
Pax-5 Protein Expression Is Regulated by Transcriptional 3′UTR Editing

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Models and Culturing Conditions
2.2. PCR, Cloning, and Quantitative RT-PCR
2.3. Reporter Gene Assays and Transfection
2.4. Polysomal Fractionation and Profiling
2.5. Data Mining and Processing
3. Results
3.1. Pax-5 3′UTR Mapping and Polyadenylation Signal Prediction
3.2. Pax-5 3′UTR Alternative Splicing in Primary B-Lymphocytes and Clinical Samples
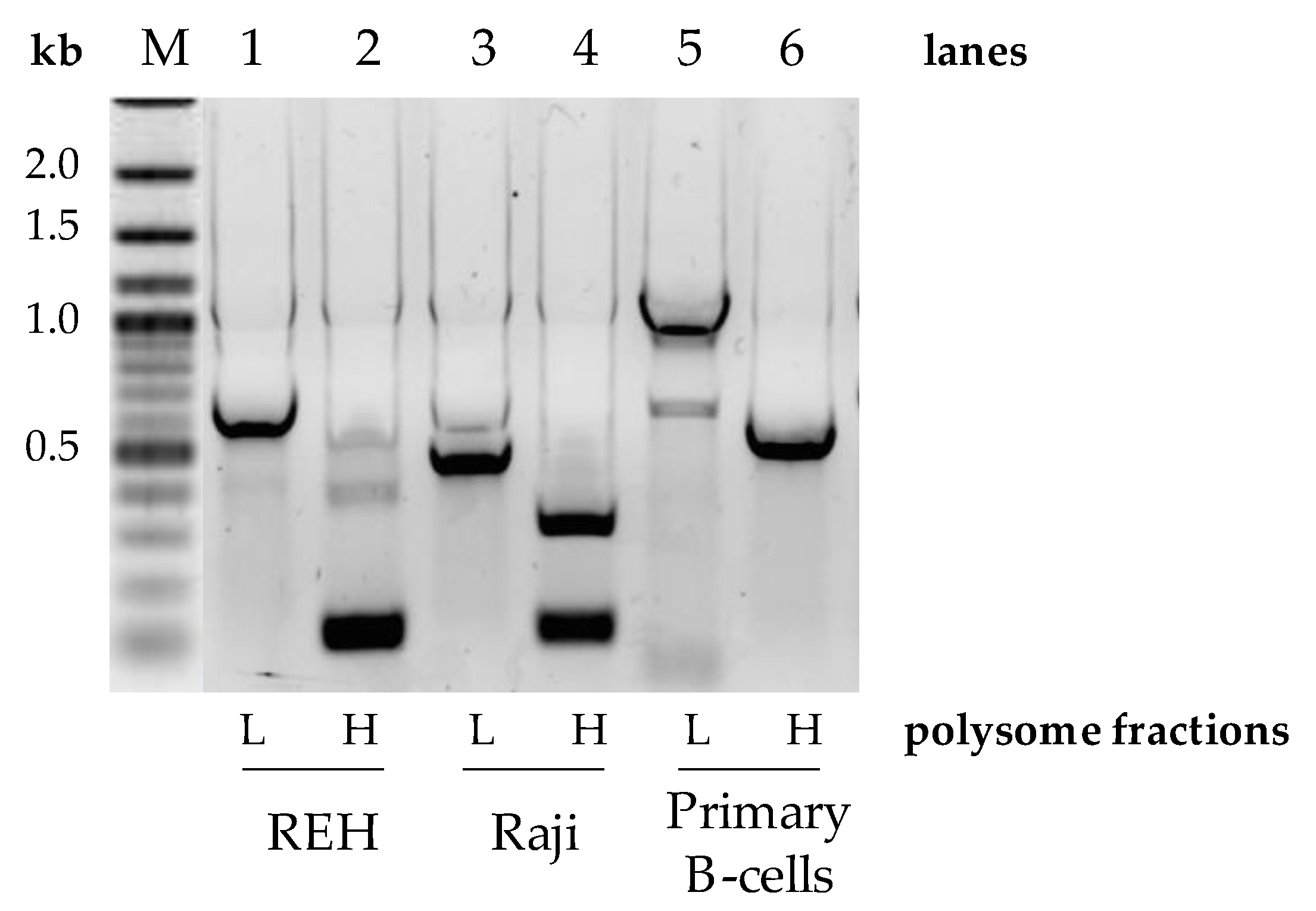
3.3. UTR Editing of the Pax-5 Transcript Affects Ribosomal Translation
3.4. Impact of 3′UTR Editing on Pax-5 Post-Transcriptional Regulation
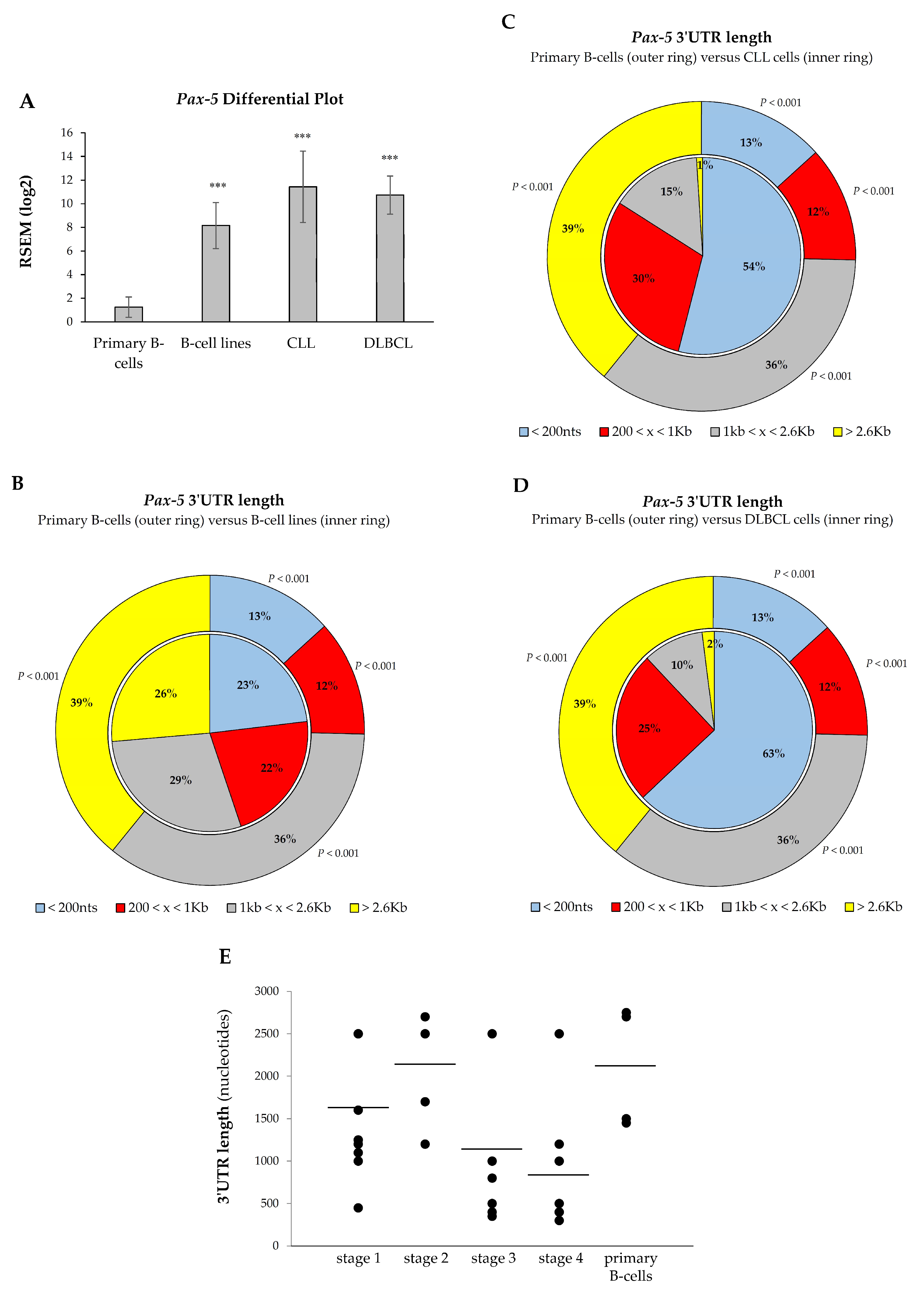
3.5. Shortening of Pax-5 3′UTR Is Prevalent in B-Cell Cancer Malignancies
3.6. Pax-5 3′UTR Shortening Relates with Hematopoietic Malignancy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Baralle, D.; Baralle, M. Splicing in action: Assessing disease causing sequence changes. J. Med. Genet. 2005, 42, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srebrow, A.; Kornblihtt, A.R. The connection between splicing and cancer. J. Cell Sci. 2006, 119, 2635–2641. [Google Scholar] [CrossRef] [Green Version]
- Barberis, A.; Widenhorn, K.; Vitelli, L.; Busslinger, M. A novel B-cell lineage-specific transcription factor present at early but not late stages of differentiation. Genes Dev. 1990, 4, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Nutt, S.L.; Heavey, B.; Rolink, A.G.; Busslinger, M. Commitment to the b-lymphoid lineage depends on the transcription factor pax5. Nature 1999, 401, 556–562. [Google Scholar] [CrossRef] [PubMed]
- Morrison, A.M.; Nutt, S.L.; Thévenin, C.; Rolink, A.; Busslinger, M. Loss- and gain-of-function mutations reveal an important role of bsap (Pax-5) at the start and end of b cell differentiation. Semin. Immunol. 1998, 10, 133–142. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, P.; Morin, P.; Ouellette, R.; Robichaud, G. The Pax-5 gene: A pluripotent regulator of B-cell differentiation and cancer disease. Cancer Res. 2011, 71, 7345–7350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwollo, P.; Arrieta, H.; Ede, K.; Molinder, K.; Desiderio, S.; Pollock, R. The Pax-5 gene is alternatively spliced during B-cell development. J. Biol. Chem. 1997, 272, 10160–10168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robichaud, G.A.; Nardini, M.; Laflamme, M.; Cuperlovic-Culf, M.; Ouellette, R.J. Human Pax-5 C-terminal isoforms possess distinct transactivation properties and are differentially modulated in normal and malignant B cells. J. Biol. Chem 2004, 279, 49956–49963. [Google Scholar] [CrossRef] [Green Version]
