
Advances in ER-Phagy and Its Diseases Relevance


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Structure and Function of the ER
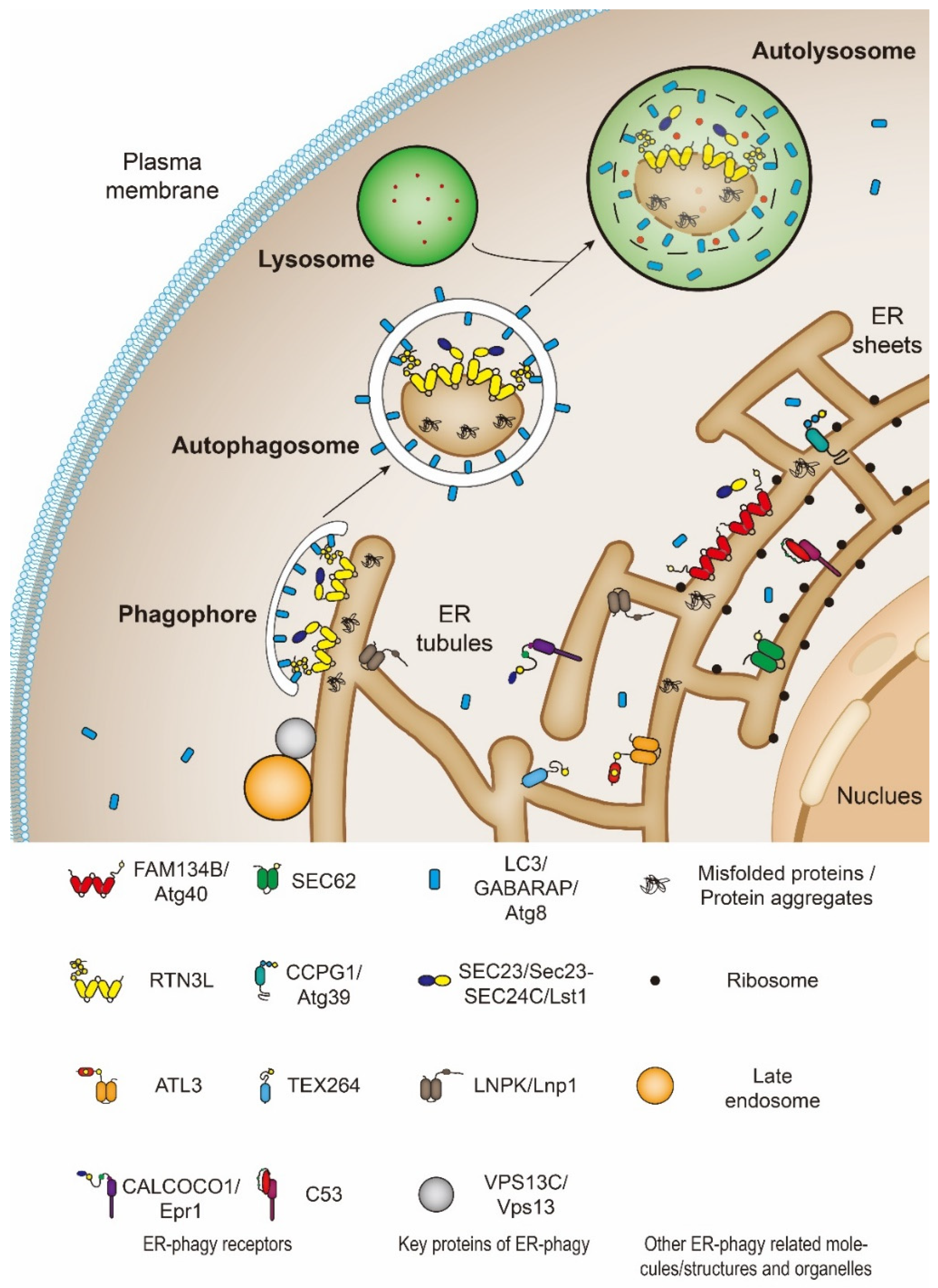
3. ER-Phagy
3.1. Definition, Classification, and Function of ER-Phagy
3.2. The Catabolic Process and Regulation Mechanism of ER-Phagy
3.3. The Catabolic Process of Micro-ER-Phagy and ERLAD
4. Key Molecules of ER-Phagy (Receptors, Cofactors, etc.)
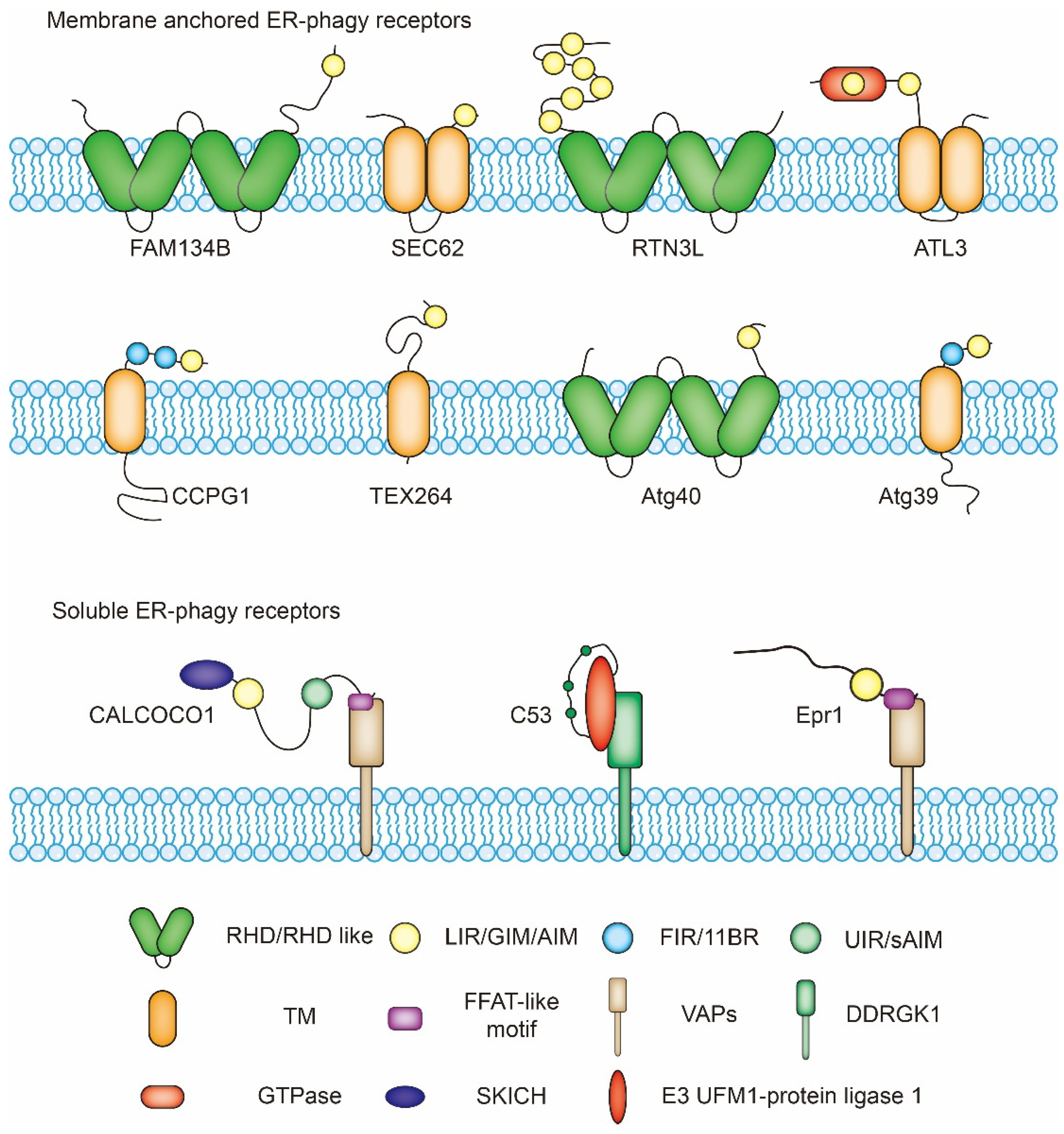
4.1. Receptors, the Key ER-Phagy Players
4.1.1. FAM134B (Family with Sequence Similarity 134 Member B)
4.1.2. RTN3L (Reticulon-3L)
4.1.3. ATL3 (Atlastin-3)
4.1.4. SEC62 (Translocation Protein SEC62)
4.1.5. CCPG1 (Cell Cycle Progression Protein 1)
4.1.6. TEX264 (Testis-Expressed Protein 264)
4.1.7. CALCOCO1 (Calcium-Binding and Coiled-Coil Domain-Containing Protein 1)
4.1.8. Atg40 (Autophagy-Related Protein 40)
4.1.9. Atg39 (Autophagy-Related Protein 39)
4.1.10. Epr1(ER-Phagy Receptor 1)
4.1.11. C53 (CDK5 Regulatory Subunit-Associated Protein 3)
4.2. Other Important ER-Phagy Players
4.2.1. SQSTM1 (Sequestosome-1)
4.2.2. SEC24C (Protein Transport Protein SEC24C)/Lst1 (Lethal with Sec13 Protein 1)
4.2.3. Lnp1 (Lunapark Family Member1)
4.2.4. Vps13 (Vacuolar Protein Sorting-Associated Protein 13)
4.3. Transcription Factors in ER-Phagy
4.3.1. TFEB and TFE3 (Transcription Factor EB/E3)
4.3.2. C/EBPβ (CCAAT/Enhancer-Binding Protein β)
4.3.3. Rpd3L (Histone Deacetylase Large Complex)
4.3.4. Mig1 and Mig2 (Regulatory Protein Mig 1 and 2)
5. The Diseases Relevance of ER-Phagy
5.1. Neurodegenerative Diseases
5.1.1. FAM134B and Hereditary Sensory and Autonomic Neuropathy (HSAN)
5.1.2. FAM134B and Niemann-Pick Type C Disease
5.1.3. ATL3/ATL1 and Neurodegenerative Diseases
5.1.4. RTN3 and Alzheimer Disease (AD)
5.1.5. VPS13C and Parkinson Disease (PD)
5.2. Cancers
5.2.1. FAM134B
5.2.2. SEC62
5.2.3. CALCOCO1
5.2.4. C53
5.3. Metabolic Diseases
5.3.1. Diseases Caused by Misfolded Procollagen
5.3.2. Liver and Lung Diseases Associated with α1-Antitrypsin Deficiency (ATD)
5.3.3. Diseases Caused by Misfolded Hormone Precursor Proteins
5.4. Viral Infection and Innate Immunity
5.4.1. Antiviral Role of FAM134B-Dependent ER-Phagy
5.4.2. Divergent Roles of Atlastins in Flavivirus Infection
5.4.3. RTN3/RTN4 and Viral Infection
5.4.4. ER-Phagy and Innate Immunity
5.5. Other Diseases
5.5.1. CCPG1-Mediated ER-Phagy and Pancreatic Diseases
5.5.2. TEX264 and Diseases
5.5.3. C53 and Development
5.5.4. LNPK and a New Recessive Neurodevelopmental Syndrome
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2020, 432, 185–205. [Google Scholar] [CrossRef]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 2015, 10, 173–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, J.H.; Walter, P.; Yen, T.S. Endoplasmic reticulum stress in disease pathogenesis. Annu. Rev. Pathol. 2008, 3, 399–425. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Fregno, I.; Guerra, C.; Molinari, M. Eat it right: ER-phagy and recovER-phagy. Biochem. Soc. Trans. 2018, 46, 699–706. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, S. ER-phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 2019, 286, 2645–2663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S.; Gallagher, C.M.; Walter, P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J. Cell Sci. 2014, 127, 4078–4088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolender, R.P.; Weibel, E.R. A morphometric study of the removal of phenobarbital-induced membranes from hepatocytes after cessation of threatment. J. Cell Biol. 1973, 56, 746–761. [Google Scholar] [CrossRef]
- Hamasaki, M.; Noda, T.; Baba, M.; Ohsumi, Y. Starvation triggers the delivery of the endoplasmic reticulum to the vacuole via autophagy in yeast. Traffic 2005, 6, 56–65. [Google Scholar] [CrossRef]
- Xu, F.; Du, W.; Zou, Q.; Wang, Y.; Zhang, X.; Xing, X.; Li, Y.; Zhang, D.; Wang, H.; Zhang, W.; et al. COPII mitigates ER stress by promoting formation of ER whorls. Cell Res. 2021, 31, 141–156. [Google Scholar] [CrossRef] [PubMed]
- Bernales, S.; McDonald, K.L.; Walter, P. Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol. 2006, 4, e423. [Google Scholar] [CrossRef] [Green Version]
