
GLP-1 Receptor Agonists in Neurodegeneration: Neurovascular Unit in the Spotlight

 , ,
, ,  and
and
Abstract
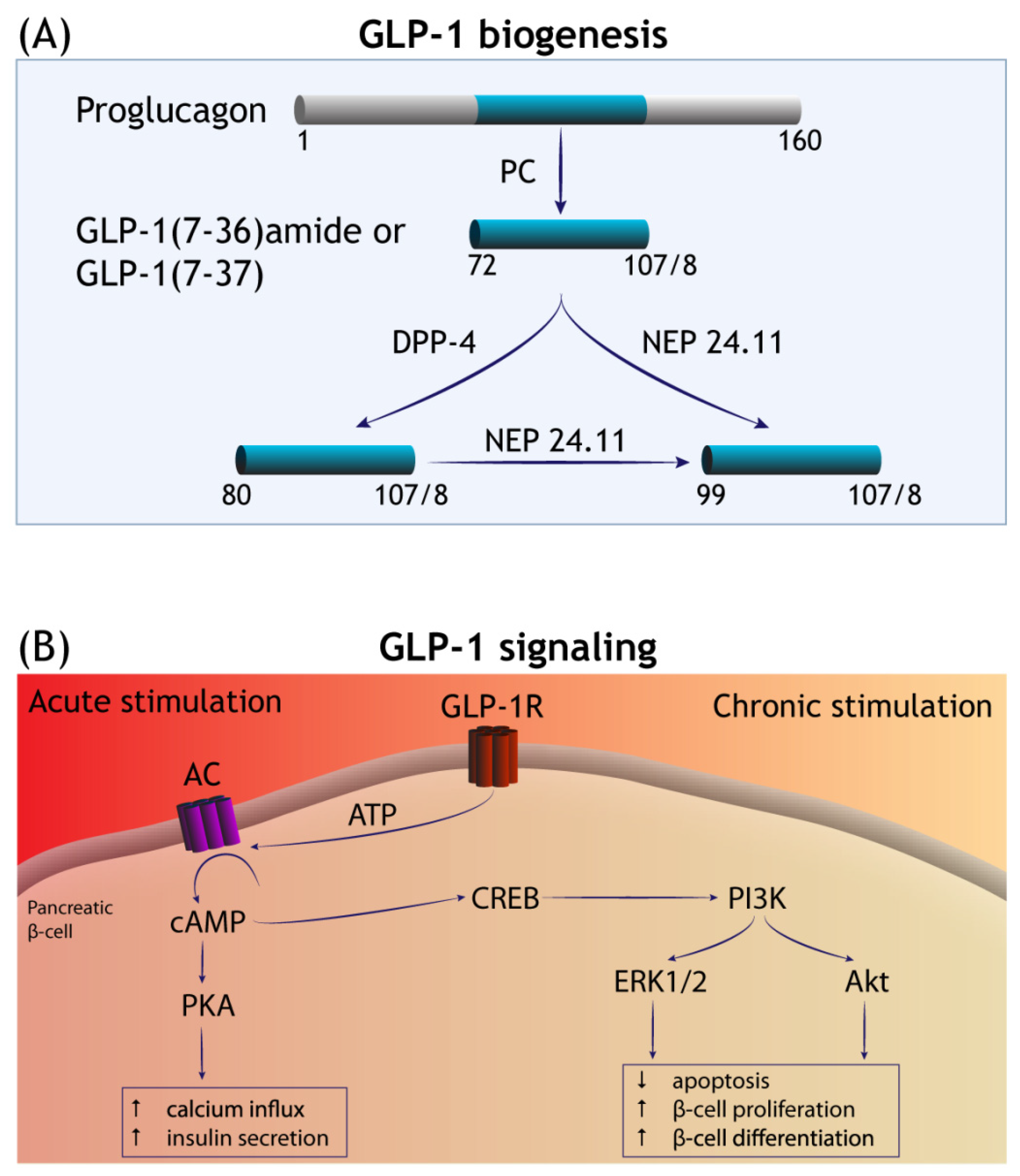
:1. Introduction
2. GLP-1RAs in Neurodegenerative Diseases
3. Neurovascular Unit (NVU) in Ageing and Neurodegeneration
3.1. Neurovascular Coupling and Brain Energy Metabolism
3.2. Structural Derangements of NVU in Ageing and Neurodegeneration
3.3. Functional Derangements of NVU in Ageing and Neurodegeneration
4. GLP-1RAs in Neurodegeneration: The Neurovascular Connection
5. Unresolved Questions and the Need for Novel Experimental Paradigms
5.1. Effects of GLP1-RAs at the NVU
5.2. Effects of GLP1-RAs in Periphery and Inter-Organ Communication
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Campbell, J.E.; Drucker, D.J. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metab. 2013, 17, 819–837. [Google Scholar] [CrossRef] [Green Version]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Meier, J.J. GLP-1 receptor agonists in the treatment of type 2 diabetes—State-of-the-art. Mol. Metab. 2021, 46, 101102. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Quast, D.R.; Wefers, J.; Pfeiffer, A.F.H. The evolving story of incretins (GIP and GLP-1) in metabolic and cardiovascular disease: A pathophysiological update. Diabetes Obes. Metab. 2021, 23 (Suppl. S3), 5–29. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, J.; Heng, J.; Newsholme, P.; Carlessi, R. Pleiotropic Effects of GLP-1 and Analogs on Cell Signaling, Metabolism, and Function. Front. Endocrinol. 2018, 9, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kieffer, T.J.; Francis Habener, J. The Glucagon-like Peptides. Endocr. Rev. 1999, 20, 876–913. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.M.; Morgan, L.M.; Tredger, J.A.; Deacon, S.; Wright, J.; Marks, V. Glucagon-like peptide-1 (7-36)amide and glucose-dependent insulinotropic polypeptide secretion in response to nutrient ingestion in man: Acute post-prandial and 24-h secretion patterns. J. Endocrinol. 1993, 138, 159–166. [Google Scholar] [CrossRef]
- Pujadas, G.; Drucker, D.J. Vascular Biology of Glucagon Receptor Superfamily Peptides: Mechanistic and Clinical Relevance. Endocr. Rev. 2016, 37, 554–583. [Google Scholar] [CrossRef] [Green Version]
- Orskov, C.; Rabenhoj, L.; Wettergren, A.; Kofod, H.; Holst, J.J. Tissue and plasma concentrations of amidated and glycine-extended glucagon-like peptide I in humans. Diabetes 1994, 43, 535–539. [Google Scholar] [CrossRef]
- Fehmann, H.C.; Habener, J.F. Functional receptors for the insulinotropic hormone glucagon-like peptide-I(7–37) on a somatostatin secreting cell line. FEBS Lett. 1991, 279, 335–340. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.; Martínez, M.D.; Roncero, I.; Chowen, J.A.; García-Cuartero, B.; Gispert, J.D.; Sanz, C.; Vázquez, P.; Maldonado, A.; De Cáceres, J.; et al. The expression of GLP-1 receptor mRNA and protein allows the effect of GLP-1 on glucose metabolism in the human hypothalamus and brainstem. J. Neurochem. 2005, 92, 798–806. [Google Scholar] [CrossRef]