- Robichaud, G.A.; Perreault, J.P.; Ouellette, R.J. Development of an isoform-specific gene suppression system: The study of the human Pax-5b transcriptional element. Nucleic Acids Res. 2008, 36, 4609–4620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.L.; Rahman, M.; Hirabayashi, Y.; Sasaki, T. Sequence analysis of 5′-flanking region of human Pax-5 gene exon 1b. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2002, 10, 100–103. [Google Scholar]
- Busslinger, M.; Klix, N.; Pfeffer, P.; Graninger, P.G.; Kozmik, Z. Deregulation of Pax-5 by translocation of the emu enhancer of the igh locus adjacent to two alternative Pax-5 promoters in a diffuse large-cell lymphoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6129–6134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cazzaniga, G.; Daniotti, M.; Tosi, S.; Giudici, G.; Aloisi, A.; Pogliani, E.; Kearney, L.; Biondi, A. The paired box domain gene pax5 is fused to etv6/tel in an acute lymphoblastic leukemia case. Cancer Res. 2001, 61, 4666–4670. [Google Scholar] [PubMed]
- Nebral, K.; Konig, M.; Harder, L.; Siebert, R.; Haas, O.A.; Strehl, S. Identification of pml as novel pax5 fusion partner in childhood acute lymphoblastic leukaemia. Br. J. Haematol. 2007, 139, 269–274. [Google Scholar] [CrossRef]
- Poppe, B.; De Paepe, P.; Michaux, L.; Dastugue, N.; Bastard, C.; Herens, C.; Moreau, E.; Cavazzini, F.; Yigit, N.; Van Limbergen, H.; et al. Pax5/igh rearrangement is a recurrent finding in a subset of aggressive b-nhl with complex chromosomal rearrangements. Genes Chromosom. Cancer 2005, 44, 218–223. [Google Scholar] [CrossRef]
- Kanteti, R.; Nallasura, V.; Loganathan, S.; Tretiakova, M.; Kroll, T.; Krishnaswamy, S.; Faoro, L.; Cagle, P.; Husain, A.; Vokes, E.; et al. Pax5 is expressed in small-cell lung cancer and positively regulates c-met transcription. Lab. Investig. A J. Tech. Methods Pathol. 2009, 89, 301–314. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, N.; Harquail, J.; Crapoulet, N.; Ouellette, R.J.; Robichaud, G.A. Pax-5 inhibits breast cancer proliferation through miR-215 up-regulation. Anticancer Res. 2018, 38, 5013–5026. [Google Scholar] [CrossRef] [Green Version]
- Harquail, J.; LeBlanc, N.; Landry, C.; Crapoulet, N.; Robichaud, G.A. Pax-5 inhibits Nf-κB activity in breast cancer cells through IKKε and mirna-155 effectors. J. Mammary Gland Biol. Neoplasia 2018, 23, 177–187. [Google Scholar] [CrossRef]
