
Brain Immune Interactions—Novel Emerging Options to Treat Acute Ischemic Brain Injury

 ,
, 
 ,
, 
{kind=link}
Abstract
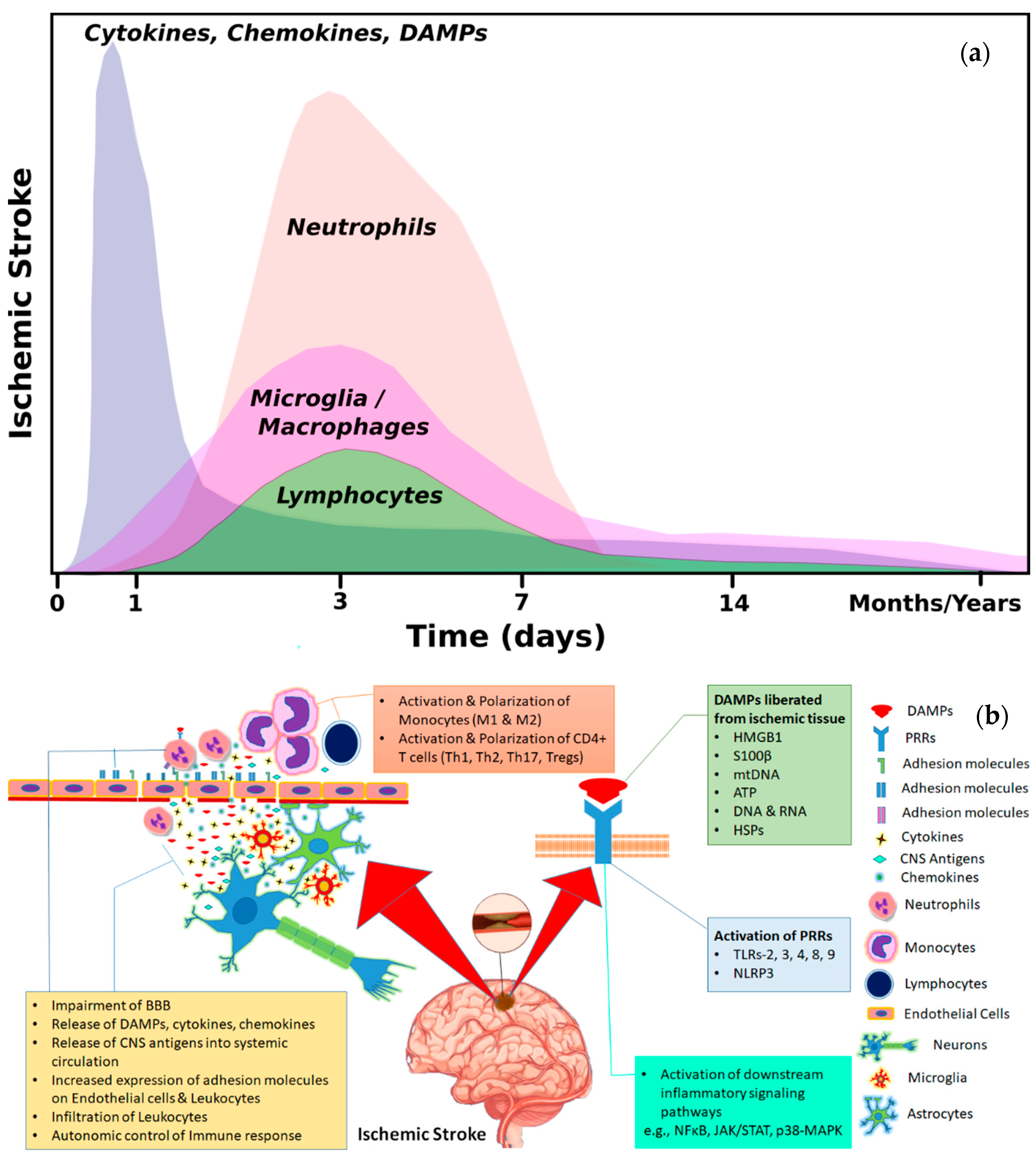
:1. Introduction
1.1. Epidemiology and Pathophysiology of Stroke
1.2. Inflammation and Immune Response after Cerebral Ischemia
2. Means of Interaction between Brain and Immune Cells
2.1. Activation of Immune System through Brain Derived Antigens
2.2. Brain–Immune Interaction through DAMPs
2.2.1. Adenosine Triphosphate (ATP)
2.2.2. S100B
2.2.3. High-Mobility Group Box-1 (HMGB1)
2.2.4. Peroxiredoxins
2.2.5. Cytokines as DAMPs
2.2.6. Extracellular-Matrix-Derived DAMPs
2.3. Brain–Immune Interaction through Immune Signaling Molecules
2.4. Brain–Immune Interaction via Autonomic Nervous System
3. Impact of Systemic Inflammation and Infiltrated Immune Cells on Ischemic Brain
3.1. Impact of Innate Immune Cells on Ischemic Brain
3.2. Impact of Adaptive Immunity (Immune Cells) on Ischemic Brain
4. Brain–Immune Interaction after Cerebral Ischemia and Therapeutic Options
4.1. Targeting DAMPs and Their Receptors
4.2. Targeting Immune-Signaling Molecules
4.3. Targeting the Autonomic Nervous System
4.4. Targeting Polarization of Microglia/Macrophages towards M2 Type Phenotype
4.5. Targeting Polarization of Specific T Cell Response towards Th2 and Regulatory T-Cells
4.6. Evidence of Stem Cell Signals to Instruct Immune Micro Milieu
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lloyd-Jones, D.; Adams, R.; Carnethon, M.; De Simone, G.; Ferguson, T.B.; Flegal, K.; Ford, E.; Furie, K.; Go, A.; Greenlund, K.; et al. Heart disease and stroke statistics—2009 update: A report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 2009, 119, e21–e181. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Iadecola, C.; Moskowitz, M.A. Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci. 1999, 22, 391–397. [Google Scholar] [CrossRef]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.C.V.; Mitchell, P.J.; Kleinig, T.J.; Dewey, H.M.; Churilov, L.; Yassi, N.; Yan, B.; Dowling, R.J.; Parsons, M.W.; Oxley, T.J.; et al. Endovascular Therapy for Ischemic Stroke with Perfusion-Imaging Selection. N. Engl. J. Med. 2015, 372, 1009–1018. [Google Scholar] [CrossRef] [Green Version]
- Saver, J.L.; Goyal, M.; Bonafe, A.; Diener, H.-C.; Levy, E.I.; Pereira, V.M.; Albers, G.W.; Cognard, C.; Cohen, D.J.; Hacke, W.; et al. Stent-Retriever Thrombectomy after Intravenous t-PA vs. t-PA Alone in Stroke. N. Engl. J. Med. 2015, 372, 2285–2295. [Google Scholar] [CrossRef] [Green Version]
- Hacke, W.; Kaste, M.; Bluhmki, E.; Brozman, M.; Dávalos, A.; Guidetti, D.; Larrue, V.; Lees, K.R.; Medeghri, Z.; Machnig, T.; et al. Thrombolysis with Alteplase 3 to 4.5 Hours after Acute Ischemic Stroke. N. Engl. J. Med. 2008, 359, 1317–1329. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, O.; Baumann, B.; de Lorenzi, R.; Muhammad, S.; Zhang, W.; Kleesiek, J.; Malfertheiner, M.; Kohrmann, M.; Potrovita, I.; Maegele, I.; et al. IKK mediates ischemia-induced neuronal death. Nat. Med. 2005, 11, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Schneider, A.; Martin-Villalba, A.; Weih, F.; Vogel, J.; Wirth, T.; Schwaninger, M. NF-kappaB is activated and promotes cell death in focal cerebral ischemia. Nat. Med. 1999, 5, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Denes, A.; Humphreys, N.; Lane, T.E.; Grencis, R.; Rothwell, N. Chronic systemic infection exacerbates ischemic brain damage via a CCL5 (regulated on activation, normal T-cell expressed and secreted)-mediated proinflammatory response in mice. J. Neurosci. 2010, 30, 10086–10095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inta, I.; Frauenknecht, K.; Dörr, H.; Kohlhof, P.; Rabsilber, T.; Auffarth, G.U.; Burkly, L.; Mittelbronn, M.; Hahm, K.; Sommer, C.; et al. Induction of the cytokine TWEAK and its receptor Fn14 in ischemic stroke. J. Neurol. Sci. 2008, 275, 117–120. [Google Scholar] [CrossRef]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef]
- Muhammad, S.; Barakat, W.; Stoyanov, S.; Murikinati, S.; Yang, H.; Tracey, K.J.; Bendszus, M.; Rossetti, G.; Nawroth, P.P.; Bierhaus, A.; et al. The HMGB1 Receptor RAGE Mediates Ischemic Brain Damage. J. Neurosci. 2008, 28, 12023–12031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muhammad, S.; Haasbach, E.; Kotchourko, M.; Strigli, A.; Krenz, A.; Ridder, D.A.; Vogel, A.B.; Marti, H.H.; Al-Abed, Y.; Planz, O.; et al. Influenza virus infection aggravates stroke outcome. Stroke 2011, 42, 783–791. [Google Scholar] [CrossRef] [Green Version]
- Murikinati, S.; Jüttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2010, 24, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Muller-Fielitz, H.; Pokorna, B.; Vollbrandt, T.; Stolting, I.; Nadrowitz, R.; et al. The beta-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Mehta, S.L.; Manhas, N.; Raghubir, R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res. Rev. 2007, 54, 34–66. [Google Scholar] [CrossRef]
