
Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis

 , , , ,
, , , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Ethics
2.2. Cloning and Expression of the Murine RTB Fusion Construct
2.3. Production and Purification of mβ-Gal:RTB
2.4. In Vivo Treatment with mβ-Gal:RTB
2.5. Western Blot Analyses
2.6. Enzyme Activity Assays
2.7. Immunohistochemistry and IF
2.8. HPTLC Analysis of GM1
2.9. GM1 Sandwich and IgG1 ELISA
2.10. Statistical Analysis
3. Results
3.1. Characterization of Murine β-Gal Fused to RTB
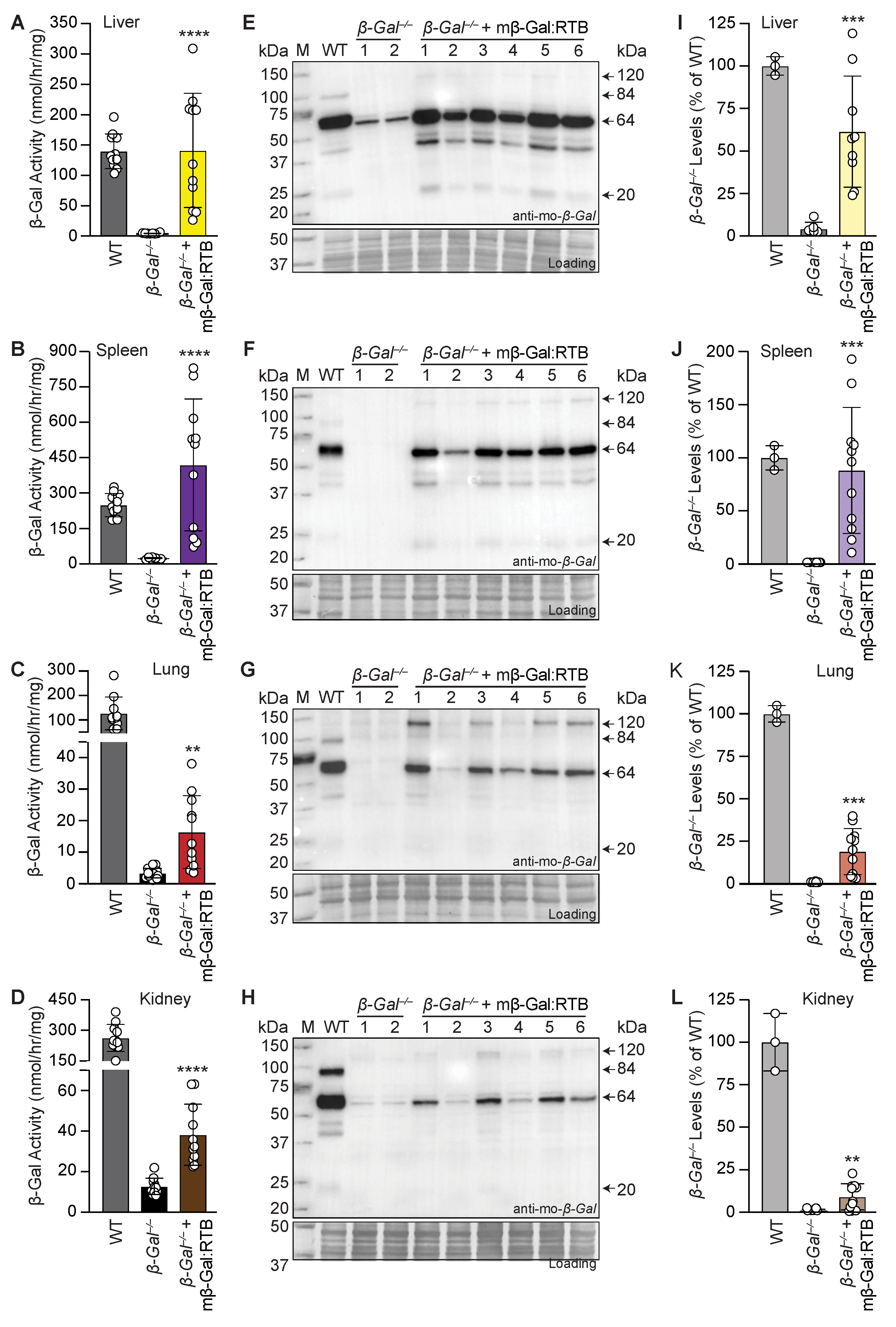
3.2. β-Gal−/− Mice Treated with mβ-Gal:RTB Show Increased β-Gal Activity and Biodistribution in Visceral Organs
3.3. Treatment with mβ-Gal:RTB Led to Increased β-Gal Activity and Decreased GM1 Levels in the Brain
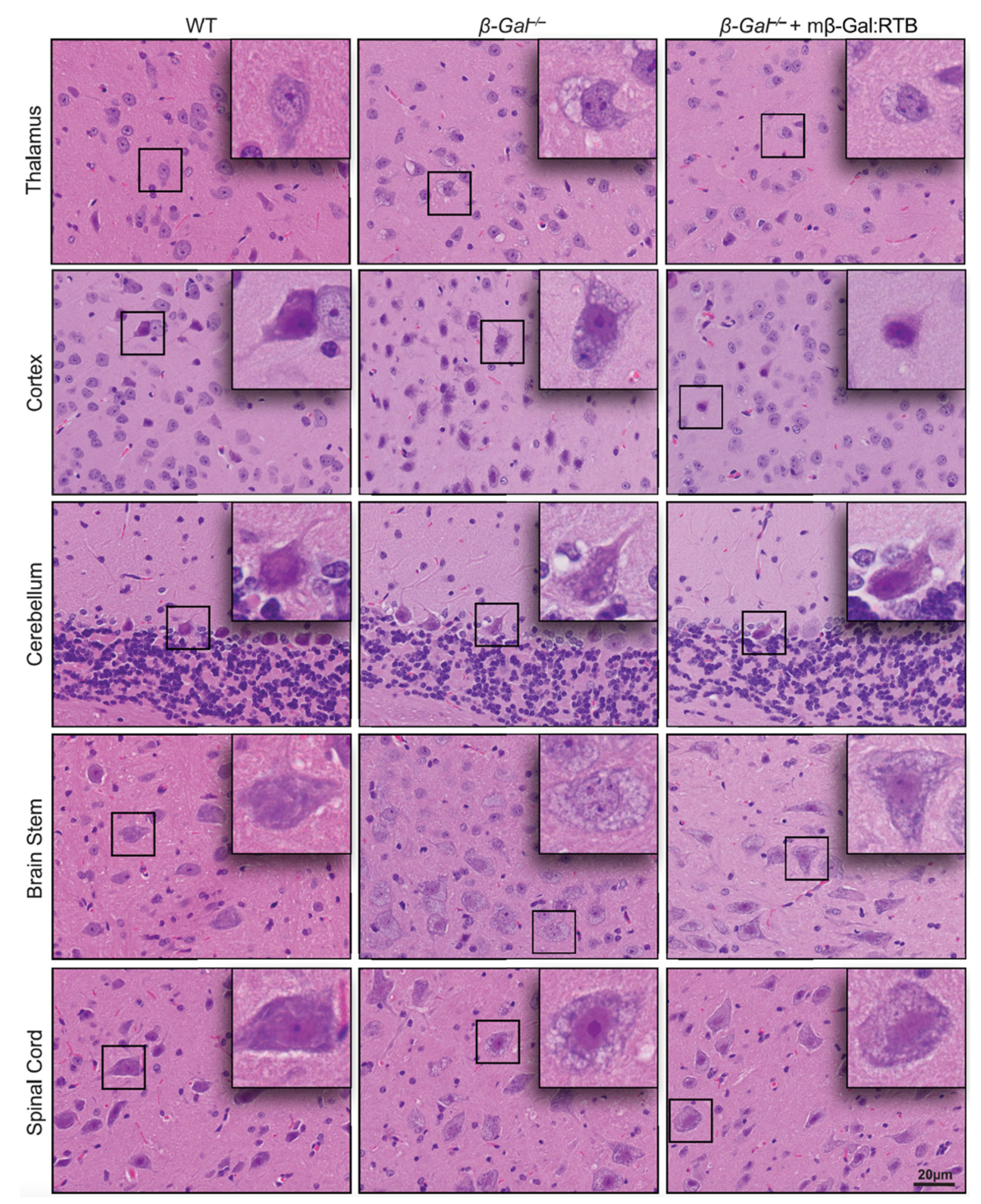
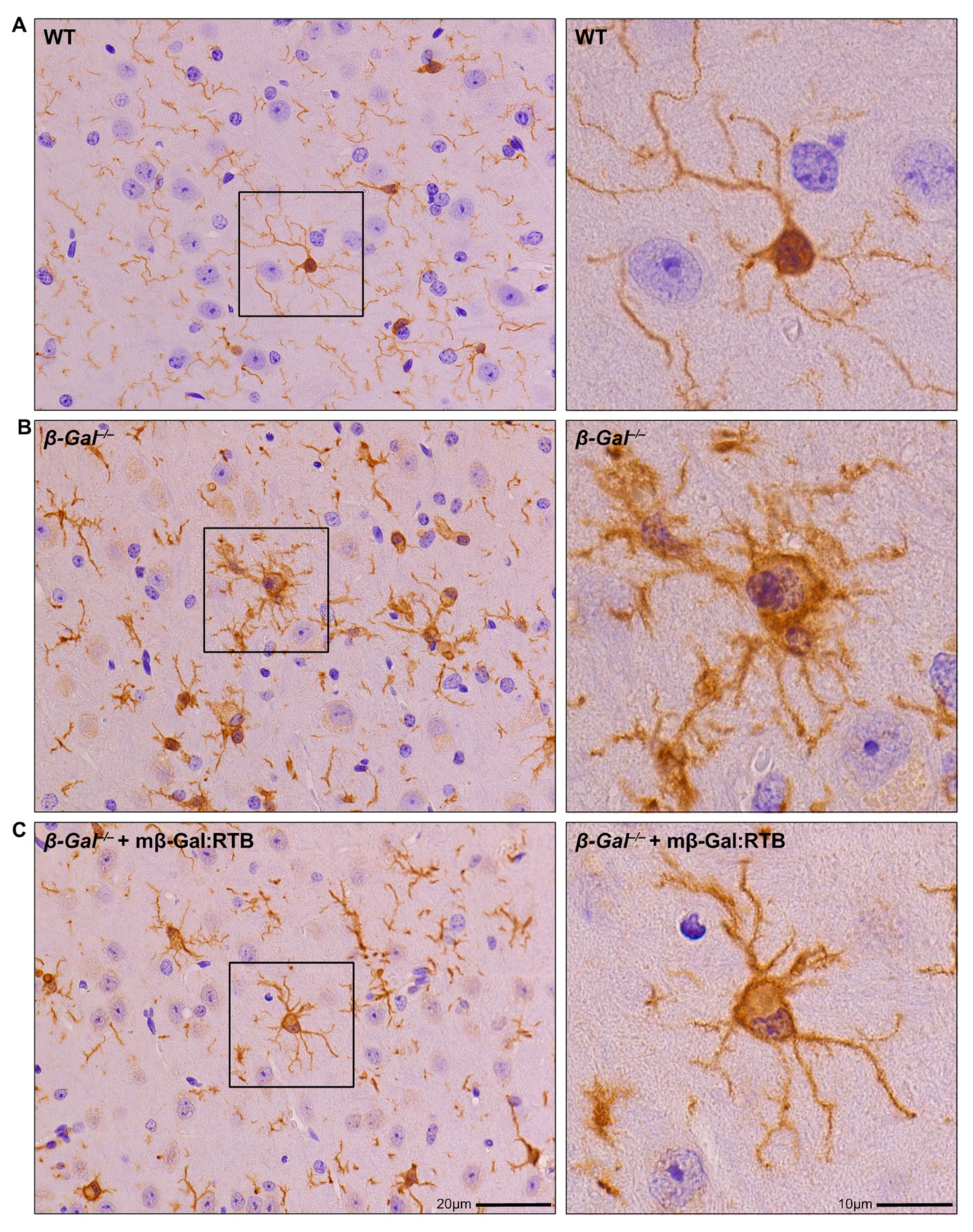
3.4. Amelioration of Phenotypic Abnormalities in mβ-Gal:RTB-Treated β-Gal−/− Brain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Platt, F.M.; D’azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Primers 2018, 4, 27. [Google Scholar] [CrossRef]
- Hasilik, A.; Neufeld, E.F. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J. Biol. Chem. 1980, 255, 4937–4945. [Google Scholar] [CrossRef]
- Lemansky, P.; Hasilik, A.; von Figura, K.; Helmy, S.; Fishman, J.; Fine, R.E.; Kedersha, N.L.; Rome, L.H. Lysosomal enzyme precursors in coated vesicles derived from the exocytic and endocytic pathways. J. Cell. Biol. 1987, 104, 1743–1748. [Google Scholar] [CrossRef]
- Trivedi, P.C.; Bartlett, J.J.; Pulinilkunnil, T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells 2020, 9, 1131. [Google Scholar] [CrossRef]
- Zhu, Y.; Conner, G.E. Intermolecular association of lysosomal protein precursors during biosynthesis. J. Biol. Chem. 1994, 269, 3846–3851. [Google Scholar] [CrossRef]