- Harquail, J.; LeBlanc, N.; Ouellette, R.J.; Robichaud, G.A. Mirnas 484 and 210 regulate Pax-5 expression and function in breast cancer cells. Carcinogenesis 2019, 40, 1010–1020. [Google Scholar] [CrossRef]
- Sachs, A. The role of poly(a) in the translation and stability of mRNA. Curr. Opin. Cell Biol. 1990, 2, 1092–1098. [Google Scholar] [CrossRef]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by micrornas. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Andreassi, C.; Riccio, A. To localize or not to localize: MRNA fate is in 3′utr ends. Trends Cell Biol. 2009, 19, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Hu, J.; Zhang, H.; Lutz, C. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Res. 2005, 33, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.; Nishida, K.; Manley, J. Mechanisms and consequences of alternative polyadenylation. Mol. Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [Green Version]
- Sandberg, R.; Neilson, J.R.; Sarma, A.; Sharp, P.A.; Burge, C.B. Proliferating cells express mRNAs with shortened 3′ untranslated regions and fewer microrna target sites. Science 2008, 320, 1643–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, C.; Bartel, D. Widespread shortening of 3′utrs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzina, S.; Harquail, J.; Jean, S.; Beauregard, A.P.; Colquhoun, C.D.; Carroll, M.; Bos, A.; Gray, C.A.; Robichaud, G.A. Deoxypodophyllotoxin isolated from juniperus communis induces apoptosis in breast cancer cells. Anticancer Agents Med. Chem. 2015, 15, 79–88. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative pcr and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Esposito, A.M.; Mateyak, M.; He, D.; Lewis, M.; Sasikumar, A.N.; Hutton, J.; Copeland, P.R.; Kinzy, T.G. Eukaryotic polyribosome profile analysis. J. Vis. Exp. 2010, 40, 1948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushwaha, G.; Dozmorov, M.; Wren, J.D.; Qiu, J.; Shi, H.; Xu, D. Hypomethylation coordinates antagonistically with hypermethylation in cancer development: A case study of leukemia. Hum. Genom. 2016, 10 (Suppl. 2), 18. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Hiatt, J.B.; Nguyen, D.K.; Ercan, S.; Sturgill, D.; Hillier, L.W.; Schlesinger, F.; Davis, C.A.; Reinke, V.J.; Gingeras, T.R.; et al. Evidence for compensatory upregulation of expressed x-linked genes in mammals, caenorhabditis elegans and drosophila melanogaster. Nat. Genet. 2011, 43, 1179–1185. [Google Scholar] [CrossRef] [Green Version]