- Hill, W.D.; Hess, D.C.; Martin-Studdard, A.; Carothers, J.J.; Zheng, J.; Hale, D.; Maeda, M.; Fagan, S.C.; Carroll, J.E.; Conway, S.J. SDF-1 (CXCL12) is upregulated in the ischemic penumbra following stroke: Association with bone marrow cell homing to injury. J. Neuropathol. Exp. Neurol. 2004, 63, 84–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chamorro, A.; Meisel, A.; Planas, A.M.; Urra, X.; van de Beek, D.; Veltkamp, R. The immunology of acute stroke. Nat. Rev. Neurol. 2012, 8, 401–410. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- An, C.; Shi, Y.; Li, P.; Hu, X.; Gan, Y.; Stetler, R.A.; Leak, R.K.; Gao, Y.; Sun, B.-L.; Zheng, P.; et al. Molecular dialogs between the ischemic brain and the peripheral immune system: Dualistic roles in injury and repair. Prog. Neurobiol. 2014, 115, 6–24. [Google Scholar] [CrossRef] [Green Version]
- Mecha, M.; Carrillo-Salinas, F.J.; Mestre, L.; Feliu, A.; Guaza, C. Viral models of multiple sclerosis: Neurodegeneration and demyelination in mice infected with Theiler’s virus. Prog. Neurobiol. 2013, 101–102, 46–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittel, B.N.; Visintin, I.; Merchant, R.M.; Janeway, C.A., Jr. Presentation of the self antigen myelin basic protein by dendritic cells leads to experimental autoimmune encephalomyelitis. J. Immunol. 1999, 163, 32–39. [Google Scholar] [PubMed]
- Bornstein, N.M.; Aronovich, B.; Korczyn, A.D.; Shavit, S.; Michaelson, D.M.; Chapman, J. Antibodies to brain antigens following stroke. Neurology 2001, 56, 529–530. [Google Scholar] [CrossRef]
- Dambinova, S.A.; Khounteev, G.A.; Izykenova, G.A.; Zavolokov, I.G.; Ilyukhina, A.Y.; Skoromets, A.A. Blood test detecting autoantibodies to N-methyl-D-aspartate neuroreceptors for evaluation of patients with transient ischemic attack and stroke. Clin. Chem. 2003, 49, 1752–1762. [Google Scholar] [CrossRef] [Green Version]
- Tsuda, M.; Shigemoto-Mogami, Y.; Koizumi, S.; Mizokoshi, A.; Kohsaka, S.; Salter, M.W.; Inoue, K. P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 2003, 424, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Planas, A.M.; Gomez-Choco, M.; Urra, X.; Gorina, R.; Caballero, M.; Chamorro, A. Brain-derived antigens in lymphoid tissue of patients with acute stroke. J. Immunol. 2012, 188, 2156–2163. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, L. Complexity of danger: The diverse nature of damage-associated molecular patterns. J. Biol. Chem. 2014, 289, 35237–35245. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Hafez, A.; Rezai Jahromi, B.; Kinfe, T.M.; Lamprecht, A.; Niemelä, M.; Muhammad, S. Role of Damage Associated Molecular Pattern Molecules (DAMPs) in Aneurysmal Subarachnoid Hemorrhage (aSAH). Int. J. Mol. Sci. 2018, 19, 2035. [Google Scholar] [CrossRef] [Green Version]
- Latini, S.; Pedata, F. Adenosine in the central nervous system: Release mechanisms and extracellular concentrations. J. Neurochem. 2001, 79, 463–484. [Google Scholar] [CrossRef] [Green Version]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Li, W.; Niu, G.; Leak, R.K.; Chen, J.; Zhang, F. ATP induces mild hypothermia in rats but has a strikingly detrimental impact on focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2013, 33, e1–e10. [Google Scholar] [CrossRef]
- Chu, K.; Yin, B.; Wang, J.; Peng, G.; Liang, H.; Xu, Z.; Du, Y.; Fang, M.; Xia, Q.; Luo, B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foerch, C.; Singer, O.C.; Neumann-Haefelin, T.; du Mesnil de Rochemont, R.; Steinmetz, H.; Sitzer, M. Evaluation of serum S100B as a surrogate marker for long-term outcome and infarct volume in acute middle cerebral artery infarction. Arch. Neurol. 2005, 62, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Mori, T.; Tan, J.; Arendash, G.W.; Koyama, N.; Nojima, Y.; Town, T. Overexpression of human S100B exacerbates brain damage and periinfarct gliosis after permanent focal ischemia. Stroke 2008, 39, 2114–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bianchi, R.; Giambanco, I.; Donato, R. S100B/RAGE-dependent activation of microglia via NF-kappaB and AP-1 Co-regulation of COX-2 expression by S100B, IL-1beta and TNF-alpha. Neurobiol. Aging 2010, 31, 665–677. [Google Scholar] [CrossRef]
- Kim, J.B.; Sig Choi, J.; Yu, Y.M.; Nam, K.; Piao, C.S.; Kim, S.W.; Lee, M.H.; Han, P.L.; Park, J.S.; Lee, J.K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shichita, T.; Hasegawa, E.; Kimura, A.; Morita, R.; Sakaguchi, R.; Takada, I.; Sekiya, T.; Ooboshi, H.; Kitazono, T.; Yanagawa, T.; et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nat. Med. 2012, 18, 911–917. [Google Scholar] [CrossRef]