- Rastall, D.P.; Amalfitano, A. Recent advances in gene therapy for lysosomal storage disorders. Appl. Clin. Genet. 2015, 8, 157–169. [Google Scholar] [CrossRef]
- Cadaoas, J.; Hu, H.; Boyle, G.; Gomero, E.; Mosca, R.; Jayashankar, K.; Machado, M.; Cullen, S.; Guzman, B.; van de Vlekkert, D.; et al. Galactosialidosis: Preclinical enzyme replacement therapy in a mouse model of the disease, a proof of concept. Mol. Ther. Methods Clin. Dev. 2021, 20, 191–203. [Google Scholar] [CrossRef]
- Sevin, C.; Deiva, K. Clinical Trials for Gene Therapy in Lysosomal Diseases With CNS Involvement. Front. Mol. Biosci. 2021, 8, 624988. [Google Scholar] [CrossRef]
- Concolino, D.; Deodato, F.; Parini, R. Enzyme replacement therapy: Efficacy and limitations. Ital. J. Pediatr. 2018, 44, 120. [Google Scholar] [CrossRef]
- Edelmann, M.J.; Maegawa, G.H.B. CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges. Front. Mol. Biosci. 2020, 7, 559804. [Google Scholar] [CrossRef]
- Chen, J.C.; Luu, A.R.; Wise, N.; Angelis, R.; Agrawal, V.; Mangini, L.; Vincelette, J.; Handyside, B.; Sterling, H.; Lo, M.J.; et al. Intracerebroventricular enzyme replacement therapy with β-galactosidase reverses brain pathologies due to GM1 gangliosidosis in mice. J. Biol. Chem. 2020, 295, 13532–13555. [Google Scholar] [CrossRef] [PubMed]
- Boado, R.J.; Lu, J.Z.; Hui, E.K.; Pardridge, W.M. Insulin receptor antibody-sulfamidase fusion protein penetrates the primate blood-brain barrier and reduces glycosoaminoglycans in Sanfilippo type A cells. Mol. Pharm. 2014, 11, 2928–2934. [Google Scholar] [CrossRef] [PubMed]
- Przybilla, M.J.; Stewart, C.; Carlson, T.W.; Ou, L.; Koniar, B.L.; Sidhu, R.; Kell, P.J.; Jiang, X.; Jarnes, J.R.; O’Sullivan, M.G.; et al. Examination of a blood-brain barrier targeting β-galactosidase-monoclonal antibody fusion protein in a murine model of GM1-gangliosidosis. Mol. Genet. Metab. Rep. 2021, 27, 100748. [Google Scholar] [CrossRef]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.J.; Koniar, B.; Whitley, C.B. RTB lectin-mediated delivery of lysosomal α-l-iduronidase mitigates disease manifestations systemically including the central nervous system. Mol. Genet. Metab. 