- Farr, O.M.; Sofopoulos, M.; Tsoukas, M.A.; Dincer, F.; Thakkar, B.; Sahin-Efe, A.; Filippaios, A.; Bowers, J.; Srnka, A.; Gavrieli, A.; et al. GLP-1 receptors exist in the parietal cortex, hypothalamus and medulla of human brains and the GLP-1 analogue liraglutide alters brain activity related to highly desirable food cues in individuals with diabetes: A crossover, randomised, placebo-controlled trial. Diabetologia 2016, 59, 954–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holt, M.K.; Llewellyn-Smith, I.J.; Reimann, F.; Gribble, F.M.; Trapp, S. Serotonergic modulation of the activity of GLP-1 producing neurons in the nucleus of the solitary tract in mouse. Mol. Metab. 2017, 6, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Mojsov, S. Tissue-specific expression of the human receptor for glucagon-like peptide-I: Brain, heart and pancreatic forms have the same deduced amino acid sequences. FEBS Lett. 1995, 358, 219–224. [Google Scholar] [CrossRef] [Green Version]
- During, M.J.; Cao, L.; Zuzga, D.S.; Francis, J.S.; Fitzsimons, H.L.; Jiao, X.; Bland, R.J.; Klugmann, M.; Banks, W.A.; Drucker, D.J.; et al. Glucagon-like peptide-1 receptor is involved in learning and neuroprotection. Nat. Med. 2003, 9, 1173–1179. [Google Scholar] [CrossRef]
- Hamilton, A.; Holscher, C. Receptors for the insulin-like peptide GLP-1 are expressed on neurons in the CNS. Neuroreport 2009, 20, 1161–1166. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C. Chapter Thirteen—The Role of GLP-1 in Neuronal Activity and Neurodegeneration. In Vitamins & Hormones; Litwack, G., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 84, pp. 331–354. [Google Scholar]
- Perry, T.; Greig, N.H. Enhancing central nervous system endogenous GLP-1 receptor pathways for intervention in Alzheimer’s disease. Curr. Alzheimer Res. 2005, 2, 377–385. [Google Scholar] [CrossRef]
- Meloni, A.R.; De Young, M.B.; Lowe, C.; Parkes, D.G. GLP-1 receptor activated insulin secretion from pancreatic beta-cells: Mechanism and glucose dependence. Diabetes Obes. Metab. 2013, 15, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef] [Green Version]
- Cendrine, C.; Rémy, B. GLP-1, the Gut-Brain, and Brain-Periphery Axes. Rev. Diabet. Stud. 2011, 8, 418. [Google Scholar]
- Anderberg, R.H.; Richard, J.E.; Eerola, K.; López-Ferreras, L.; Banke, E.; Hansson, C.; Nissbrandt, H.; Berqquist, F.; Gribble, F.M.; Reimann, F.; et al. Glucagon-like Peptide 1 and Its Analogs Act in the Dorsal Raphe and Modulate Central Serotonin to Reduce Appetite and Body Weight. Diabetes 2017, 66, 1062–1073. [Google Scholar] [CrossRef] [Green Version]
- Holscher, C. Central effects of GLP-1: New opportunities for treatments of neurodegenerative diseases. J. Endocrinol. 2014, 221, T31–T41. [Google Scholar] [CrossRef] [Green Version]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Gentilella, R.; Pechtner, V.; Corcos, A.; Consoli, A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: Are they all the same? Diabetes Metab. Res. Rev. 2019, 35, e3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M.A.; Meier, J.J. Management Of Endocrine Disease: Are all GLP-1 agonists equal in the treatment of type 2 diabetes? Eur. J. Endocrinol. 2019, 181, R211–R234. [Google Scholar] [CrossRef] [PubMed]
