
Muscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Presentation of Bone Disease in Cystinosis
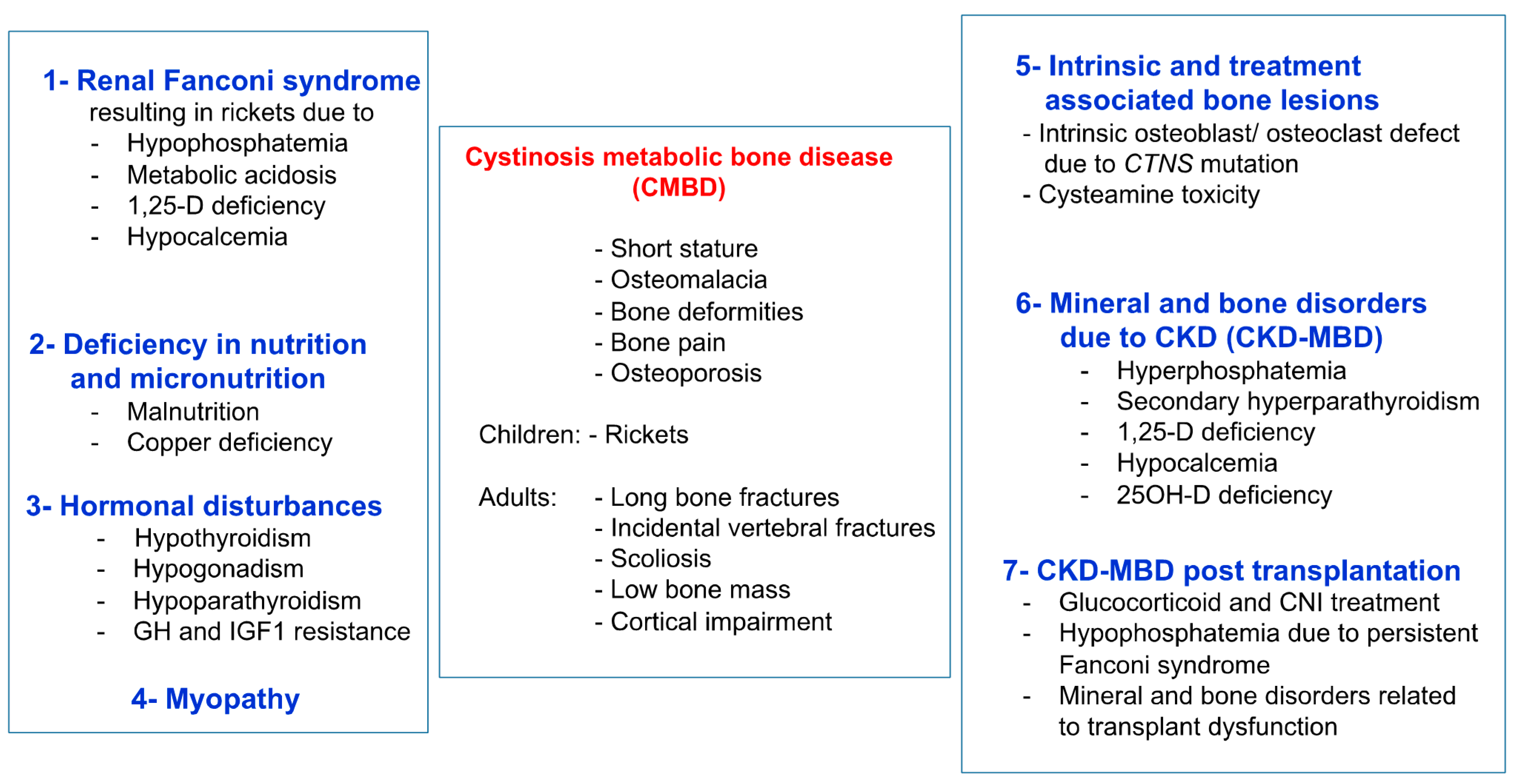
3. Cystinosis Metabolic Bone Disease
3.1. Fanconi Syndrome
3.2. Deficiency in Nutrition and Micronutrition
3.3. Hormonal Disturbances
3.4. Myopathy
3.5. Mineral and Bone Disorders Due to CKD (CKD-MBD)
3.6. CKD-MBD Post Kidney Transplantation
3.7. Intrinsic Bone Defect
3.8. Cysteamine Toxicity
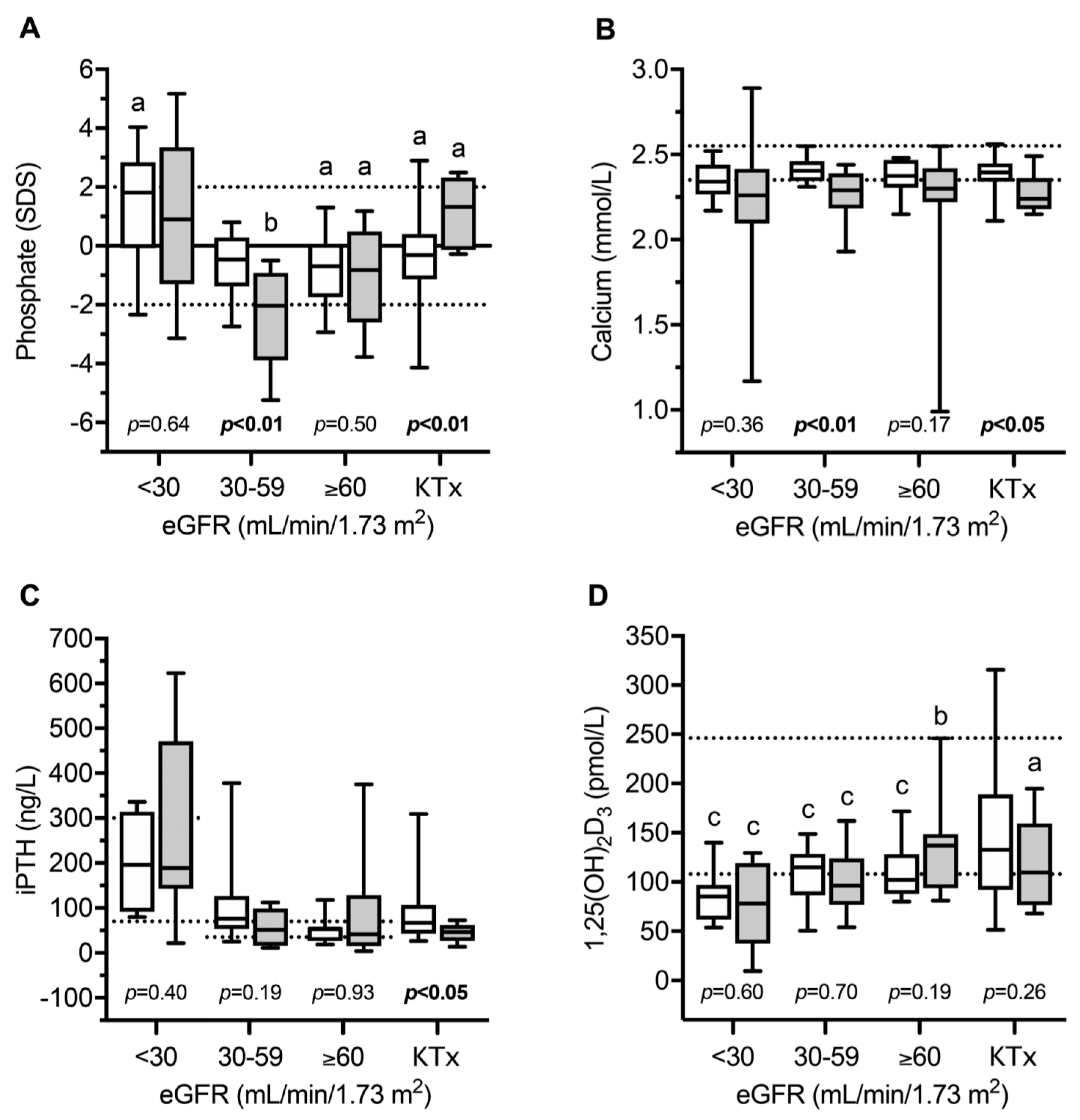
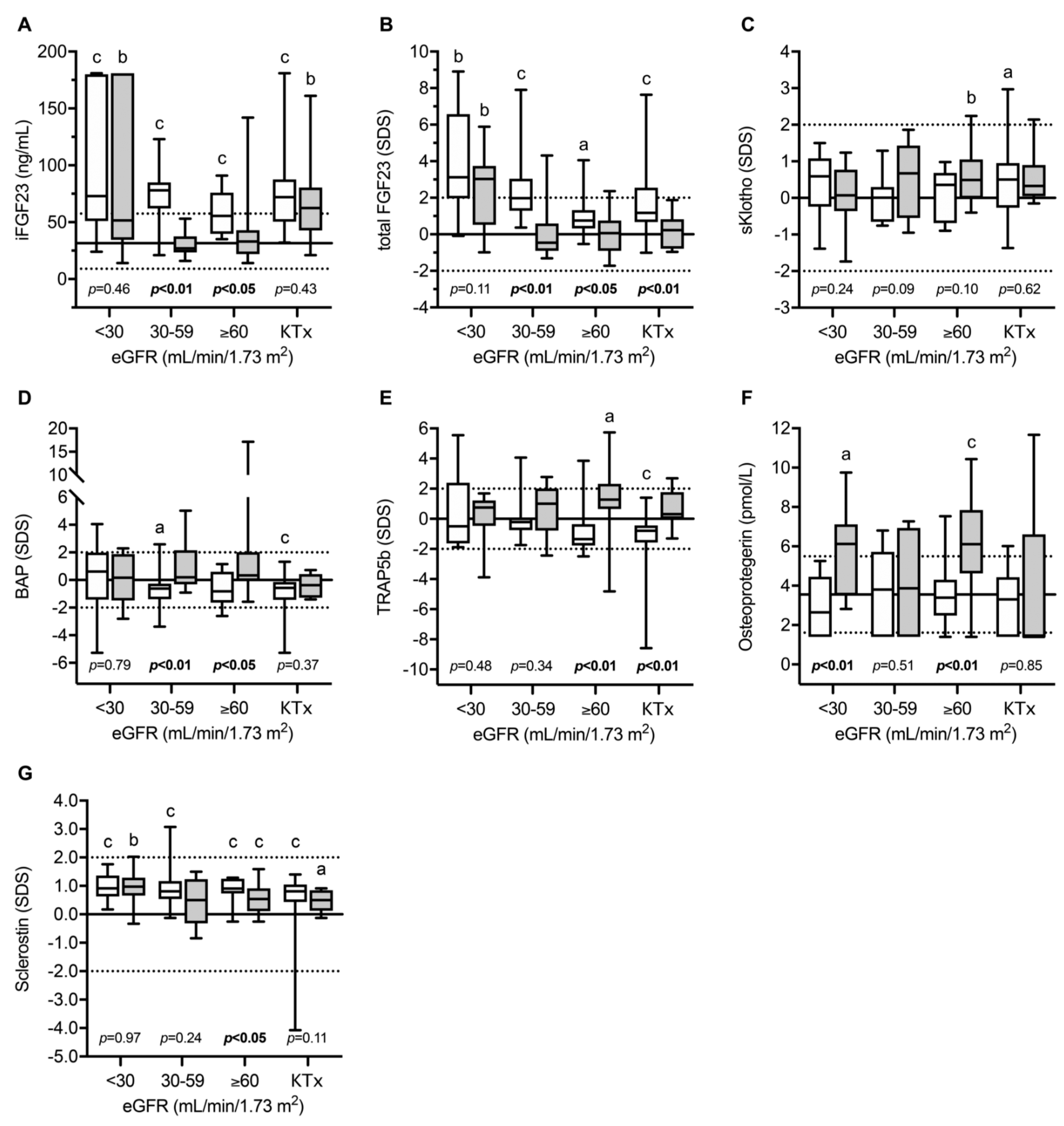
4. Bone and Mineral Metabolism in INC Patients
5. Towards an Interplay between Bone, Adipocytes, and Muscles
6. Conclusions: Perspectives in Research
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Callen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef]
- Markello, T.C.; Bernardini, I.M.; Gahl, W.A. Improved renal function in children with cystinosis treated with cysteamine. N. Engl. J. Med. 1993, 328, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Machuca-Gayet, I.; Quinaux, T.; Bertholet-Thomas, A.; Gaillard, S.; Claramunt-Taberner, D.; Acquaviva-Bourdain, C.; Bacchetta, J. Bone Disease in Nephropathic Cystinosis: Beyond Renal Osteodystrophy. Int. J. Mol. Sci. 2020, 21, 3109. [Google Scholar] [CrossRef]
- Bacchetta, J.; Greco, M.; Bertholet-Thomas, A.; Nobili, F.; Zustin, J.; Cochat, P.; Emma, F.; Boivin, G. Skeletal implications and management of cystinosis: Three case reports and literature review. BoneKEy Rep. 2016, 5, 828. [Google Scholar] [CrossRef] [Green Version]
- Bertholet-Thomas, A.; Claramunt-Taberner, D.; Gaillard, S.; Deschênes, G.; Sornay-Rendu, E.; Szulc, P.; Cohen-Solal, M.; Pelletier, S.; Carlier, M.C.; Cochat, P.; et al. Teenagers and young adults with nephropathic cystinosis display significant bone disease and cortical impairment. Pediatr. Nephrol. 2018, 33, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, P.; Ferreira, C.; Nesterova, G.; Roberts, M.S.; Tella, S.H.; de Castro, L.F.; Brown, S.M.; Whitaker, A.; Pereira, R.C.; Bulas, D.; et al. Skeletal Consequences of Nephropathic Cystinosis. J. Bone Miner. Res. 2018, 33, 1870–1880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langman, C.B. Bone Complications of Cystinosis. J. Pediatr. 2017, 183S, S2–S4. [Google Scholar] [CrossRef]
