of rat liver mitochondria. Eur J. Biochem. 1969, 7, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Wong, Y.C.; Kim, S.; Peng, W.; Krainc, D. Regulation and Function of Mitochondria-Lysosome Membrane Contact Sites in Cellular Homeostasis. Trends Cell Biol. 2019, 29, 500–513. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef] [Green Version]
- Myerowitz, R.; Puertollano, R.; Raben, N. Impaired autophagy: The collateral damage of lysosomal storage disorders. EBioMedicine 2021, 63, 103166. [Google Scholar] [CrossRef]
- Bajaj, L.; Lotfi, P.; Pal, R.; Ronza, A.D.; Sharma, J.; Sardiello, M. Lysosome biogenesis in health and disease. J. Neurochem. 2019, 148, 573–589. [Google Scholar] [CrossRef] [Green Version]
- Awad, O.; Sarkar, C.; Panicker, L.M.; Miller, D.; Zeng, X.; Sgambato, J.A.; Lipinski, M.M.; Feldman, R.A. Altered TFEB-mediated lysosomal biogenesis in Gaucher disease iPSC-derived neuronal cells. Hum. Mol. Genet. 2015, 24, 5775–5788. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Liou, B.; Lin, Y.; Fannin, V.; Zhang, W.; Feldman, R.A.; Setchell, K.D.R.; Grabowski, G.A.; Sun, Y. Substrate Reduction Therapy Reverses Mitochondrial, mTOR, and Autophagy Alterations in a Cell Model of Gaucher Disease. Cells 2021, 10, 2286. https://doi.org/10.3390/cells10092286
Peng Y, Liou B, Lin Y, Fannin V, Zhang W, Feldman RA, Setchell KDR, Grabowski GA, Sun Y. Substrate Reduction Therapy Reverses Mitochondrial, mTOR, and Autophagy Alterations in a Cell Model of Gaucher Disease. Cells. 2021; 10(9):2286. https://doi.org/10.3390/cells10092286
Chicago/Turabian StylePeng, Yanyan, Benjamin Liou, Yi Lin, Venette Fannin, Wujuan Zhang, Ricardo A. Feldman, Kenneth D. R. Setchell, Gregory A. Grabowski, and Ying Sun. 2021. "Substrate Reduction Therapy Reverses Mitochondrial, mTOR, and Autophagy Alterations in a Cell Model of Gaucher Disease" Cells 10, no. 9: 2286. https://doi.org/10.3390/cells10092286
APA StylePeng, Y., Liou, B., Lin, Y., Fannin, V., Zhang, W., Feldman, R. A., Setchell, K. D. R., Grabowski, G. A., & Sun, Y. (2021). Substrate Reduction Therapy Reverses Mitochondrial, mTOR, and Autophagy Alterations in a Cell Model of Gaucher Disease. Cells, 10(9), 2286. https://doi.org/10.3390/cells10092286