2018, 123, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Condori, J.; Acosta, W.; Ayala, J.; Katta, V.; Flory, A.; Martin, R.; Radin, J.; Cramer, C.L.; Radin, D.N. Enzyme replacement for GM1-gangliosidosis: Uptake, lysosomal activation, and cellular disease correction using a novel β-galactosidase:RTB lectin fusion. Mol. Genet. Metab. 2016, 117, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Acosta, W.; Cramer, C.L. Targeting Macromolecules to CNS and Other Hard-to-Treat Organs Using Lectin-Mediated Delivery. Int. J. Mol. Sci. 2020, 21, 971. [Google Scholar] [CrossRef]
- Nicoli, E.R.; Annunziata, I.; D’azzo, A.; Platt, F.M.; Tifft, C.J.; Stepien, K.M. GM1 Gangliosidosis-A Mini-Review. Front. Genet. 2021, 12, 734878. [Google Scholar] [CrossRef]
- Rha, A.K.; Maguire, A.S.; Martin, D.R. GM1 Gangliosidosis: Mechanisms and Management. Appl. Clin. Genet. 2021, 14, 209–233. [Google Scholar] [CrossRef]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef]
- Bonten, E.J.; Campos, Y.; Zaitsev, V.; Nourse, A.; Waddell, B.; Lewis, W.; Taylor, G.; d’Azzo, A. Heterodimerization of the sialidase NEU1 with the chaperone protective protein/cathepsin A prevents its premature oligomerization. J. Biol. Chem. 2009, 284, 28430–28441. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Morreau, H.; Rottier, R.; Davis, D.; Bonten, E.; Gillemans, N.; Wenger, D.; Grosveld, F.G.; Doherty, P.; Suzuki, K.; et al. Mouse model for the lysosomal disorder galactosialidosis and correction of the phenotype with overexpressing erythroid precursor cells. Genes Dev. 1995, 9, 2623–2634. [Google Scholar] [CrossRef] [PubMed]