- Kunze, A.; Zierath, D.; Tanzi, P.; Cain, K.; Becker, K. Peroxiredoxin 5 (PRX5) Is Correlated Inversely to Systemic Markers of Inflammation in Acute Stroke. Stroke 2014, 45, 608–610. [Google Scholar] [CrossRef]
- Richard, S.; Lapierre, V.; Girerd, N.; Bonnerot, M.; Burkhard, P.R.; Lagerstedt, L.; Bracard, S.; Debouverie, M.; Turck, N.; Sanchez, J.-C. Diagnostic performance of peroxiredoxin 1 to determine time-of-onset of acute cerebral infarction. Sci. Rep. 2016, 6, 38300. [Google Scholar] [CrossRef]
- Hirsiger, S.; Simmen, H.P.; Werner, C.M.; Wanner, G.A.; Rittirsch, D. Danger signals activating the immune response after trauma. Mediat. Inflamm. 2012, 315941. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-J.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 2007, 13, 851–856. [Google Scholar] [CrossRef]
- Kim, B.; Lee, Y.; Kim, E.; Kwak, A.; Ryoo, S.; Bae, S.H.; Azam, T.; Kim, S.; Dinarello, C.A. The Interleukin-1α Precursor is Biologically Active and is Likely a Key Alarmin in the IL-1 Family of Cytokines. Front. Immunol. 2013, 4, 391. [Google Scholar] [CrossRef] [Green Version]
- Luheshi, N.M.; Kovács, K.J.; Lopez-Castejon, G.; Brough, D.; Denes, A. Interleukin-1α expression precedes IL-1β after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflam. 2011, 8, 186. [Google Scholar] [CrossRef] [Green Version]
- Salmeron, K.E.; Maniskas, M.E.; Edwards, D.N.; Wong, R.; Rajkovic, I.; Trout, A.; Rahman, A.A.; Hamilton, S.; Fraser, J.F.; Pinteaux, E.; et al. Interleukin 1 alpha administration is neuroprotective and neuro-restorative following experimental ischemic stroke. J. Neuroinflam. 2019, 16, 222. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Liu, H.; Zhang, H.; Ye, Q.; Wang, J.; Yang, B.; Mao, L.; Zhu, W.; Leak, R.K.; Xiao, B.; et al. ST2/IL-33-Dependent Microglial Response Limits Acute Ischemic Brain Injury. J. Neurosci. 2017, 37, 4692–4704. [Google Scholar] [CrossRef]
- Gülke, E.; Gelderblom, M.; Magnus, T. Danger signals in stroke and their role on microglia activation after ischemia. Adv. Neurol. Disord. 2018, 11, 1756286418774254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Potts-Kant, E.N.; Garantziotis, S.; Foster, W.M.; Hollingsworth, J.W. Hyaluronan signaling during ozone-induced lung injury requires TLR4, MyD88, and TIRAP. PLoS ONE 2011, 6, e27137. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.-C.; Yeh, S.-J.; Tsai, L.-K.; Hu, C.-J.; Lien, L.-M.; Peng, G.-S.; Yang, W.-S.; Chiou, H.-Y.; Jeng, J.-S. Association between plasma levels of hyaluronic acid and functional outcome in acute stroke patients. J. Neuroinflam. 2014, 11, 101. [Google Scholar] [CrossRef] [Green Version]
- Al’Qteishat, A.; Gaffney, J.; Krupinski, J.; Rubio, F.; West, D.; Kumar, S.; Kumar, P.; Mitsios, N.; Slevin, M. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain A J. Neurol. 2006, 129, 2158–2176. [Google Scholar] [CrossRef] [PubMed]
- Al Qteishat, A.; Gaffney, J.J.; Krupinski, J.; Slevin, M. Hyaluronan expression following middle cerebral artery occlusion in the rat. Neuroreport 2006, 17, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Katarzyna Greda, A.; Nowicka, D. Hyaluronidase inhibition accelerates functional recovery from stroke in the mouse brain. J. Neurochem. 2021, 157, 781–801. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Castano, D.; Dzyubenko, E.; Borbor, M.; Vasileiadou, P.; Kleinschnitz, C.; Roll, L.; Faissner, A.; Hermann, D.M. Tenascin-C preserves microglia surveillance and restricts leukocyte and, more specifically, T cell infiltration of the ischemic brain. Brain Behav. Immun. 2021, 91, 639–648. [Google Scholar] [CrossRef]
- Shiba, M.; Suzuki, H. Lessons from tenascin-C knockout mice and potential clinical application to subarachnoid hemorrhage. Neural. Regen. Res. 2019, 14, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Chelluboina, B.; Chokkalla, A.K.; Mehta, S.L.; Bathula, S.; Dempsey, R.J.; Vemuganti, R. Abstract P776: Post-Stroke Tenascin-C Induction Mediates the Ischemic Pathogenesis. Stroke 2021, 52, AP776. [Google Scholar] [CrossRef]
- Zang, N.; Lin, Z.; Huang, K.; Pan, Y.; Wu, Y.; Wu, Y.; Wang, S.; Wang, D.; Ji, Z.; Pan, S. Biomarkers of Unfavorable Outcome in Acute Ischemic Stroke Patients with Successful Recanalization by Endovascular Thrombectomy. Cerebrovasc. Dis. 2020, 49, 583–592. [Google Scholar] [CrossRef]
- Lee, S.C.; Liu, W.; Dickson, D.W.; Brosnan, C.F.; Berman, J.W. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 beta. J. Immunol. 1993, 150, 2659–2667. [Google Scholar]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Devel. 2014, 8, 2221–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobowale, O.A.; Parry-Jones, A.R.; Smith, C.J.; Tyrrell, P.J.; Rothwell, N.J.; Allan, S.M. Interleukin-1 in Stroke: From Bench to Bedside. Stroke 2016, 47, 2160–2167. [Google Scholar] [CrossRef]