- Carlessi, R.; Chen, Y.; Rowlands, J.; Cruzat, V.F.; Keane, K.N.; Egan, L.; Mamotte, C.; Stokes, R.; Gunton, J.E.; Bittencourt, P.I.H.; et al. GLP-1 receptor signalling promotes beta-cell glucose metabolism via mTOR-dependent HIF-1alpha activation. Sci. Rep. 2017, 7, 2661. [Google Scholar] [CrossRef] [Green Version]
- Park, M.K. Subchapter 17C—Glucagon-like Peptide-1. In Handbook of Hormones; Takei, Y., Ando, H., Tsutsui, K., Eds.; Academic Press: San Diego, CA, USA, 2016; p. 135-e117C-135. [Google Scholar]
- Cornu, M.; Thorens, B. GLP-1 protects β-cells against apoptosis by enhancing the activity of an IGF-2/IGF1-receptor autocrine loop. Islets 2009, 1, 280–282. [Google Scholar] [CrossRef]
- Arden, C. A role for Glucagon-like Peptide-1 in the regulation of beta-cell autophagy. Peptides 2018, 100, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Zummo, F.P.; Cullen, K.S.; Honkanen-Scott, M.; Shaw, J.A.M.; Lovat, P.E.; Arden, C. Glucagon-like Peptide 1 Protects Pancreatic Beta-Cells from Death by Increasing Autophagic Flux and Restoring Lysosomal Function. Diabetes 2017, 66, 1272–1285. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Sha, S.; Sun, L.; Zhang, J.; Dong, M. GLP-1 analogue improves hepatic lipid accumulation by inducing autophagy via AMPK/mTOR pathway. Biochem. Biophys. Res. Commun. 2016, 476, 196–203. [Google Scholar] [CrossRef]
- Tsunekawa, S.; Yamamoto, N.; Tsukamoto, K.; Itoh, Y.; Kaneko, Y.; Kimura, T.; Ariyoshi, Y.; Miura, Y.; Oiso, Y.; Niki, I. Protection of pancreatic beta-cells by exendin-4 may involve the reduction of endoplasmic reticulum stress; in vivo and in vitro studies. J. Endocrinol. 2007, 193, 65–74. [Google Scholar] [CrossRef]
- Yusta, B.; Baggio, L.L.; Estall, J.L.; Koehler, J.A.; Holland, D.P.; Li, H.; Pipeleers, D.; Ling, Z.; Drucker, D.J. GLP-1 receptor activation improves beta cell function and survival following induction of endoplasmic reticulum stress. Cell Metab. 2006, 4, 391–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, D.A.; Ladriere, L.; Ortis, F.; Igoillo-Esteve, M.; Gurzov, E.N.; Lupi, R.; Marchetti, P.; Eizirik, D.L.; Cnop, M. Glucagon-like peptide-1 agonists protect pancreatic beta-cells from lipotoxic endoplasmic reticulum stress through upregulation of BiP and JunB. Diabetes 2009, 58, 2851–2862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacco, F.; Du, X.; Carratu, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation after Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.D.; Finan, B.; Bloom, S.R.; D’Alessio, D.; Drucker, D.J.; Flatt, P.R.; Fritsche, A.; Gribble, F.; Grill, H.J.; Habener, J.F.; et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019, 30, 72–130. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Serino, M.; Cabou, C. A role for the gut-to-brain GLP-1-dependent axis in the control of metabolism. Curr. Opin. Pharmacol. 2009, 9, 744–752. [Google Scholar] [CrossRef]
- Marlet, I.R.; Olmestig, J.N.E.; Vilsboll, T.; Rungby, J.; Kruuse, C. Neuroprotective Mechanisms of Glucagon-like Peptide-1-Based Therapies in Ischaemic Stroke: A Systematic Review Based on Pre-Clinical Studies. Basic Clin. Pharm. Toxicol. 2018, 122, 559–569. [Google Scholar] [CrossRef] [Green Version]