- Lambourne, M.D.; Potter, M.A. Murine β-galactosidase stability is not dependent on temperature or protective protein/cathepsin A. Mol. Genet. Metab. 2011, 104, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Eikelberg, D.; Lehmbecker, A.; Brogden, G.; Tongtako, W.; Hahn, K.; Habierski, A.; Hennermann, J.B.; Naim, H.Y.; Felmy, F.; Baumgärtner, W.; et al. Axonopathy and Reduction of Membrane Resistance: Key Features in a New Murine Model of Human G(M1)-Gangliosidosis. J. Clin. Med. 2020, 9, 1004. [Google Scholar] [CrossRef]
- Hahn, C.N.; del Pilar Martin, M.; Schröder, M.; Vanier, M.T.; Hara, Y.; Suzuki, K.; Suzuki, K.; d’Azzo, A. Generalized CNS disease and massive GM1-ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum. Mol. Genet. 1997, 6, 205–211. [Google Scholar] [CrossRef]
- Liu, S.; Feng, Y.; Huang, Y.; Jiang, X.; Tang, C.; Tang, F.; Zeng, C.; Liu, L. A GM1 gangliosidosis mutant mouse model exhibits activated microglia and disturbed autophagy. Exp. Biol. Med. 2021, 246, 1330–1341. [Google Scholar] [CrossRef]
- Matsuda, J.; Suzuki, O.; Oshima, A.; Ogura, A.; Noguchi, Y.; Yamamoto, Y.; Asano, T.; Takimoto, K.; Sukegawa, K.; Suzuki, Y.; et al. Beta-galactosidase-deficient mouse as an animal model for GM1-gangliosidosis. Glycoconj. J. 1997, 14, 729–736. [Google Scholar] [CrossRef]
- Przybilla, M.J.; Ou, L.; Tăbăran, A.F.; Jiang, X.; Sidhu, R.; Kell, P.J.; Ory, D.S.; O’Sullivan, M.G.; Whitley, C.B. Comprehensive behavioral and biochemical outcomes of novel murine models of GM1-gangliosidosis and Morquio syndrome type B. Mol. Genet. Metab. 2019, 126, 139–150. [Google Scholar] [CrossRef]
- Jeyakumar, M.; Thomas, R.; Elliot-Smith, E.; Smith, D.A.; van der Spoel, A.C.; d’Azzo, A.; Perry, V.H.; Butters, T.D.; Dwek, R.A.; Platt, F.M. Central nervous system inflammation is a hallmark of pathogenesis in mouse models of GM1 and GM2 gangliosidosis. Brain 2003, 126, 974–987. [Google Scholar] [CrossRef]