- Hotter, B.; Hoffmann, S.; Ulm, L.; Meisel, C.; Fiebach, J.B.; Meisel, A. IL-6 Plasma Levels Correlate With Cerebral Perfusion Deficits and Infarct Sizes in Stroke Patients Without Associated Infections. Front. Neurol. 2019, 10, 83. [Google Scholar] [CrossRef]
- Suzuki, S.; Tanaka, K.; Suzuki, N. Ambivalent Aspects of Interleukin-6 in Cerebral Ischemia: Inflammatory versus Neurotrophic Aspects. J. Cereb. Blood Flow Metab. 2009, 29, 464–479. [Google Scholar] [CrossRef]
- Losy, J.; Zaremba, J. Monocyte chemoattractant protein-1 is increased in the cerebrospinal fluid of patients with ischemic stroke. Stroke 2001, 32, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagami, S.; Tamura, M.; Hayashi, M.; Endo, N.; Tanabe, H.; Katsuura, Y.; Komoriya, K. Differential production of MCP-1 and cytokine-induced neutrophil chemoattractant in the ischemic brain after transient focal ischemia in rats. J. Leukoc. Biol. 1999, 65, 744–749. [Google Scholar] [CrossRef] [PubMed]
- Verslegers, M.; Lemmens, K.; Van Hove, I.; Moons, L. Matrix metalloproteinase-2 and -9 as promising benefactors in development, plasticity and repair of the nervous system. Prog. Neurobiol. 2013, 105, 60–78. [Google Scholar] [CrossRef]
- Zaremba, J.; Ilkowski, J.; Losy, J. Serial measurements of levels of the chemokines CCL2, CCL3 and CCL5 in serum of patients with acute ischaemic stroke. Folia Neuropathol. 2006, 44, 282–289. [Google Scholar] [PubMed]
- Muhammad, S.; Planz, O.; Schwaninger, M. Increased Plasma Matrix Metalloproteinase-9 Levels Contribute to Intracerebral Hemorrhage during Thrombolysis after Concomitant Stroke and Influenza Infection. Cerebrovasc. Dis. Extra 2016, 6, 50–59. [Google Scholar] [CrossRef]
- Hughes, P.M.; Allegrini, P.R.; Rudin, M.; Perry, V.H.; Mir, A.K.; Wiessner, C. Monocyte chemoattractant protein-1 deficiency is protective in a murine stroke model. J. Cereb. Blood Flow Metab. 2002, 22, 308–317. [Google Scholar] [CrossRef] [Green Version]
- Schilling, M.; Strecker, J.K.; Ringelstein, E.B.; Schabitz, W.R.; Kiefer, R. The role of CC chemokine receptor 2 on microglia activation and blood-borne cell recruitment after transient focal cerebral ischemia in mice. Brain. Res. 2009, 1289, 79–84. [Google Scholar] [CrossRef]
- Kim, J.S.; Gautam, S.C.; Chopp, M.; Zaloga, C.; Jones, M.L.; Ward, P.A.; Welch, K.M. Expression of monocyte chemoattractant protein-1 and macrophage inflammatory protein-1 after focal cerebral ischemia in the rat. J. Neuroimmunol. 1995, 56, 127–134. [Google Scholar] [CrossRef]
- Domac, F.M.; Misirli, H. The role of neutrophils and interleukin-8 in acute ischemic stroke. Neuroscience 2008, 13, 136–141. [Google Scholar]
- Villa, P.; Triulzi, S.; Cavalieri, B.; Di Bitondo, R.; Bertini, R.; Barbera, S.; Bigini, P.; Mennini, T.; Gelosa, P.; Tremoli, E.; et al. The interleukin-8 (IL-8/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol. Med. 2007, 13, 125–133. [Google Scholar] [CrossRef]
- Okada, Y.; Copeland, B.R.; Mori, E.; Tung, M.M.; Thomas, W.S.; del Zoppo, G.J. P-selectin and intercellular adhesion molecule-1 expression after focal brain ischemia and reperfusion. Stroke 1994, 25, 202–211. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Chopp, M.; Zhang, Z.; Jiang, N.; Powers, C. The expression of P- and E-selectins in three models of middle cerebral artery occlusion. Brain Res. 1998, 785, 207–214. [Google Scholar] [CrossRef]
- Edwards, D.N.; Bix, G.J. The Inflammatory Response After Ischemic Stroke: Targeting β2 and β1 Integrins. Front. Neurosci. 2019, 13, 540. [Google Scholar] [CrossRef]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Prass, K.; Meisel, C.; Höflich, C.; Braun, J.; Halle, E.; Wolf, T.; Ruscher, K.; Victorov, I.V.; Priller, J.; Dirnagl, U.; et al. Stroke-induced Immunodeficiency Promotes Spontaneous Bacterial Infections and Is Mediated by Sympathetic Activation Reversal by Poststroke T Helper Cell Type 1–like Immunostimulation. J. Exp. Med. 2003, 198, 725–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdes-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [Green Version]
- Pavlov, V.A.; Ochani, M.; Yang, L.H.; Gallowitsch-Puerta, M.; Ochani, K.; Lin, X.; Levi, J.; Parrish, W.R.; Rosas-Ballina, M.; Czura, C.J.; et al. Selective alpha7-nicotinic acetylcholine receptor agonist GTS-21 improves survival in murine endotoxemia and severe sepsis. Crit. Care Med. 2007, 35, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Ay, I.; Sorensen, A.G.; Ay, H. Vagus nerve stimulation reduces infarct size in rat focal cerebral ischemia: An unlikely role for cerebral blood flow. Brain Res. 2011, 1392, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.; Jenne, C.N.; Lee, W.Y.; Leger, C.; Kubes, P. Functional innervation of hepatic iNKT cells is immunosuppressive following stroke. Science 2011, 334, 101–105. [Google Scholar] [CrossRef]