- Raju, S.; Kretzmer, L.Z.; Koues, O.I.; Payton, J.E.; Oltz, E.M.; Cashen, A.; Polic, B.; Schreiber, R.D.; Shaw, A.S.; Markiewicz, M.A. Nkg2d-nkg2d ligand interaction inhibits the outgrowth of naturally arising low-grade b cell lymphoma in vivo. J. Immunol. 2016, 196, 4805–4813. [Google Scholar] [CrossRef] [Green Version]
- Koues, O.I.; Kowalewski, R.A.; Chang, L.W.; Pyfrom, S.C.; Schmidt, J.A.; Luo, H.; Sandoval, L.E.; Hughes, T.B.; Bednarski, J.J.; Cashen, A.F.; et al. Enhancer sequence variants and transcription-factor deregulation synergize to construct pathogenic regulatory circuits in B-cell lymphoma. Immunity 2015, 42, 186–198. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. Tophat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.; Gao, G. Cpc2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef]
- Lin, M.F.; Jungreis, I.; Kellis, M. Phylocsf: A comparative genomics method to distinguish protein coding and non-coding regions. Bioinformatics 2011, 27, i275–i282. [Google Scholar] [CrossRef]
- Liu, H.; Han, H.; Li, J.; Wong, L. Dnafsminer: A web-based software toolbox to recognize two types of functional sites in DNA sequences. Bioinformatics 2005, 21, 671–673. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Kumar, M.; Raghava, G.P. Prediction of polyadenylation signals in human DNA sequences using nucleotide frequencies. In Silico Biol. 2009, 9, 135–148. [Google Scholar] [CrossRef]
- Salamov, A.A.; Solovyev, V.V. Recognition of 3′-processing sites of human mRNA precursors. Comput. Appl. Biosci. 1997, 13, 23–28. [Google Scholar] [CrossRef]
- Chang, T.H.; Huang, H.Y.; Hsu, J.B.; Weng, S.L.; Horng, J.T.; Huang, H.D. An enhanced computational platform for investigating the roles of regulatory RNA and for identifying functional RNA motifs. BMC Bioinform. 2013, 14 (Suppl. 2), S4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakheet, T.; Williams, B.R.; Khabar, K.S. Ared 3.0: The large and diverse au-rich transcriptome. Nucleic Acids Res. 2006, 34, D111–D114. [Google Scholar] [CrossRef] [PubMed]
- Betel, D.; Wilson, M.; Gabow, A.; Marks, D.S.; Sander, C. The microrna.Org resource: Targets and expression. Nucleic Acids Res. 2008, 36, D149–D153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Wang, X. Mirdb: An online database for prediction of functional microrna targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microrna target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, M.; Li, H.; Wang, S.; Tang, S.; Kong, R.M.; Yu, R. Ultra-sensitive label-free electrochemical detection of the acute leukaemia gene Pax-5a based on enzyme-assisted cycle amplification. Biosens. Bioelectron. 2019, 143, 111593. [Google Scholar] [CrossRef] [PubMed]