- Sano, R.; Annunziata, I.; Patterson, A.; Moshiach, S.; Gomero, E.; Opferman, J.; Forte, M.; d’Azzo, A. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca(2+)-dependent mitochondrial apoptosis. Mol. Cell 2009, 36, 500–511. [Google Scholar] [CrossRef]
- Sano, R.; Tessitore, A.; Ingrassia, A.; d’Azzo, A. Chemokine-induced recruitment of genetically modified bone marrow cells into the CNS of GM1-gangliosidosis mice corrects neuronal pathology. Blood 2005, 106, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Tessitore, A.; del, P.M.M.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; d’Azzo, A. GM1-ganglioside-mediated activation of the unfolded protein response causes neuronal death in a neurodegenerative gangliosidosis. Mol. Cell 2004, 15, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Baek, R.C.; Broekman, M.L.; Leroy, S.G.; Tierney, L.A.; Sandberg, M.A.; d’Azzo, A.; Seyfried, T.N.; Sena-Esteves, M. AAV-mediated gene delivery in adult GM1-gangliosidosis mice corrects lysosomal storage in CNS and improves survival. PLoS ONE 2010, 5, e13468. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ma, W.; Feng, Y.; Zhang, Y.; Jia, X.; Tang, C.; Tang, F.; Wu, X.; Huang, Y. AAV9-coGLB1 Improves Lysosomal Storage and Rescues Central Nervous System Inflammation in a Mutant Mouse Model of GM1 Gangliosidosis. Curr. Gene Ther. 2022, 22, 352–365. [Google Scholar] [CrossRef] [PubMed]
- Tsunogai, T.; Ohashi, T.; Shimada, Y.; Higuchi, T.; Kimura, A.; Watabe, A.M.; Kato, F.; Ida, H.; Kobayashi, H. Hematopoietic stem cell gene therapy ameliorates CNS involvement in murine model of GM1-gangliosidosis. Mol. Ther. Methods Clin. Dev. 2022, 25, 448–460. [Google Scholar] [CrossRef]
- Medrano, G.; Reidy, M.J.; Liu, J.; Ayala, J.; Dolan, M.C.; Cramer, C.L. Rapid system for evaluating bioproduction capacity of complex pharmaceutical proteins in plants. Methods Mol. Biol. 2009, 483, 51–67. [Google Scholar] [CrossRef]