- Ajmo, C.T., Jr.; Collier, L.A.; Leonardo, C.C.; Hall, A.A.; Green, S.M.; Womble, T.A.; Cuevas, J.; Willing, A.E.; Pennypacker, K.R. Blockade of adrenoreceptors inhibits the splenic response to stroke. Exp. Neurol. 2009, 218, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Rupalla, K.; Allegrini, P.R.; Sauer, D.; Wiessner, C. Time course of microglia activation and apoptosis in various brain regions after permanent focal cerebral ischemia in mice. Acta Neuropathol. 1998, 96, 172–178. [Google Scholar] [CrossRef]
- Schilling, M.; Besselmann, M.; Leonhard, C.; Mueller, M.; Ringelstein, E.B.; Kiefer, R. Microglial activation precedes and predominates over macrophage infiltration in transient focal cerebral ischemia: A study in green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2003, 183, 25–33. [Google Scholar] [CrossRef]
- Morrison, H.W.; Filosa, J.A. A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J. Neuroinflamm. 2013, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C.; Buckwalter, M.S.; Anrather, J. Immune responses to stroke: Mechanisms, modulation, and therapeutic potential. J. Clin. Investig. 2020, 130, 2777–2788. [Google Scholar] [CrossRef] [PubMed]
- Planas, A.M. Role of Immune Cells Migrating to the Ischemic Brain. Stroke 2018, 49, 2261–2267. [Google Scholar] [CrossRef]
- Iadecola, C.; Anrather, J. The immunology of stroke: From mechanisms to translation. Nat. Med. 2011, 17, 796–808. [Google Scholar] [CrossRef]
- Schilling, M.; Besselmann, M.; Muller, M.; Strecker, J.K.; Ringelstein, E.B.; Kiefer, R. Predominant phagocytic activity of resident microglia over hematogenous macrophages following transient focal cerebral ischemia: An investigation using green fluorescent protein transgenic bone marrow chimeric mice. Exp. Neurol. 2005, 196, 290–297. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Yang, G.; Li, G. Inflammatory mechanisms in ischemic stroke: Role of inflammatory cells. J. Leukoc. Biol. 2010, 87, 779–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connolly, E.S., Jr.; Winfree, C.J.; Prestigiacomo, C.J.; Kim, S.C.; Choudhri, T.F.; Hoh, B.L.; Naka, Y.; Solomon, R.A.; Pinsky, D.J. Exacerbation of cerebral injury in mice that express the P-selectin gene: Identification of P-selectin blockade as a new target for the treatment of stroke. Circ. Res. 1997, 81, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Connolly, E.S., Jr.; Winfree, C.J.; Springer, T.A.; Naka, Y.; Liao, H.; Yan, S.D.; Stern, D.M.; Solomon, R.A.; Gutierrez-Ramos, J.C.; Pinsky, D.J. Cerebral protection in homozygous null ICAM-1 mice after middle cerebral artery occlusion. Role of neutrophil adhesion in the pathogenesis of stroke. J. Clin. Investig. 1996, 97, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Shiga, Y.; Onodera, H.; Kogure, K.; Yamasaki, Y.; Yashima, Y.; Syozuhara, H.; Sendo, F. Neutrophil as a mediator of ischemic edema formation in the brain. Neurosci. Lett. 1991, 125, 110–112. [Google Scholar] [CrossRef]
- Felger, J.C.; Abe, T.; Kaunzner, U.W.; Gottfried-Blackmore, A.; Gal-Toth, J.; McEwen, B.S.; Iadecola, C.; Bulloch, K. Brain dendritic cells in ischemic stroke: Time course, activation state, and origin. Brain Behav. Immun. 2010, 24, 724–737. [Google Scholar] [CrossRef] [Green Version]
- Brait, V.H.; Jackman, K.A.; Walduck, A.K.; Selemidis, S.; Diep, H.; Mast, A.E.; Guida, E.; Broughton, B.R.; Drummond, G.R.; Sobey, C.G. Mechanisms contributing to cerebral infarct size after stroke: Gender, reperfusion, T lymphocytes, and Nox2-derived superoxide. J. Cereb. Blood Flow Metab. 2010, 30, 1306–1317. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Schwab, N.; Kraft, P.; Hagedorn, I.; Dreykluft, A.; Schwarz, T.; Austinat, M.; Nieswandt, B.; Wiendl, H.; Stoll, G. Early detrimental T-cell effects in experimental cerebral ischemia are neither related to adaptive immunity nor thrombus formation. Blood 2010, 115, 3835–3842. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T lymphocytes and interferon-gamma in ischemic stroke. Circulation 2006, 113, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liesz, A.; Zhou, W.; Mracsko, E.; Karcher, S.; Bauer, H.; Schwarting, S.; Sun, L.; Bruder, D.; Stegemann, S.; Cerwenka, A.; et al. Inhibition of lymphocyte trafficking shields the brain against deleterious neuroinflammation after stroke. Brain A J. Neurol. 2011, 134, 704–720. [Google Scholar] [CrossRef] [Green Version]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.K.; Lee, E.V.; White, R.F.; Jonak, Z.L.; Feuerstein, G.Z.; Barone, F.C. Reperfusion following focal stroke hastens inflammation and resolution of ischemic injured tissue. Brain Res. Bull. 1994, 35, 387–392. [Google Scholar] [CrossRef]