- Torlakovic, E.; Slipicevic, A.; Robinson, C.; DeCoteau, J.F.; Alfsen, G.C.; Vyberg, M.; Chibbar, R.; Flørenes, V.A. Pax-5 expression in nonhematopoietic tissues. Am. J. Clin. Pathol. 2006, 126, 798–804. [Google Scholar] [CrossRef] [PubMed]
- Krenacs, L.; Himmelmann, A.; Quintanilla-Martinez, L.; Fest, T.; Riva, A.; Wellmann, A.; Bagdi, E.; Kehrl, J.; Jaffe, E.; Raffeld, M. Transcription factor B-cell-specific activator protein (bsap) is differentially expressed in b cells and in subsets of B-cell lymphomas. Blood 1998, 92, 1308–1316. [Google Scholar] [CrossRef] [PubMed]
- Cobaleda, C.; Schebesta, A.; Delogu, A.; Busslinger, M. Pax5: The guardian of b cell identity and function. Nat. Immunol. 2007, 8, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Powell, S.K.; Plummer, R.S.; Young, K.P.; Ruggeri, B.A. Pax genes: Roles in development, pathophysiology, and cancer. Biochem. Pharmacol. 2007, 73, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Benzina, S.; Beauregard, A.-P.; Guerrette, R.; Jean, S.; Faye, M.D.; Laflamme, M.; Maïcas, E.; Crapoulet, N.; Ouellette, R.J.; Robichaud, G.A. Pax-5 is a potent regulator of E-cadherin and breast cancer malignant processes. Oncotarget 2017, 8, 12052–12066. [Google Scholar] [CrossRef] [Green Version]
- Vidal, L.; Perry, J.; Vouyovitch, C.; Pandey, V.; Brunet-Dunand, S.; Mertani, H.; Liu, D.-X.; Lobie, P. Pax5alpha enhances the epithelial behavior of human mammary carcinoma cells. Mol. Cancer Res. MCR 2010, 8, 444–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borson, N.D.; Lacy, M.Q.; Wettstein, P.J. Altered mRNA expression of pax5 and blimp-1 in b cells in multiple myeloma. Blood 2002, 100, 4629–4639. [Google Scholar] [CrossRef]
- Modrek, B.; Resch, A.; Grasso, C.; Lee, C. Genome-wide detection of alternative splicing in expressed sequences of human genes. Nucleic Acids Res. 2001, 29, 2850–2859. [Google Scholar] [CrossRef] [Green Version]
- Danckwardt, S.; Kaufmann, I.; Gentzel, M.; Foerstner, K.U.; Gantzert, A.S.; Gehring, N.H.; Neu-Yilik, G.; Bork, P.; Keller, W.; Wilm, M.; et al. Splicing factors stimulate polyadenylation via uses at non-canonical 3′ end formation signals. EMBO J. 2007, 26, 2658–2669. [Google Scholar] [CrossRef] [Green Version]