- Langman, C.B. Oh cystinosin: Let me count the ways! Kidney Int. 2019, 96, 275–277. [Google Scholar] [CrossRef]
- Santos, F.; Díaz-Anadón, L.; Ordóñez, F.A.; Haffner, D. Bone Disease in CKD in Children. Calcif. Tissue Int. 2021, 108, 423–438. [Google Scholar] [CrossRef]
- Claramunt-Taberner, D.; Flammier, S.; Gaillard, S.; Cochat, P.; Peyruchaud, O.; Machuca-Gayet, I.; Bacchetta, J. Bone disease in nephropathic cystinosis is related to cystinosin-induced osteoclastic dysfunction. Nephrol. Dial. Transpl. 2018, 33, 1525–1532. [Google Scholar] [CrossRef]
- Battafarano, G.; Rossi, M.; Rega, L.R.; Di Giovamberardino, G.; Pastore, A.; D’Agostini, M.; Porzio, O.; Nevo, N.; Emma, F.; Taranta, A.; et al. Intrinsic Bone Defects in Cystinotic Mice. Am. J. Pathol. 2019, 189, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Ewert, A.; Leifheit-Nestler, M.; Hohenfellner, K.; Büscher, A.; Kemper, M.J.; Oh, J.; Billing, H.; Thumfart, J.; Stangl, G.; Baur, A.C.; et al. Bone and mineral metabolism in children with nephropathic cystinosis compared to other CKD entities. J. Clin. Endocrinol. Metab. 2020, 105, e2738–e2752. [Google Scholar] [CrossRef] [PubMed]
- Florenzano, P.; Jimenez, M.; Ferreira, C.R.; Nesterova, G.; Roberts, M.S.; Tella, S.H.; Fernandez de Castro, L.; Gafni, R.I.; Wolf, M.; Jüppner, H.; et al. Nephropathic Cystinosis: A Distinct Form of CKD-Mineral and Bone Disorder that Provides Novel Insights into the Regulation of FGF23. J. Am. Soc. Nephrol. JASN 2020, 31, 2184–2192. [Google Scholar] [CrossRef]
- Quinaux, T.; Bertholet-Thomas, A.; Servais, A.; Boyer, O.; Vrillon, I.; Hogan, J.; Lemoine, S.; Gaillard, S.; Alioli, C.; Vasseur, S.; et al. Response to Cysteamine in Osteoclasts Obtained from Patients with Nephropathic Cystinosis: A Genotype/Phenotype Correlation. Cells 2021, 10, 2498. [Google Scholar] [CrossRef]