- Khaminets, A.; Heinrich, T.; Mari, M.; Grumati, P.; Huebner, A.K.; Akutsu, M.; Liebmann, L.; Stolz, A.; Nietzsche, S.; Koch, N.; et al. Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 2015, 522, 354–358. [Google Scholar] [CrossRef]
- Mochida, K.; Oikawa, Y.; Kimura, Y.; Kirisako, H.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015, 522, 359–362. [Google Scholar] [CrossRef]
- Fregno, I.; Fasana, E.; Bergmann, T.J.; Raimondi, A.; Loi, M.; Solda, T.; Galli, C.; D’Antuono, R.; Morone, D.; Danieli, A.; et al. ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J. 2018, 37, e99259. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev. Cell 2018, 44, 217–232.e211. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Ordureau, A.; Paulo, J.A.; Shoemaker, C.J.; Denic, V.; Harper, J.W. TEX264 Is an Endoplasmic Reticulum-Resident ATG8-Interacting Protein Critical for ER Remodeling during Nutrient Stress. Mol. Cell 2019, 74, 891–908.e810. [Google Scholar] [CrossRef]
- Wild, P.; Farhan, H.; McEwan, D.G.; Wagner, S.; Rogov, V.V.; Brady, N.R.; Richter, B.; Korac, J.; Waidmann, O.; Choudhary, C.; et al. Phosphorylation of the Autophagy Receptor Optineurin Restricts Salmonella Growth. Science 2011, 333, 228–233. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ni, H.M.; Guo, F.; Ding, Y.; Shi, Y.H.; Lahiri, P.; Frohlich, L.F.; Rulicke, T.; Smole, C.; Schmidt, V.C.; et al. Sequestosome 1/p62 Protein Is Associated with Autophagic Removal of Excess Hepatic Endoplasmic Reticulum in Mice. J. Biol. Chem. 2016, 291, 18663–18674. [Google Scholar] [CrossRef] [Green Version]
- Ji, C.H.; Kim, H.Y.; Heo, A.J.; Lee, S.H.; Lee, M.J.; Kim, S.B.; Srinivasrao, G.; Mun, S.R.; Cha-Molstad, H.; Ciechanover, A.; et al. The N-Degron Pathway Mediates ER-phagy. Mol. Cell 2019, 75, 1058–1072.e1059. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, J.; Fu, T.; Wu, P.; Peng, C.; Gong, X.; Wang, Y.; Zhang, M.; Li, Y.; Wang, Y.; et al. Phosphorylation regulates the binding of autophagy receptors to FIP200 Claw domain for selective autophagy initiation. Nat. Commun. 2021, 12, 1570. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Zorca, C.E.; Ragheb, M.A.; Rashidi, F.B.; El-Din, D.S.S. Fine-tuning ER-phagy by post-translational modifications. Bioessays 2021, 43, e2000212. [Google Scholar] [CrossRef]
- Omari, S.; Makareeva, E.; Roberts-Pilgrim, A.; Mirigian, L.; Jarnik, M.; Ott, C.; Lippincott-Schwartz, J.; Leikin, S. Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc. Natl. Acad. Sci. USA 2018, 115, E10099–E10108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fregno, I.; Fasana, E.; Solda, T.; Galli, C.; Molinari, M. N-glycan processing selects ERAD-resistant misfolded proteins for ER-to-lysosome-associated degradation. EMBO J. 2021, 40, e107240. [Google Scholar] [CrossRef]
- Grumati, P.; Morozzi, G.; Holper, S.; Mari, M.; Harwardt, M.I.; Yan, R.; Muller, S.; Reggiori, F.; Heilemann, M.; Dikic, I. Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 2017, 6, e25555. [Google Scholar] [CrossRef]
- Chino, H.; Hatta, T.; Natsume, T.; Mizushima, N. Intrinsically Disordered Protein TEX264 Mediates ER-phagy. Mol. Cell 2019, 74, 909–921.e906. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, Y.; Chai, P.; Zheng, P.; Teng, J.; Chen, J. ATL3 Is a Tubular ER-Phagy Receptor for GABARAP-Mediated Selective Autophagy. Curr. Biol. 2019, 29, 846–855.e846. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, F.; Noack, J.; Bergmann, T.J.; Cebollero, E.; Pisoni, G.B.; Fasana, E.; Fregno, I.; Galli, C.; Loi, M.; Solda, T.; et al. Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat. Cell Biol. 2016, 18, 1173–1184. [Google Scholar] [CrossRef] [Green Version]
- Nthiga, T.M.; Shrestha, B.K.; Sjottem, E.; Bruun, J.A.; Larsen, K.B.; Bhujabal, Z.; Lamark, T.; Johansen, T. CALCOCO1 acts with VAMP-associated proteins to mediate ER-phagy. EMBO J. 2020, 39, e103649. [Google Scholar] [CrossRef]
- Stefely, J.A.; Zhang, Y.; Freiberger, E.C.; Kwiecien, N.W.; Thomas, H.E.; Davis, A.M.; Lowry, N.D.; Vincent, C.E.; Shishkova, E.; Clark, N.A.; et al. Mass spectrometry proteomics reveals a function for mammalian CALCOCO1 in MTOR-regulated selective autophagy. Autophagy 2020, 16, 2219–2237. [Google Scholar] [CrossRef]