- Ralph, S.J.; Weissenberger, A.; Bonev, V.; King, L.D.; Bonham, M.D.; Ferguson, S.; Smith, A.D.; Goodman-Jones, A.A.; Espinet, A.J. Phase I/II parallel double-blind randomized controlled clinical trial of perispinal etanercept for chronic stroke: Improved mobility and pain alleviation. Expert Opin. Investig. Drugs 2020, 29, 311–326. [Google Scholar] [CrossRef]
- Wu, M.-H.; Huang, C.-C.; Chio, C.-C.; Tsai, K.-J.; Chang, C.-P.; Lin, N.-K.; Lin, M.-T. Inhibition of Peripheral TNF-α and Downregulation of Microglial Activation by Alpha-Lipoic Acid and Etanercept Protect Rat Brain Against Ischemic Stroke. Mol. Neurobiol. 2016, 53, 4961–4971. [Google Scholar] [CrossRef]
- Yamashita, T.; Sawamoto, K.; Suzuki, S.; Suzuki, N.; Adachi, K.; Kawase, T.; Mihara, M.; Ohsugi, Y.; Abe, K.; Okano, H. Blockade of interleukin-6 signaling aggravates ischemic cerebral damage in mice: Possible involvement of Stat3 activation in the protection of neurons. J. Neurochem. 2005, 94, 459–468. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, J.; Kang, W.; Dong, Z.; Wang, H. Tocilizumab inhibits neuronal cell apoptosis and activates STAT3 in cerebral infarction rat model. Bosn. J. Basic. Med. Sci. 2016, 16, 145–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudobenko, J.; Chauhan, A.; McCullough, L. Abstract 128: Amelioration of Ischemic Stroke Damage Through Inhibition of Interleukin-6 Signaling With Tocilizumab Requires Sex Specific Dosing. Stroke 2019, 50, A128. [Google Scholar] [CrossRef]
- Huang, J.; Choudhri, T.F.; Winfree, C.J.; McTaggart, R.A.; Kiss, S.; Mocco, J.; Kim, L.J.; Protopsaltis, T.S.; Zhang, Y.; Pinsky, D.J.; et al. Postischemic cerebrovascular E-selectin expression mediates tissue injury in murine stroke. Stroke 2000, 31, 3047–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Abe, K.; Tojo, S.J.; Kitagawa, H.; Kimura, K.; Mizugaki, M.; Itoyama, Y. Reduction of ischemic brain injury by anti-P-selectin monoclonal antibody after permanent middle cerebral artery occlusion in rat. Neurol. Res. 1999, 21, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Veltkamp, R.; Gill, D. Clinical Trials of Immunomodulation in Ischemic Stroke. Neurother. J. Am. Soc. Exp. Neurother. 2016, 13, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434. [Google Scholar] [CrossRef]
- Streit, W.J.; Hurley, S.D.; McGraw, T.S.; Semple-Rowland, S.L. Comparative evaluation of cytokine profiles and reactive gliosis supports a critical role for interleukin-6 in neuron-glia signaling during regeneration. J. Neurosci. Res. 2000, 61, 10–20. [Google Scholar] [CrossRef]
- Lu, Y.; Wahl, L.M. Production of matrix metalloproteinase-9 by activated human monocytes involves a phosphatidylinositol-3 kinase/Akt/IKKalpha/NF-kappaB pathway. J. Leukoc. Biol. 2005, 78, 259–265. [Google Scholar] [CrossRef] [Green Version]
- Moldovan, L.; Moldovan, N.I. Role of monocytes and macrophages in angiogenesis. EXS 2005, 127–146. [Google Scholar]
- Scharfman, H.; Goodman, J.; Macleod, A.; Phani, S.; Antonelli, C.; Croll, S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp. Neurol. 2005, 192, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Unoki, N.; Murakami, T.; Nishijima, K.; Ogino, K.; van Rooijen, N.; Yoshimura, N. SDF-1/CXCR4 contributes to the activation of tip cells and microglia in retinal angiogenesis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3362–3371. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.P.; Sailor, K.A.; Lang, B.T.; Park, S.W.; Vemuganti, R.; Dempsey, R.J. Monocyte chemoattractant protein-1 plays a critical role in neuroblast migration after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2007, 27, 1213–1224. [Google Scholar] [CrossRef]
- Kodama, H.; Inoue, T.; Watanabe, R.; Yasutomi, D.; Kawakami, Y.; Ogawa, S.; Mikoshiba, K.; Ikeda, Y.; Kuwana, M. Neurogenic potential of progenitors derived from human circulating CD14+ monocytes. Immunol. Cell Biol. 2006, 84, 209–217. [Google Scholar] [CrossRef]
- Lalive, P.H.; Paglinawan, R.; Biollaz, G.; Kappos, E.A.; Leone, D.P.; Malipiero, U.; Relvas, J.B.; Moransard, M.; Suter, T.; Fontana, A. TGF-beta-treated microglia induce oligodendrocyte precursor cell chemotaxis through the HGF-c-Met pathway. Eur. J. Immunol. 2005, 35, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Mitrovic, B.; Ignarro, L.J.; Vinters, H.V.; Akers, M.A.; Schmid, I.; Uittenbogaart, C.; Merrill, J.E. Nitric oxide induces necrotic but not apoptotic cell death in oligodendrocytes. Neuroscience 1995, 65, 531–539. [Google Scholar] [CrossRef]
- Liu, X.; Liu, J.; Zhao, S.; Zhang, H.; Cai, W.; Cai, M.; Ji, X.; Leak, R.K.; Gao, Y.; Chen, J.; et al. Interleukin-4 Is Essential for Microglia/Macrophage M2 Polarization and Long-Term Recovery After Cerebral Ischemia. Stroke 2016, 47, 498–504. [Google Scholar] [CrossRef]