- Gonzalez, A.; Cheung, W.W.; Perens, E.A.; Oliveira, E.A.; Gertler, A.; Mak, R.H. A Leptin Receptor Antagonist Attenuates Adipose Tissue Browning and Muscle Wasting in Infantile Nephropathic Cystinosis-Associated Cachexia. Cells 2021, 10, 1954. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.W.; Cherqui, S.; Ding, W.; Esparza, M.; Zhou, P.; Shao, J.; Lieber, R.L.; Mak, R.H. Muscle wasting and adipose tissue browning in infantile nephropathic cystinosis. J. Cachexia Sarcopenia Muscle 2016, 7, 152–164. [Google Scholar] [CrossRef]
- Hohenfellner, K.; Rauch, F.; Ariceta, G.; Awan, A.; Bacchetta, J.; Bergmann, C.; Bechtold, S.; Cassidy, N.; Deschenes, G.; Elenberg, E.; et al. Management of bone disease in cystinosis: Statement from an international conference. J. Inherit. Metab. Dis. 2019, 42, 1019–1029. [Google Scholar] [CrossRef] [Green Version]
- Bertholet-Thomas, A.; Bacchetta, J.; Tasic, V.; Cochat, P. Nephropathic cystinosis--a gap between developing and developed nations. N. Engl. J. Med. 2014, 370, 1366–1367. [Google Scholar] [CrossRef]
- Nesterova, G.; Gahl, W.A. Cystinosis: The evolution of a treatable disease. Pediatr. Nephrol. 2013, 28, 51–59. [Google Scholar] [CrossRef] [Green Version]
- Besouw, M.T.P.; Bowker, R.; Dutertre, J.P.; Emma, F.; Gahl, W.A.; Greco, M.; Lilien, M.R.; McKiernan, J.; Nobili, F.; Schneider, J.A.; et al. Cysteamine toxicity in patients with cystinosis. J. Pediatr. 2011, 159, 1004–1011. [Google Scholar] [CrossRef] [PubMed]
- Drube, J.; Wan, M.; Bonthuis, M.; Wühl, E.; Bacchetta, J.; Santos, F.; Grenda, R.; Edefonti, A.; Harambat, J.; Shroff, R.; et al. Clinical practice recommendations for growth hormone treatment in children with chronic kidney disease. Nat. Rev. Nephrol. 2019, 15, 577–589. [Google Scholar] [CrossRef] [Green Version]
- Tiosano, D.; Hochberg, Z. Hypophosphatemia: The common denominator of all rickets. J. Bone Miner. Metab. 2009, 27, 392–401. [Google Scholar] [CrossRef]