- Rubinsztein, D.C. Receptors for selective recycling. Nature 2015, 522, 291–292. [Google Scholar] [CrossRef] [PubMed]
- Nakatogawa, H.; Mochida, K. Reticulophagy and nucleophagy: New findings and unsolved issues. Autophagy 2015, 11, 2377–2378. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Du, L.L. Epr1, a UPR-upregulated soluble autophagy receptor for reticulophagy. Autophagy 2020, 16, 2112–2113. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Zou, C.X.; Liu, X.M.; Jiang, Z.D.; Yu, Z.Q.; Suo, F.; Du, T.Y.; Dong, M.Q.; He, W.; Du, L.L. A UPR-Induced Soluble ER-Phagy Receptor Acts with VAPs to Confer ER Stress Resistance. Mol. Cell 2020, 79, 963–977.e963. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Bhaskara, R.M.; Grumati, P.; Garcia-Pardo, J.; Kalayil, S.; Covarrubias-Pinto, A.; Chen, W.; Kudryashev, M.; Dikic, I.; Hummer, G. Curvature induction and membrane remodeling by FAM134B reticulon homology domain assist selective ER-phagy. Nat. Commun. 2019, 10, 2370. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, X.; Ding, X.; Du, M.; Li, B.; Weng, X.; Zhang, J.; Li, L.; Tian, R.; Zhu, Q.; et al. FAM134B oligomerization drives endoplasmic reticulum membrane scission for ER-phagy. EMBO J. 2020, 39, e102608. [Google Scholar] [CrossRef]
- Liang, J.R.; Lingeman, E.; Ahmed, S.; Corn, J.E. Atlastins remodel the endoplasmic reticulum for selective autophagy. J. Cell Biol. 2018, 217, 3354–3367. [Google Scholar] [CrossRef] [Green Version]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef]
- Kumar, D.; Lak, B.; Suntio, T.; Vihinen, H.; Belevich, I.; Viita, T.; Xiaonan, L.; Vartiainen, A.; Vartiainen, M.; Varjosalo, M.; et al. RTN4B interacting protein FAM134C promotes ER membrane curvature and has a functional role in autophagy. Mol. Biol. Cell 2021, 32, 1158–1170. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, X.; Marshall, R.S.; Paez-Valencia, J.; Lacey, P.; Vierstra, R.D.; Otegui, M.S. Reticulon proteins modulate autophagy of the endoplasmic reticulum in maize endosperm. Elife 2020, 9, e51918. [Google Scholar] [CrossRef]
- Li, J.; Abosmaha, E.; Coffin, C.S.; Labonte, P.; Bukong, T.N. Reticulon-3 modulates the incorporation of replication competent hepatitis C virus molecules for release inside infectious exosomes. PLoS ONE 2020, 15, e0239153. [Google Scholar] [CrossRef]
- Wu, H.; Voeltz, G.K. Reticulon-3 Promotes Endosome Maturation at ER Membrane Contact Sites. Dev. Cell 2021, 56, 52–66.e57. [Google Scholar] [CrossRef]
- Wang, S.; Tukachinsky, H.; Romano, F.B.; Rapoport, T.A. Cooperation of the ER-shaping proteins atlastin, lunapark, and reticulons to generate a tubular membrane network. Elife 2016, 5, e18605. [Google Scholar] [CrossRef]
- Liu, N.; Zhao, H.; Zhao, Y.G.; Hu, J.; Zhang, H. Atlastin 2/3 regulate ER targeting of the ULK1 complex to initiate autophagy. J. Cell Biol. 2021, 220, e202012091. [Google Scholar] [CrossRef]
- Krols, M.; Detry, S.; Asselbergh, B.; Almeida-Souza, L.; Kremer, A.; Lippens, S.; De Rycke, R.; De Winter, V.; Muller, F.J.; Kurth, I.; et al. Sensory-Neuropathy-Causing Mutations in ATL3 Cause Aberrant ER Membrane Tethering. Cell Rep. 2018, 23, 2026–2038. [Google Scholar] [CrossRef]
- Muriel, M.P.; Dauphin, A.; Namekawa, M.; Gervais, A.; Brice, A.; Ruberg, M. Atlastin-1, the dynamin-like GTPase responsible for spastic paraplegia SPG3A, remodels lipid membranes and may form tubules and vesicles in the endoplasmic reticulum. J. Neurochem. 2009, 110, 1607–1616. [Google Scholar] [CrossRef]
- Zhu, P.P.; Patterson, A.; Lavoie, B.; Stadler, J.; Shoeb, M.; Patel, R.; Blackstone, C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 2003, 278, 49063–49071. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shibata, Y.; Zhu, P.P.; Voss, C.; Rismanchi, N.; Prinz, W.A.; Rapoport, T.A.; Blackstone, C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell 2009, 138, 549–561. [Google Scholar] [CrossRef] [Green Version]