- Crawley, A.; Ramsay, S.L.; Byers, S.; Hopwood, J.; Meikle, P.J. Monitoring dose response of enzyme replacement therapy in feline mucopolysaccharidosis type VI by tandem mass spectrometry. Pediatr. Res. 2004, 55, 585–591. [Google Scholar] [CrossRef]
- Morreau, H.; Galjart, N.J.; Gillemans, N.; Willemsen, R.; van der Horst, G.T.; d’Azzo, A. Alternative splicing of beta-galactosidase mRNA generates the classic lysosomal enzyme and a beta-galactosidase-related protein. J. Biol. Chem. 1989, 264, 20655–20663. [Google Scholar] [CrossRef]
- Morrone, A.; Morreau, H.; Zhou, X.Y.; Zammarchi, E.; Kleijer, W.J.; Galjaard, H.; d’Azzo, A. Insertion of a T next to the donor splice site of intron 1 causes aberrantly spliced mRNA in a case of infantile GM1-gangliosidosis. Hum. Mutat. 1994, 3, 112–120. [Google Scholar] [CrossRef]
- Radin, D.N.; Acosta, W. Materials and Methods for Mitigating Immune-Sensitization. U.S. Patent US20190046637A1, 14 February 2019. [Google Scholar]
- Porter, A.G.; Jänicke, R.U. Emerging roles of caspase-3 in apoptosis. Cell Death Differ. 1999, 6, 99–104. [Google Scholar] [CrossRef]
- Elliot-Smith, E.; Speak, A.O.; Lloyd-Evans, E.; Smith, D.A.; van der Spoel, A.C.; Jeyakumar, M.; Butters, T.D.; Dwek, R.A.; d’Azzo, A.; Platt, F.M. Beneficial effects of substrate reduction therapy in a mouse model of GM1 gangliosidosis. Mol. Genet. Metab. 2008, 94, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Tifft, C.J. A Phase 1/2 Study of Intravenous Gene Transfer With an AAV9 Vector Expressing Human Beta-Galactosidase in Type I and Type II GM1 Gangliosidosis. Available online: https://clinicaltrials.gov/ct2/show/NCT03952637 (accessed on 1 July 2022).
- Zimran, A.; Elstein, D. Management of Gaucher disease: Enzyme replacement therapy. Pediatr. Endocrinol. Rev. 2014, 12 (Suppl. S1), 82–87. [Google Scholar] [PubMed]