- Akman, B.H.; Can, T.; Erson-Bensan, A.E. Estrogen-induced upregulation and 3′-utr shortening of cdc6. Nucleic Acids Res. 2012, 40, 10679–10688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Leva, G.; Garofalo, M.; Croce, C.M. Micrornas in cancer. Annu. Rev. Pathol. 2014, 9, 287–314. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D. Micrornas: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, M.; Wang, Z.; Han, S.; Tang, X.; Ge, Y.; Zhou, L.; Zhou, C.; Yuan, Q.; Yang, M. Silencing of long noncoding RNA malat1 by miR-101 and miR-217 inhibits proliferation, migration, and invasion of esophageal squamous cell carcinoma cells. J. Biol. Chem. 2015, 290, 3925–3935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Li, D.; Zhang, W. Tumor suppressor role of miR-217 in human epithelial ovarian cancer by targeting igf1r. Oncol. Rep. 2016, 35, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Lai, X.; Yu, S.; Chen, S.; Ma, Y.; Zhang, Y.; Li, H.; Zhu, X.; Yao, L.; Zhang, J. Exosomal miR-221/222 enhances tamoxifen resistance in recipient er-positive breast cancer cells. Breast Cancer Res. Treat. 2014, 147, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Pena-Chilet, M.; Martinez, M.T.; Perez-Fidalgo, J.A.; Peiro-Chova, L.; Oltra, S.S.; Tormo, E.; Alonso-Yuste, E.; Martinez-Delgado, B.; Eroles, P.; Climent, J.; et al. Microrna profile in very young women with breast cancer. BMC Cancer 2014, 14, 529. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.Y.; Shyu, A.B. Au-rich elements: Characterization and importance in mRNA degradation. Trends Biochem. Sci. 1995, 20, 465–470. [Google Scholar] [CrossRef]
- Khabar, K.S. Hallmarks of cancer and AU-rich elements. Wiley Interdiscip. Rev. RNA 2017, 8, e1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by micrornas: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beauregard, A.-P.; Hannay, B.; Gharib, E.; Crapoulet, N.; Finn, N.; Guerrette, R.; Ouellet, A.; Robichaud, G.A. Pax-5 Protein Expression Is Regulated by Transcriptional 3′UTR Editing. Cells 2022, 11, 76. https://doi.org/10.3390/cells11010076
Beauregard A-P, Hannay B, Gharib E, Crapoulet N, Finn N, Guerrette R, Ouellet A, Robichaud GA. Pax-5 Protein Expression Is Regulated by Transcriptional 3′UTR Editing. Cells. 2022; 11(1):76. https://doi.org/10.3390/cells11010076
Chicago/Turabian StyleBeauregard, Annie-Pier, Brandon Hannay, Ehsan Gharib, Nicolas Crapoulet, Nicholas Finn, Roxann Guerrette, Amélie Ouellet, and Gilles A. Robichaud. 2022. "Pax-5 Protein Expression Is Regulated by Transcriptional 3′UTR Editing" Cells 11, no. 1: 76. https://doi.org/10.3390/cells11010076
APA StyleBeauregard, A. -P., Hannay, B., Gharib, E., Crapoulet, N., Finn, N., Guerrette, R., Ouellet, A., & Robichaud, G. A. (2022). Pax-5 Protein Expression Is Regulated by Transcriptional 3′UTR Editing. Cells, 11(1), 76. https://doi.org/10.3390/cells11010076