- Hu, X.; Wu, F.; Sun, S.; Yu, W.; Hu, J. Human atlastin GTPases mediate differentiated fusion of endoplasmic reticulum membranes. Protein Cell 2015, 6, 307–311. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.P.; Cooley, R.B.; Kelly, C.M.; Miller, K.; Andersen, O.S.; Rusinova, R.; Sondermann, H. Timing and Reset Mechanism of GTP Hydrolysis-Driven Conformational Changes of Atlastin. Structure 2017, 25, 997–1010.e1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, S.; Benedix, J.; Fedeles, S.V.; Schorr, S.; Schirra, C.; Schauble, N.; Jalal, C.; Greiner, M.; Hassdenteufel, S.; Tatzelt, J.; et al. Different effects of Sec61alpha, Sec62 and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 2012, 125, 1958–1969. [Google Scholar] [CrossRef] [Green Version]
- Linxweiler, M.; Schick, B.; Zimmermann, R. Let’s talk about Secs: Sec61, Sec62 and Sec63 in signal transduction, oncology and personalized medicine. Signal Transduct. Target. Ther. 2017, 2, 17002. [Google Scholar] [CrossRef]
- Hassdenteufel, S.; Johnson, N.; Paton, A.W.; Paton, J.C.; High, S.; Zimmermann, R. Chaperone-Mediated Sec61 Channel Gating during ER Import of Small Precursor Proteins Overcomes Sec61 Inhibitor-Reinforced Energy Barrier. Cell Rep. 2018, 23, 1373–1386. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Ye, H.; Cui, Y.; Jiang, L. AtSec62 is critical for plant development and is involved in ER-phagy in Arabidopsis thaliana. J. Integr. Plant Biol. 2020, 62, 181–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, L.; de Escauriaza, M.D.; Lajoie, P.; Theis, M.; Jung, M.; Muller, A.; Burgard, C.; Greiner, M.; Snapp, E.L.; Dudek, J.; et al. Evolutionary gain of function for the ER membrane protein Sec62 from yeast to humans. Mol. Biol. Cell 2010, 21, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.C.; Liegeois, N.; Horecka, J.; DePinho, R.A.; Sprague, G.F., Jr.; Tyers, M.; Elledge, S.J. Human CPR (cell cycle progression restoration) genes impart a Far- phenotype on yeast cells. Genetics 1997, 147, 1063–1076. [Google Scholar] [CrossRef]
- Fielden, J.; Wiseman, K.; Torrecilla, I.; Li, S.; Hume, S.; Chiang, S.C.; Ruggiano, A.; Singh, A.N.; Freire, R.; Hassanieh, S.; et al. TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat. Commun. 2020, 11, 1274. [Google Scholar] [CrossRef] [Green Version]
- Nthiga, T.M.; Shrestha, B.K.; Bruun, J.-A.; Larsen, K.B.; Lamark, T.; Johansen, T. Regulation of Golgi turnover by CALCOCO1-mediated selective autophagy. J. Cell Biol. 2021, 220, e202006128. [Google Scholar] [CrossRef]
- Mochida, K.; Yamasaki, A.; Matoba, K.; Kirisako, H.; Noda, N.N.; Nakatogawa, H. Super-assembly of ER-phagy receptor Atg40 induces local ER remodeling at contacts with forming autophagosomal membranes. Nat. Commun. 2020, 11, 3306. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Parashar, S.; Zahoor, M.; Needham, P.G.; Mari, M.; Zhu, M.; Chen, S.; Ho, H.C.; Reggiori, F.; Farhan, H.; et al. A COPII subunit acts with an autophagy receptor to target endoplasmic reticulum for degradation. Science 2019, 365, 53–60. [Google Scholar] [CrossRef]
- Cui, Y.; Parashar, S.; Ferro-Novick, S. A new role for a COPII cargo adaptor in autophagy. Autophagy 2020, 16, 376–378. [Google Scholar] [CrossRef]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Yorimitsu, T.; Klionsky, D.J. Atg11 links cargo to the vesicle-forming machinery in the cytoplasm to vacuole targeting pathway. Mol. Biol. Cell 2005, 16, 1593–1605. [Google Scholar] [CrossRef] [Green Version]
- Otto, F.B.; Thumm, M. Mechanistic dissection of macro- and micronucleophagy. Autophagy 2021, 17, 626–639. [Google Scholar] [CrossRef]
- Tomioka, Y.; Kotani, T.; Kirisako, H.; Oikawa, Y.; Kimura, Y.; Hirano, H.; Ohsumi, Y.; Nakatogawa, H. TORC1 inactivation stimulates autophagy of nucleoporin and nuclear pore complexes. J. Cell Biol. 2020, 219, e201910063. [Google Scholar] [CrossRef]
- Wang, J.; He, X.; Luo, Y.; Yarbrough, W.G. A novel ARF-binding protein (LZAP) alters ARF regulation of HDM2. Biochem. J. 2006, 393, 489–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephani, M.; Picchianti, L.; Gajic, A.; Beveridge, R.; Skarwan, E.; Sanchez de Medina Hernandez, V.; Mohseni, A.; Clavel, M.; Zeng, Y.; Naumann, C.; et al. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. Elife 2020, 9, e58396. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Novick, P.; Ferro-Novick, S. ER network formation requires a balance of the dynamin-like GTPase Sey1p and the Lunapark family member Lnp1p. Nat. Cell Biol. 2012, 14, 707–716. [Google Scholar] [CrossRef]