- Albini, A.; Marchisone, C.; Del Grosso, F.; Benelli, R.; Masiello, L.; Tacchetti, C.; Bono, M.; Ferrantini, M.; Rozera, C.; Truini, M.; et al. Inhibition of angiogenesis and vascular tumor growth by interferon-producing cells: A gene therapy approach. Am. J. Pathol. 2000, 156, 1381–1393. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Goumans, M.J.; Sjostrand, L.J.; van Rooijen, M.A.; Ward, D.; Leveen, P.; Xu, X.; ten Dijke, P.; Mummery, C.L.; Karlsson, S. Abnormal angiogenesis but intact hematopoietic potential in TGF-beta type I receptor-deficient mice. EMBO J. 2001, 20, 1663–1673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, J.S. Recalibrating the Relevance of Adult Neurogenesis. Trends Neurosci. 2019, 42, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Naik, S.; Larsen, S.B.; Cowley, C.J.; Fuchs, E. Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 2018, 175, 908–920. [Google Scholar] [CrossRef] [Green Version]
- Grund-Gröschke, S.; Stockmaier, G.; Aberger, F. Hedgehog/GLI signaling in tumor immunity-new therapeutic opportunities and clinical implications. Cell Commun. Signal. 2019, 17, 172. [Google Scholar] [CrossRef] [Green Version]
- Haseeb, M.; Pirzada, R.H.; Ain, Q.U.; Choi, S. Wnt Signaling in the Regulation of Immune Cell and Cancer Therapeutics. Cells 2019, 8, 1380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosla, R.; Vyas, A.K.; Trehanpati, N. Dichotomy of Notch signalling in regulating tumour immune surveillance. Scand. J. Immunol. 2019, 89, e12744. [Google Scholar] [CrossRef] [PubMed]
- Boshuizen, M.C.S.; Steinberg, G.K. Stem Cell-Based Immunomodulation After Stroke: Effects on Brain Repair Processes. Stroke 2018, 49, 1563–1570. [Google Scholar] [CrossRef]
- Jin, Y.; Raviv, N.; Barnett, A.; Bambakidis, N.C.; Filichia, E.; Luo, Y. The shh signaling pathway is upregulated in multiple cell types in cortical ischemia and influences the outcome of stroke in an animal model. PLoS ONE 2015, 10, e0124657. [Google Scholar] [CrossRef]
- Wei, Z.; Chigurupati, S.; Arumugam, T.V.; Jo, D.G.; Li, H.; Chan, S.L. Notch activation enhances the microglia-mediated inflammatory response associated with focal cerebral ischemia. Stroke 2011, 42, 2589–2594. [Google Scholar] [CrossRef]
- Marchetti, B.; Pluchino, S. Wnt your brain be inflamed? Yes, it Wnt! Trends. Mol. Med. 2013, 19, 144–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Zhang, Z. Microglia and Wnt Pathways: Prospects for Inflammation in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 110. [Google Scholar] [CrossRef]
- Lengfeld, J.E.; Lutz, S.E.; Smith, J.R.; Diaconu, C.; Scott, C.; Kofman, S.B.; Choi, C.; Walsh, C.M.; Raine, C.S.; Agalliu, I.; et al. Endothelial Wnt/β-catenin signaling reduces immune cell infiltration in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2017, 114, E1168–E1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, L.; Kan, E.M.; Kaur, C.; Dheen, S.T.; Hao, A.; Lu, J.; Ling, E.-A. Notch-1 signaling regulates microglia activation via NF-κB pathway after hypoxic exposure in vivo and in vitro. PLoS ONE 2013, 8, e78439. [Google Scholar] [CrossRef] [Green Version]
- Marei, H.E.; Hasan, A.; Rizzi, R.; Althani, A.; Afifi, N.; Cenciarelli, C.; Caceci, T.; Shuaib, A. Potential of Stem Cell-Based Therapy for Ischemic Stroke. Front. Neurol. 2018, 9, 34. [Google Scholar] [CrossRef]
- Thored, P.; Heldmann, U.; Gomes-Leal, W.; Gisler, R.; Darsalia, V.; Taneera, J.; Nygren, J.M.; Jacobsen, S.E.; Ekdahl, C.T.; Kokaia, Z.; et al. Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 2009, 57, 835–849. [Google Scholar] [CrossRef]
- Wang, J.; Xie, L.; Yang, C.; Ren, C.; Zhou, K.; Wang, B.; Zhang, Z.; Wang, Y.; Jin, K.; Yang, G.Y. Activated regulatory T cell regulates neural stem cell proliferation in the subventricular zone of normal and ischemic mouse brain through interleukin 10. Front. Cell Neurosci. 2015, 9, 361. [Google Scholar] [CrossRef]
- Nakano-Doi, A.; Nakagomi, T.; Fujikawa, M.; Nakagomi, N.; Kubo, S.; Lu, S.; Yoshikawa, H.; Soma, T.; Taguchi, A.; Matsuyama, T. Bone marrow mononuclear cells promote proliferation of endogenous neural stem cells through vascular niches after cerebral infarction. Stem Cells 2010, 28, 1292–1302. [Google Scholar] [CrossRef] [PubMed]
- Borlongan, C.V. Concise Review: Stem Cell Therapy for Stroke Patients: Are We There Yet? Stem Cells Transl. Med. 2019, 8, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Shichita, T.; Ito, M.; Yoshimura, A. Post-ischemic inflammation regulates neural damage and protection. Front. Cell Neurosci. 2014, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Di Raimondo, D.; Pecoraro, R.; Casuccio, A.; Di Bona, D.; Aiello, A.; Accardi, G.; Arnao, V.; Clemente, G.; Corte, V.D.; et al. HLA and killer cell immunoglobulin-like receptor (KIRs) genotyping in patients with acute ischemic stroke. J. Neuroinflamm. 2019, 16, 88. [Google Scholar] [CrossRef] [Green Version]