- Haffner, D.; Leifheit-Nestler, M.; Grund, A.; Schnabel, D. Rickets guidance: Part I-diagnostic workup. Pediatr. Nephrol. 2021. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Bagga, A.; Sinha, A. Renal Tubular Acidosis. Indian J. Pediatr. 2020, 87, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Besouw, M.T.P.; Van Dyck, M.; Cassiman, D.; Claes, K.J.; Levtchenko, E.N. Management dilemmas in pediatric nephrology: Cystinosis. Pediatr. Nephrol. 2015, 30, 1349–1360. [Google Scholar] [CrossRef]
- Besouw, M.T.P.; Schneider, J.; Janssen, M.C.; Greco, M.; Emma, F.; Cornelissen, E.A.; Desmet, K.; Skovby, F.; Nobili, F.; Lilien, M.R.; et al. Copper deficiency in patients with cystinosis with cysteamine toxicity. J. Pediatr. 2013, 163, 754–760. [Google Scholar] [CrossRef] [PubMed]
- Nesterova, G.; Gahl, W. Nephropathic cystinosis: Late complications of a multisystemic disease. Pediatr. Nephrol. 2008, 23, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Winkler, L.; Offner, G.; Krull, F.; Brodehl, J. Growth and pubertal development in nephropathic cystinosis. Eur. J. Pediatr. 1993, 152, 244–249. [Google Scholar] [CrossRef]
- Chik, C.L.; Friedman, A.; Merriam, G.R.; Gahl, W.A. Pituitary-testicular function in nephropathic cystinosis. Ann. Intern. Med. 1993, 119, 568–575. [Google Scholar] [CrossRef]
- Rohayem, J.; Haffner, D.; Cremers, J.F.; Huss, S.; Wistuba, J.; Weitzel, D.; Kliesch, S.; Hohenfellner, K. Testicular function in males with infantile nephropathic cystinosis. Hum. Reprod. 2021, 36, 1191–1204. [Google Scholar] [CrossRef]
- Wühl, E.; Haffner, D.; Offner, G.; Broyer, M.; van’t Hoff, W.; Mehls, O.; European Study Group on Growth Hormone Treatment in Children with Nephropathic Cystinosis. Long-term treatment with growth hormone in short children with nephropathic cystinosis. J. Pediatr. 2001, 138, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, B.; Cronin, M.J.; Reichert, M.; Haffner, D.; Wingen, A.M.; Blum, W.F.; Mehls, O. Reduced concentration of serum growth hormone (GH)-binding protein in children with chronic renal failure: Correlation with GH insensitivity. The European Study Group for Nutritional Treatment of Chronic Renal Failure in Childhood. The German Study Group for Growth Hormone Treatment in Chronic Renal Failure. J. Clin. Endocrinol. Metab. 1997, 82, 1007–1013. [Google Scholar] [PubMed] [Green Version]