- Case, L.E.; Beckemeyer, A.A.; Kishnani, P.S. Infantile Pompe disease on ERT: Update on clinical presentation, musculoskeletal management, and exercise considerations. Am. J. Med. Genet. C Semin. Med. Genet. 2012, 160c, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Ceccarini, M.R.; Codini, M.; Conte, C.; Patria, F.; Cataldi, S.; Bertelli, M.; Albi, E.; Beccari, T. Alpha-Mannosidosis: Therapeutic Strategies. Int. J. Mol. Sci. 2018, 19, 1500. [Google Scholar] [CrossRef]
- Dornelles, A.D.; de Camargo Pinto, L.L.; de Paula, A.C.; Steiner, C.E.; Lourenço, C.M.; Kim, C.A.; Horovitz, D.D.; Ribeiro, E.M.; Valadares, E.R.; Goulart, I.; et al. Enzyme replacement therapy for Mucopolysaccharidosis Type I among patients followed within the MPS Brazil Network. Genet. Mol. Biol. 2014, 37, 23–29. [Google Scholar] [CrossRef]
- Fox, J.E.; Volpe, L.; Bullaro, J.; Kakkis, E.D.; Sly, W.S. First human treatment with investigational rhGUS enzyme replacement therapy in an advanced stage MPS VII patient. Mol. Genet. Metab. 2015, 114, 203–208. [Google Scholar] [CrossRef]
- Geraets, R.D.; Koh, S.y.; Hastings, M.L.; Kielian, T.; Pearce, D.A.; Weimer, J.M. Moving towards effective therapeutic strategies for Neuronal Ceroid Lipofuscinosis. Orphanet J. Rare Dis. 2016, 11, 40. [Google Scholar] [CrossRef]
- Lampe, C.; Bosserhoff, A.K.; Burton, B.K.; Giugliani, R.; de Souza, C.F.; Bittar, C.; Muschol, N.; Olson, R.; Mendelsohn, N.J. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: An international case series. J. Inherit. Metab. Dis. 2014, 37, 823–829. [Google Scholar] [CrossRef]
- Lin, H.Y.; Chuang, C.K.; Chen, M.R.; Lin, S.M.; Hung, C.L.; Chang, C.Y.; Chiu, P.C.; Tsai, W.H.; Niu, D.M.; Tsai, F.J.; et al. Cardiac structure and function and effects of enzyme replacement therapy in patients with mucopolysaccharidoses I, II, IVA and VI. Mol. Genet. Metab. 2016, 117, 431–437. [Google Scholar] [CrossRef]
- Pastores, G.M.; Hughes, D.A. Lysosomal Acid Lipase Deficiency: Therapeutic Options. Drug Des. Dev. Ther. 2020, 14, 591–601. [Google Scholar] [CrossRef]