- Chen, S.; Cui, Y.; Parashar, S.; Novick, P.J.; Ferro-Novick, S. ER-phagy requires Lnp1, a protein that stabilizes rearrangements of the ER network. Proc. Natl. Acad. Sci. USA 2018, 115, E6237–E6244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Desai, T.; McNew, J.A.; Gerard, P.; Novick, P.J.; Ferro-Novick, S. Lunapark stabilizes nascent three-way junctions in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2015, 112, 418–423. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Mari, M.; Parashar, S.; Liu, D.; Cui, Y.; Reggiori, F.; Novick, P.J.; Ferro-Novick, S. Vps13 is required for the packaging of the ER into autophagosomes during ER-phagy. Proc. Natl. Acad. Sci. USA 2020, 117, 18530–18539. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinque, L.; De Leonibus, C.; Iavazzo, M.; Krahmer, N.; Intartaglia, D.; Salierno, F.G.; De Cegli, R.; Di Malta, C.; Svelto, M.; Lanzara, C.; et al. MiT/TFE factors control ER-phagy via transcriptional regulation of FAM134B. EMBO J. 2020, 39, e105696. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Panda, S.; Lin, J.D. Temporal orchestration of circadian autophagy rhythm by C/EBPbeta. EMBO J. 2011, 30, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, S.; Akira, S.; Kishimoto, T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc. Natl. Acad. Sci. USA 1992, 89, 1473–1476. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Shiozaki, Y.; Keenan, A.L.; Miyazaki-Anzai, S.; Miyazaki, M. An N-terminal-truncated isoform of FAM134B (FAM134B-2) regulates starvation-induced hepatic selective ER-phagy. Life Sci. Alliance 2019, 2, e201900340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartholomew, C.R.; Suzuki, T.; Du, Z.; Backues, S.K.; Jin, M.; Lynch-Day, M.A.; Umekawa, M.; Kamath, A.; Zhao, M.; Xie, Z.; et al. Ume6 transcription factor is part of a signaling cascade that regulates autophagy. Proc. Natl. Acad. Sci. USA 2012, 109, 11206–11210. [Google Scholar] [CrossRef] [Green Version]
- Jin, M.; He, D.; Backues, S.K.; Freeberg, M.A.; Liu, X.; Kim, J.K.; Klionsky, D.J. Transcriptional regulation by Pho23 modulates the frequency of autophagosome formation. Curr. Biol. 2014, 24, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaniel, S.L.; Strahl, B.D. Stress-free with Rpd3: A unique chromatin complex mediates the response to oxidative stress. Mol. Cell Biol. 2013, 33, 3726–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, T.; Muroi, K.; Irie, K. Snf1 AMPK positively regulates ER-phagy via expression control of Atg39 autophagy receptor in yeast ER stress response. PLoS Genet. 2020, 16, e1009053. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Klenerman, D.; Rubinsztein, D.C. Developing Therapies for Neurodegenerative Disorders: Insights from Protein Aggregation and Cellular Stress Responses. Annu. Rev. Cell Dev. Biol. 2020, 36, 165–189. [Google Scholar] [CrossRef] [PubMed]
- Davidson, G.; Murphy, S.; Polke, J.; Laura, M.; Salih, M.; Muntoni, F.; Blake, J.; Brandner, S.; Davies, N.; Horvath, R.; et al. Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J. Neurol. 2012, 259, 1673–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakil, S.M.; Monies, D.; Hagos, S.; Al-Ajlan, F.; Finsterer, J.; Qahtani, A.A.; Ramzan, K.; Humaidy, R.A.; Al-Muhaizea, M.A.; Meyer, B.; et al. Exome Sequencing: Mutilating Sensory Neuropathy with Spastic Paraplegia due to a Mutation in FAM134B Gene. Case Rep. Genet. 2018, 2018, 9468049. [Google Scholar] [CrossRef]
- Kurth, I.; Pamminger, T.; Hennings, J.C.; Soehendra, D.; Huebner, A.K.; Rotthier, A.; Baets, J.; Senderek, J.; Topaloglu, H.; Farrell, S.A.; et al. Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat. Genet. 2009, 41, 1179–1181. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Chen, J.; Zhang, B. Critical roles of FAM134B in ER-phagy and diseases. Cell Death Dis. 2020, 11, 983. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.L.; Krus, K.L.; Lieberman, A.P. Lysosome and endoplasmic reticulum quality control pathways in Niemann-Pick type C disease. Brain Res. 2016, 1649, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.L.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat. Commun. 2018, 9, 3671. [Google Scholar] [CrossRef]