- Vester, U.; Schubert, M.; Offner, G.; Brodehl, J. Distal myopathy in nephropathic cystinosis. Pediatr. Nephrol. 2000, 14, 36–38. [Google Scholar] [CrossRef]
- Sadjadi, R.; Sullivan, S.; Grant, N.; Thomas, S.E.; Doyle, M.; Hammond, C.; Duong, R.; Corre, C.; David, W.; Eichler, F. Clinical myopathy in patients with nephropathic cystinosis. Muscle Nerve 2020, 61, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Sonies, B.C.; Almajid, P.; Kleta, R.; Bernardini, I.; Gahl, W.A. Swallowing dysfunction in 101 patients with nephropathic cystinosis: Benefit of long-term cysteamine therapy. Medicine 2005, 84, 137–146. [Google Scholar] [CrossRef] [PubMed]
- Anikster, Y.; Lacbawan, F.; Brantly, M.; Gochuico, B.L.; Avila, N.A.; Travis, W.; Gahl, W.A. Pulmonary dysfunction in adults with nephropathic cystinosis. Chest 2001, 119, 394–401. [Google Scholar] [CrossRef] [Green Version]
- Charnas, L.R.; Luciano, C.A.; Dalakas, M.; Gilliatt, R.W.; Bernardini, I.; Ishak, K.; Cwik, V.A.; Fraker, D.; Brushart, T.A.; Gahl, W.A. Distal vacuolar myopathy in nephropathic cystinosis. Ann. Neurol. 1994, 35, 181–188. [Google Scholar] [CrossRef]
- Iyob-Tessema, H.; Wang, C.S.; Kennedy, S.; Reyes, L.; Shin, S.; Greenbaum, L.A.; Hogan, J. Grip Strength in Adults and Children with Cystinosis. Kidney Int. Rep. 2021, 6, 389–395. [Google Scholar] [CrossRef]
- Frost, H.M.; Schönau, E. The “muscle-bone unit” in children and adolescents: A 2000 overview. J. Pediatr. Endocrinol. Metab. JPEM 2000, 13, 571–590. [Google Scholar] [CrossRef]
- Graciolli, F.G.; Neves, K.R.; Barreto, F.; Barreto, D.V.; Dos Reis, L.M.; Canziani, M.E.; Sabbagh, Y.; Carvalho, A.B.; Jorgetti, V.; Elias, R.M.; et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney Int. 2017, 91, 1436–1446. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Ginsberg, C.; Seifert, M.; Agapova, O.; Sugatani, T.; Register, T.C.; Freedman, B.I.; Monier-Faugere, M.