- Gong, G.; Xiang, L.; Yuan, L.; Hu, L.; Wu, W.; Cai, L.; Yin, L.; Dong, H. Protective Effect of Glycyrrhizin, a Direct HMGB1 Inhibitor, on Focal Cerebral Ischemia/Reperfusion-Induced Inflammation, Oxidative Stress, and Apoptosis in Rats. PLoS ONE 2014, 9, e89450. [Google Scholar] [CrossRef] [Green Version]
- Shichita, T.; Ito, M.; Morita, R.; Komai, K.; Noguchi, Y.; Ooboshi, H.; Koshida, R.; Takahashi, S.; Kodama, T.; Yoshimura, A. MAFB prevents excess inflammation after ischemic stroke by accelerating clearance of damage signals through MSR1. Nat. Med. 2017, 23, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Lampl, Y.; Boaz, M.; Gilad, R.; Lorberboym, M.; Dabby, R.; Rapoport, A.; Anca-Hershkowitz, M.; Sadeh, M. Minocycline treatment in acute stroke: An open-label, evaluator-blinded study. Neurology 2007, 69, 1404–1410. [Google Scholar] [CrossRef]
- Padma Srivastava, M.; Bhasin, A.; Bhatia, R.; Garg, A.; Gaikwad, S.; Prasad, K.; Singh, M.; Tripathi, M. Efficacy of minocycline in acute ischemic stroke: A single-blinded, placebo-controlled trial. Neurol. India 2012, 60, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Amantea, D. Polarizing the immune system towards neuroprotection in brain ischemia. Neural. Regen. Res. 2016, 11, 81–82. [Google Scholar] [CrossRef]
- Wang, J.; Xing, H.; Wan, L.; Jiang, X.; Wang, C.; Wu, Y. Treatment targets for M2 microglia polarization in ischemic stroke. Biomed. Pharmacother. 2018, 105, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.P.; Zhao, Y.; Qin, X.Y.; Wan, L.Y.; Fan, X.X. Non-invasive Vagus Nerve Stimulation Protects Against Cerebral Ischemia/Reperfusion Injury and Promotes Microglial M2 Polarization Via Interleukin-17A Inhibition. J. Mol. Neurosci. MN 2019, 67, 217–226. [Google Scholar] [CrossRef]
- Wei, Y.; Yemisci, M.; Kim, H.-H.; Yung, L.M.; Shin, H.K.; Hwang, S.-K.; Guo, S.; Qin, T.; Alsharif, N.; Brinkmann, V.; et al. Fingolimod provides long-term protection in rodent models of cerebral ischemia. Ann. Neurol. 2011, 69, 119–129. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, S.; Nitsch, R. The role of T helper cells in neuroprotection and regeneration. J. Neuroimmunol. 2007, 184, 100–112. [Google Scholar] [CrossRef]
- Gee, J.M.; Kalil, A.; Shea, C.; Becker, K.J. Lymphocytes. Stroke 2007, 38, 783–788. [Google Scholar] [CrossRef]
- Becker, K.J.; Kindrick, D.L.; Lester, M.P.; Shea, C.; Ye, Z.-C. Sensitization to brain antigens after stroke is augmented by lipopolysaccharide. J. Cereb. Blood Flow Metab. 2005, 25, 1634–1644. [Google Scholar] [CrossRef] [Green Version]
- Ayer, R.E.; Ostrowski, R.P.; Sugawara, T.; Ma, Q.; Jafarian, N.; Tang, J.; Zhang, J.H. Statin-induced T-lymphocyte modulation and neuroprotection following experimental subarachnoid hemorrhage. Acta Neurochir. Suppl. 2013, 115, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.J.; Hulme, S.; Vail, A.; Heal, C.; Parry-Jones, A.R.; Scarth, S.; Hopkins, K.; Hoadley, M.; Allan, S.M.; Rothwell, N.J.; et al. SCIL-STROKE (Subcutaneous Interleukin-1 Receptor Antagonist in Ischemic Stroke): A Randomized Controlled Phase 2 Trial. Stroke 2018, 49, 1210–1216. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, S.R.; Guresir, E.; Vatter, H.; Kinfe, T.M.; Dietrich, D.; Lamprecht, A.; Muhammad, S. Aneurysmal subarachnoid hemorrhage lead to systemic upregulation of IL-23/IL-17 inflammatory axis. Cytokine 2017, 97, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Konoeda, F.; Shichita, T.; Yoshida, H.; Sugiyama, Y.; Muto, G.; Hasegawa, E.; Morita, R.; Suzuki, N.; Yoshimura, A. Therapeutic effect of IL-12/23 and their signaling pathway blockade on brain ischemia model. Biochem. Biophys. Res. Commun. 2010, 402, 500–506. [Google Scholar] [CrossRef]
- Chio, C.C.; Lin, J.W.; Chang, M.W.; Wang, C.C.; Kuo, J.R.; Yang, C.Z.; Chang, C.P. Therapeutic evaluation of etanercept in a model of traumatic brain injury. J. Neurochem. 2010, 115, 921–929. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muhammad, S.; Chaudhry, S.R.; Kahlert, U.D.; Niemelä, M.; Hänggi, D. Brain Immune Interactions—Novel Emerging Options to Treat Acute Ischemic Brain Injury. Cells 2021, 10, 2429. https://doi.org/10.3390/cells10092429
Muhammad S, Chaudhry SR, Kahlert UD, Niemelä M, Hänggi D. Brain Immune Interactions—Novel Emerging Options to Treat Acute Ischemic Brain Injury. Cells. 2021; 10(9):2429. https://doi.org/10.3390/cells10092429
Chicago/Turabian StyleMuhammad, Sajjad, Shafqat Rasul Chaudhry, Ulf Dietrich Kahlert, Mika Niemelä, and Daniel Hänggi. 2021. "Brain Immune Interactions—Novel Emerging Options to Treat Acute Ischemic Brain Injury" Cells 10, no. 9: 2429. https://doi.org/10.3390/cells10092429
APA StyleMuhammad, S., Chaudhry, S. R., Kahlert, U. D., Niemelä, M., & Hänggi, D. (2021). Brain Immune Interactions—Novel Emerging Options to Treat Acute Ischemic Brain Injury. Cells, 10(9), 2429. https://doi.org/10.3390/cells10092429