- Kornak, U.; Mademan, I.; Schinke, M.; Voigt, M.; Krawitz, P.; Hecht, J.; Barvencik, F.; Schinke, T.; Giesselmann, S.; Beil, F.T.; et al. Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3. Brain 2014, 137, 683–692. [Google Scholar] [CrossRef] [Green Version]
- Fischer, D.; Schabhuttl, M.; Wieland, T.; Windhager, R.; Strom, T.M.; Auer-Grumbach, M. A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain 2014, 137, e286. [Google Scholar] [CrossRef] [Green Version]
- Behrendt, L.; Kurth, I.; Kaether, C. A disease causing ATLASTIN 3 mutation affects multiple endoplasmic reticulum-related pathways. Cell Mol. Life Sci. 2019, 76, 1433–1445. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Alvarado, D.; Rainier, S.; Lemons, R.; Hedera, P.; Weber, C.H.; Tukel, T.; Apak, M.; Heiman-Patterson, T.; Ming, L.; et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat. Genet. 2001, 29, 326–331. [Google Scholar] [CrossRef] [PubMed]
- McCorquodale, D.S., 3rd; Ozomaro, U.; Huang, J.; Montenegro, G.; Kushman, A.; Citrigno, L.; Price, J.; Speziani, F.; Pericak-Vance, M.A.; Zuchner, S. Mutation screening of spastin, atlastin, and REEP1 in hereditary spastic paraplegia. Clin. Genet. 2011, 79, 523–530. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; He, W.; Wang, K.; Han, H.; Xiao, T.; Chen, X.; Zhou, B.; Tan, J.; Xia, K.; Tang, B.; et al. Identification of rare RTN3 variants in Alzheimer’s disease in Han Chinese. Hum. Genet. 2018, 137, 141–150. [Google Scholar] [CrossRef]
- He, W.; Lu, Y.; Qahwash, I.; Hu, X.Y.; Chang, A.; Yan, R. Reticulon family members modulate BACE1 activity and amyloid-beta peptide generation. Nat. Med. 2004, 10, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Ge, Y.; Sharoar, M.G.; He, W.; Xiang, R.; Zhang, Z.; Hu, X.; Yan, R. Impact of RTN3 deficiency on expression of BACE1 and amyloid deposition. J. Neurosci. 2014, 34, 13954–13962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, S.; Drouet, V.; Majounie, E.; Deramecourt, V.; Jacoupy, M.; Nicolas, A.; Cormier-Dequaire, F.; Hassoun, S.M.; Pujol, C.; Ciura, S.; et al. Loss of VPS13C Function in Autosomal-Recessive Parkinsonism Causes Mitochondrial Dysfunction and Increases PINK1/Parkin-Dependent Mitophagy. Am. J. Hum. Genet. 2016, 98, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Viswanath, P.; Radoul, M.; Izquierdo-Garcia, J.L.; Ong, W.Q.; Luchman, H.A.; Cairncross, J.G.; Huang, B.; Pieper, R.O.; Phillips, J.J.; Ronen, S.M. 2-Hydroxyglutarate-Mediated Autophagy of the Endoplasmic Reticulum Leads to an Unusual Downregulation of Phospholipid Biosynthesis in Mutant IDH1 Gliomas. Cancer Res. 2018, 78, 2290–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Gao, W.; Zhou, L.; Chen, Y.; Qin, S.; Zhang, L.; Liu, J.; He, Y.; Lei, Y.; Chen, H.N.; et al. Repurposing Brigatinib for the Treatment of Colorectal Cancer Based on Inhibition of ER-phagy. Theranostics 2019, 9, 4878–4892. [Google Scholar] [CrossRef]
- Liao, Y.; Duan, B.; Zhang, Y.; Zhang, X.; Xia, B. Excessive ER-phagy mediated by the autophagy receptor FAM134B results in ER stress, the unfolded protein response, and cell death in HeLa cells. J. Biol. Chem. 2019, 294, 20009–20023. [Google Scholar] [CrossRef]
- Islam, F.; Gopalan, V.; Wahab, R.; Smith, R.A.; Qiao, B.; Lam, A.K. Stage dependent expression and tumor suppressive function of FAM134B (JK1) in colon cancer. Mol. Carcinog. 2017, 56, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, X.; Hua, T.; Hong, T. Integrated diagnostic network construction reveals a 4-gene panel and 5 cancer hallmarks driving breast cancer heterogeneity. Sci. Rep. 2017, 7, 6827. [Google Scholar] [CrossRef] [PubMed]
- Islam, F.; Chaousis, S.; Wahab, R.; Gopalan, V.; Lam, A.K. Protein interactions of FAM134B with EB1 and APC/beta-catenin in vitro in colon carcinoma. Mol. Carcinog. 2018, 57, 1480–1491. [Google Scholar] [CrossRef] [PubMed]