- Spada, M.; Baron, R.; Elliott, P.M.; Falissard, B.; Hilz, M.J.; Monserrat, L.; Tøndel, C.; Tylki-Szymańska, A.; Wanner, C.; Germain, D.P. The effect of enzyme replacement therapy on clinical outcomes in paediatric patients with Fabry disease—A systematic literature review by a European panel of experts. Mol. Genet. Metab. 2019, 126, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Sawamoto, K.; Shimada, T.; Bober, M.B.; Kubaski, F.; Yasuda, E.; Mason, R.W.; Khan, S.; Alméciga-Díaz, C.J.; Barrera, L.A.; et al. Enzyme replacement therapy for treating mucopolysaccharidosis type IVA (Morquio A syndrome): Effect and limitations. Expert Opin Orphan Drugs 2015, 3, 1279–1290. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weesner, J.A.; Annunziata, I.; Yang, T.; Acosta, W.; Gomero, E.; Hu, H.; van de Vlekkert, D.; Ayala, J.; Qiu, X.; Fremuth, L.E.; et al. Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis. Cells 2022, 11, 2579. https://doi.org/10.3390/cells11162579
Weesner JA, Annunziata I, Yang T, Acosta W, Gomero E, Hu H, van de Vlekkert D, Ayala J, Qiu X, Fremuth LE, et al. Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis. Cells. 2022; 11(16):2579. https://doi.org/10.3390/cells11162579
Chicago/Turabian StyleWeesner, Jason Andrew, Ida Annunziata, Tianhong Yang, Walter Acosta, Elida Gomero, Huimin Hu, Diantha van de Vlekkert, Jorge Ayala, Xiaohui Qiu, Leigh Ellen Fremuth, and et al. 2022. "Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis" Cells 11, no. 16: 2579. https://doi.org/10.3390/cells11162579
APA StyleWeesner, J. A., Annunziata, I., Yang, T., Acosta, W., Gomero, E., Hu, H., van de Vlekkert, D., Ayala, J., Qiu, X., Fremuth, L. E., Radin, D. N., Cramer, C. L., & d’Azzo, A. (2022). Preclinical Enzyme Replacement Therapy with a Recombinant β-Galactosidase-Lectin Fusion for CNS Delivery and Treatment of GM1-Gangliosidosis. Cells, 11(16), 2579. https://doi.org/10.3390/cells11162579