-C.; Malluche, H.; Hruska, K.A. CKD-induced wingless/integration1 inhibitors and phosphorus cause the CKD-mineral and bone disorder. J. Am. Soc. Nephrol. JASN 2014, 25, 1760–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-López, N.; Panizo, S.; Alonso-Montes, C.; Román-García, P.; Rodríguez, I.; Martínez-Salgado, C.; Dusso, A.S.; Naves, M.; Cannata-Andía, J.B. Direct inhibition of osteoblastic Wnt pathway by fibroblast growth factor 23 contributes to bone loss in chronic kidney disease. Kidney Int. 2016, 90, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattineni, J.; Alphonse, P.; Zhang, Q.; Mathews, N.; Bates, C.M.; Baum, M. Regulation of renal phosphate transport by FGF23 is mediated by FGFR1 and FGFR4. Am. J. Physiol. Renal Physiol. 2014, 306, F351–F358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, B.; Faul, C. FGF23 Actions on Target Tissues—With and without Klotho. Front. Endocrinol. 2018, 9, 189. [Google Scholar] [CrossRef]
- Robling, A.G.; Niziolek, P.J.; Baldridge, L.A.; Condon, K.W.; Allen, M.R.; Alam, I.; Mantila, S.M.; Gluhak-Heinrich, J.; Bellido, T.M.; Harris, S.E.; et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 2008, 283, 5866–5875. [Google Scholar] [CrossRef] [Green Version]
- Haffner, D.; Leifheit-Nestler, M. CKD-MBD post kidney transplantation. Pediatr. Nephrol. Berl. Ger. 2021, 36, 41–50. [Google Scholar] [CrossRef]
- Tönshoff, B. Immunosuppressive therapy post-transplantation in children: What the clinician needs to know. Expert Rev. Clin. Immunol. 2020, 16, 139–154. [Google Scholar] [CrossRef]
- Conforti, A.; Taranta, A.; Biagini, S.; Starc, N.; Pitisci, A.; Bellomo, F.; Cirillo, V.; Locatelli, F.; Bernardo, M.E.; Emma, F. Cysteamine treatment restores the in vitro ability to differentiate along the osteoblastic lineage of mesenchymal stromal cells isolated from bone marrow of a cystinotic patient. J. Transl. Med. 2015, 13, 143. [Google Scholar] [CrossRef] [Green Version]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; van de Hoek, G.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef]