- Linxweiler, M.; Linxweiler, J.; Barth, M.; Benedix, J.; Jung, V.; Kim, Y.J.; Bohle, R.M.; Zimmermann, R.; Greiner, M. Sec62 bridges the gap from 3q amplification to molecular cell biology in non-small cell lung cancer. Am. J. Pathol. 2012, 180, 473–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, M.; Kreutzer, B.; Lang, S.; Jung, V.; Cavalie, A.; Unteregger, G.; Zimmermann, R.; Wullich, B. Sec62 protein level is crucial for the ER stress tolerance of prostate cancer. Prostate 2011, 71, 1074–1083. [Google Scholar] [CrossRef]
- Linxweiler, M.; Bochen, F.; Schick, B.; Wemmert, S.; Al Kadah, B.; Greiner, M.; Hasenfus, A.; Bohle, R.-M.; Juhasz-Böss, I.; Solomayer, E.-F.; et al. Identification of SEC62 as a potential marker for 3q amplification and cellular migration in dysplastic cervical lesions. BMC Cancer 2016, 16, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haiman, C.A.; Garcia, R.R.; Hsu, C.; Xia, L.; Ha, H.; Sheng, X.; Marchand, L.L.; Kolonel, L.N.; Henderson, B.E.; Stallcup, M.R.; et al. Screening and association testing of common coding variation in steroid hormone receptor co-activator and co-repressor genes in relation to breast cancer risk: The Multiethnic Cohort. BMC Cancer 2009, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Luo, S.; Li, H. Cdk5 activator-binding protein C53 regulates apoptosis induced by genotoxic stress via modulating the G2/M DNA damage checkpoint. J. Biol. Chem. 2005, 280, 20651–20659. [Google Scholar] [CrossRef] [Green Version]
- Mak, G.W.; Chan, M.M.; Leong, V.Y.; Lee, J.M.; Yau, T.O.; Ng, I.O.; Ching, Y.P. Overexpression of a novel activator of PAK4, the CDK5 kinase-associated protein CDK5RAP3, promotes hepatocellular carcinoma metastasis. Cancer Res. 2011, 71, 2949–2958. [Google Scholar] [CrossRef] [Green Version]
- Ishida, Y.; Yamamoto, A.; Kitamura, A.; Lamande, S.R.; Yoshimori, T.; Bateman, J.F.; Kubota, H.; Nagata, K. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol. Biol. Cell 2009, 20, 2744–2754. [Google Scholar] [CrossRef] [Green Version]
- Ferro-Novick, S.; Reggiori, F.; Brodsky, J.L. ER-Phagy, ER Homeostasis, and ER Quality Control: Implications for Disease. Trends Biochem. Sci. 2021, 46, 630–639. [Google Scholar] [CrossRef]
- Kruse, K.B.; Brodsky, J.L.; McCracken, A.A. Characterization of an ERAD gene as VPS30/ATG6 reveals two alternative and functionally distinct protein quality control pathways: One for soluble Z variant of human alpha-1 proteinase inhibitor (A1PiZ) and another for aggregates of A1PiZ. Mol. Biol. Cell 2006, 17, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, C.N.; Williams, J.M.; Knupp, J.; Arunagiri, A.; Arvan, P.; Tsai, B. Cells Deploy a Two-Pronged Strategy to Rectify Misfolded Proinsulin Aggregates. Mol. Cell 2019, 75, 442–456.e444. [Google Scholar] [CrossRef]
- Chen, Y.J.; Williams, J.M.; Arvan, P.; Tsai, B. Reticulon protects the integrity of the ER membrane during ER escape of large macromolecular protein complexes. J. Cell Biol. 2020, 219, e201908182. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. Beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Dougherty, J.D.; Nair, V.; Robertson, S.J.; Best, S.M. FAM134B, the Selective Autophagy Receptor for Endoplasmic Reticulum Turnover, Inhibits Replication of Ebola Virus Strains Makona and Mayinga. J. Infect. Dis. 2016, 214, S319–S325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [Green Version]
- Neufeldt, C.J.; Cortese, M.; Scaturro, P.; Cerikan, B.; Wideman, J.G.; Tabata, K.; Moraes, T.; Oleksiuk, O.; Pichlmair, A.; Bartenschlager, R. ER-shaping atlastin proteins act as central hubs to promote flavivirus replication and virion assembly. Nat. Microbiol. 2019, 4, 2416–2429. [Google Scholar] [CrossRef] [PubMed]
- Monel, B.; Rajah, M.M.; Hafirassou, M.L.; Ahmed, S.S.; Burlaud-Gaillard, J.; Zhu, P.-P.; Nevers, Q.; Buchrieser, J.; Porrot, F.; Meunier, C.; et al. Atlastin Endoplasmic Reticulum-Shaping Proteins Facilitate Zika Virus Replication. J. Virol. 2019, 93, e01047-19. [Google Scholar] [CrossRef] [PubMed]
- Aktepe, T.E.; Liebscher, S.; Prier, J.E.; Simmons, C.P.; Mackenzie, J.M. The Host Protein Reticulon 3.1A Is Utilized by Flaviviruses to Facilitate Membrane Remodelling. Cell Rep. 2017, 21, 1639–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.J.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823.e813. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Gotoh, M.; Ichikawa, H.; Arai, E.; Chiku, S.; Sakamoto, H.; Fujimoto, H.; Hiramoto, M.; Nammo, T.; Yasuda, K.; Yoshida, T.; et al. Comprehensive exploration of novel chimeric transcripts in clear cell renal cell carcinomas using whole transcriptome analysis. Genes Chromosomes Cancer 2014, 53, 1018–1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stingele, J.; Bellelli, R.; Boulton, S.J. Mechanisms of DNA-protein crosslink repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 563–573. [Google Scholar] [CrossRef]
- Balakirev, M.Y.; Mullally, J.E.; Favier, A.; Assard, N.; Sulpice, E.; Lindsey, D.F.; Rulina, A.V.; Gidrol, X.; Wilkinson, K.D. Wss1 metalloprotease partners with Cdc48/Doa1 in processing genotoxic SUMO conjugates. Elife 2015, 4, e06763. [Google Scholar] [CrossRef] [PubMed]
- Quintero, M.; Liu, S.; Xia, Y.; Huang, Y.; Zou, Y.; Li, G.; Hu, L.; Singh, N.; Blumberg, R.; Cai, Y.; et al. Cdk5rap3 is essential for intestinal Paneth cell development and maintenance. Cell Death Dis. 2021, 12, 131. [Google Scholar] [CrossRef]
- Yang, R.; Wang, H.; Kang, B.; Chen, B.; Shi, Y.; Yang, S.; Sun, L.; Liu, Y.; Xiao, W.; Zhang, T.; et al. CDK5RAP3, a UFL1 substrate adaptor, is crucial for liver development. Development 2019, 146, dev.169235. [Google Scholar] [CrossRef] [Green Version]
- Breuss, M.W.; Nguyen, A.; Song, Q.; Nguyen, T.; Stanley, V.; James, K.N.; Musaev, D.; Chai, G.; Wirth, S.A.; Anzenberg, P.; et al. Mutations in LNPK, Encoding the Endoplasmic Reticulum Junction Stabilizer Lunapark, Cause a Recessive Neurodevelopmental Syndrome. Am. J. Hum. Genet. 2018, 103, 296–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anding, A.L.; Baehrecke, E.H. Cleaning House: Selective Autophagy of Organelles. Dev. Cell 2017, 41, 10–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Molinari, M. ER-phagy responses in yeast, plants, and mammalian cells and their crosstalk with UPR and ERAD. Dev. Cell 2021, 56, 949–966. [Google Scholar] [CrossRef]
- Beese, C.J.; Brynjolfsdottir, S.H.; Frankel, L.B. Selective Autophagy of the Protein Homeostasis Machinery: Ribophagy, Proteaphagy and ER-Phagy. Front. Cell Dev. Biol. 2019, 7, 373. [Google Scholar] [CrossRef] [Green Version]
- Gatica, D.; Lahiri, V.; Klionsky, D.J. Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 2018, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.R.; Lingeman, E.; Luong, T.; Ahmed, S.; Muhar, M.; Nguyen, T.; Olzmann, J.A.; Corn, J.E. A Genome-wide ER-phagy Screen Highlights Key Roles of Mitochondrial Metabolism and ER-Resident UFMylation. Cell 2020, 180, 1160–1177.e1120. [Google Scholar] [CrossRef] [PubMed]
- Zielke, S.; Kardo, S.; Zein, L.; Mari, M.; Covarrubias-Pinto, A.; Kinzler, M.N.; Meyer, N.; Stolz, A.; Fulda, S.; Reggiori, F.; et al. ATF4 links ER stress with reticulophagy in glioblastoma cells. Autophagy 2020, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Wang, Y.; Luo, Z.; Li, Y.; Duan, Y. New findings of silica nanoparticles induced ER autophagy in human colon cancer cell. Sci. Rep. 2017, 7, 42591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordani, M.; Somoza, A. Targeting autophagy using metallic nanoparticles: A promising strategy for cancer treatment. Cell Mol. Life Sci. 2019, 76, 1215–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Levine, B.; Green, D.R.; Kroemer, G. Pharmacological modulation of autophagy: Therapeutic potential and persisting obstacles. Nat. Rev. Drug Discov. 2017, 16, 487–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Qian, X.; Cui, Y. Advances in ER-Phagy and Its Diseases Relevance. Cells 2021, 10, 2328. https://doi.org/10.3390/cells10092328
He L, Qian X, Cui Y. Advances in ER-Phagy and Its Diseases Relevance. Cells. 2021; 10(9):2328. https://doi.org/10.3390/cells10092328
Chicago/Turabian StyleHe, Lingang, Xuehong Qian, and Yixian Cui. 2021. "Advances in ER-Phagy and Its Diseases Relevance" Cells 10, no. 9: 2328. https://doi.org/10.3390/cells10092328
APA StyleHe, L., Qian, X., & Cui, Y. (2021). Advances in ER-Phagy and Its Diseases Relevance. Cells, 10(9), 2328. https://doi.org/10.3390/cells10092328