- Cheung, W.W.; Hao, S.; Wang, Z.; Ding, W.; Zheng, R.; Gonzalez, A.; Zhan, J.-Y.; Zhou, P.; Li, S.; Esparza, M.C.; et al. Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia. J. Cachexia Sarcopenia Muscle 2020, 11, 120–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, J.S.; Hewison, M. Update in vitamin D. J. Clin. Endocrinol. Metab. 2010, 95, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prencipe, G.; Caiello, I.; Cherqui, S.; Whisenant, T.; Petrini, S.; Emma, F.; De Benedetti, F. Inflammasome activation by cystine crystals: Implications for the pathogenesis of cystinosis. J. Am. Soc. Nephrol. JASN 2014, 25, 1163–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozen, S. Update in familial Mediterranean fever. Curr. Opin. Rheumatol. 2021, 33, 398–402. [Google Scholar] [CrossRef]
- Cheung, W.W.; Hao, S.; Zheng, R.; Wang, Z.; Gonzalez, A.; Zhou, P.; Hoffman, H.M.; Mak, R.H. Targeting interleukin-1 for reversing fat browning and muscle wasting in infantile nephropathic cystinosis. J. Cachexia Sarcopenia Muscle 2021, 12, 1296–1311. [Google Scholar] [CrossRef]
- Feng, W.; Guo, J.; Li, M. RANKL-independent modulation of osteoclastogenesis. J. Oral Biosci. 2019, 61, 16–21. [Google Scholar] [CrossRef]
- Syres, K.; Harrison, F.; Tadlock, M.; Jester, J.V.; Simpson, J.; Roy, S.; Salomon, D.R.; Cherqui, S. Successful treatment of the murine model of cystinosis using bone marrow cell transplantation. Blood 2009, 114, 2542–2552. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haffner, D.; Leifheit-Nestler, M.; Alioli, C.; Bacchetta, J. Muscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts. Cells 2022, 11, 170. https://doi.org/10.3390/cells11010170
Haffner D, Leifheit-Nestler M, Alioli C, Bacchetta J. Muscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts. Cells. 2022; 11(1):170. https://doi.org/10.3390/cells11010170
Chicago/Turabian StyleHaffner, Dieter, Maren Leifheit-Nestler, Candide Alioli, and Justine Bacchetta. 2022. "Muscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts" Cells 11, no. 1: 170. https://doi.org/10.3390/cells11010170
APA StyleHaffner, D., Leifheit-Nestler, M., Alioli, C., & Bacchetta, J. (2022). Muscle and Bone Impairment in Infantile Nephropathic Cystinosis: New Concepts. Cells, 11(1), 170. https://doi.org/10.3390/cells11010170