- Maskery, M.P.; Holscher, C.; Jones, S.P.; Price, C.I.; Strain, W.D.; Watkins, C.L.; Werring, D.J.; Emsley, H.C. Glucagon-like peptide-1 receptor agonists as neuroprotective agents for ischemic stroke: A systematic scoping review. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2021, 41, 14–30. [Google Scholar] [CrossRef]
- Gray, F.o.; Duyckaerts, C.; De Girolami, U.; Escourolle, R.; Gray, F.O. Escourolle & Poirier’s Manual of Basic Neuropathology, 5th ed.; Oxford University Press: Oxford, UK, 2014; p. xiii. 406p. [Google Scholar]
- Knopman, D.S.; Amieva, H.; Petersen, R.C.; Chetelat, G.; Holtzman, D.M.; Hyman, B.T.; Nixon, R.A.; Jones, D.T. Alzheimer disease. Nat. Rev. Dis. Primers 2021, 7, 33. [Google Scholar] [CrossRef]
- Dickson, D.W. Neuropathology of Parkinson Disease. Parkinsonism Relat. Disord. 2018, 46 (Suppl. S1), S30–S33. [Google Scholar] [CrossRef]
- Reeve, A.; Simcox, E.; Turnbull, D. Ageing and Parkinson’s disease: Why is advancing age the biggest risk factor? Ageing Res. Rev. 2014, 14, 19–30. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Parkinson’s Disease; Oxford University Press: Oxford, UK; New York, NY, USA, 2010; p. xiv. 116p. [Google Scholar]
- Anang, J.B.; Gagnon, J.F.; Bertrand, J.A.; Romenets, S.R.; Latreille, V.; Panisset, M.; Montplaisir, J.; Postuma, R.B. Predictors of dementia in Parkinson disease: A prospective cohort study. Neurology 2014, 83, 1253–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Karran, E. The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Del Tredici, K. Neuroanatomy and pathology of sporadic Parkinson’s disease. In Advances in Anatomy Embryology Cell Biology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 201, pp. 1–119. [Google Scholar]
- Li, Y.; Chigurupati, S.; Holloway, H.W.; Mughal, M.; Tweedie, D.; Bruestle, D.A.; Mattson, M.P.; Wang, Y.; Harvey, B.K.; Ray, B.; et al. Exendin-4 ameliorates motor neuron degeneration in cellular and animal models of amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e32008. [Google Scholar] [CrossRef]
- Holst, J.J.; Burcelin, R.; Nathanson, E. Neuroprotective properties of GLP-1: Theoretical and practical applications. Curr. Med. Res. Opin. 2011, 27, 547–558. [Google Scholar] [CrossRef]
- Beal, M.F. Oxidative damage as an early marker of Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2005, 26, 585–586. [Google Scholar] [CrossRef]
- Badawi, G.A.; Abd El Fattah, M.A.; Zaki, H.F.; El Sayed, M.I. Sitagliptin and liraglutide reversed nigrostriatal degeneration of rodent brain in rotenone-induced Parkinson’s disease. Inflammopharmacology 2017, 25, 369–382. [Google Scholar] [CrossRef]
- Bader, M.; Li, Y.; Tweedie, D.; Shlobin, N.A.; Bernstein, A.; Rubovitch, V.; Tovar, Y.R.L.B.; DiMarchi, R.D.; Hoffer, B.J.; Greig, N.H.; et al. Neuroprotective Effects and Treatment Potential of Incretin Mimetics in a Murine Model of Mild Traumatic Brain Injury. Front. Cell Dev. Biol. 2019, 7, 356. [Google Scholar] [CrossRef] [Green Version]
- Bertilsson, G.; Patrone, C.; Zachrisson, O.; Andersson, A.; Dannaeus, K.; Heidrich, J.; Kortesmaa, J.; Mercer, A.; Nielsen, E.; Rönnholm, H.; et al. Peptide hormone exendin-4 stimulates subventricular zone neurogenesis in the adult rodent brain and induces recovery in an animal model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 326–338. [Google Scholar] [CrossRef]
- Chen, S.; Sun, J.; Zhao, G.; Guo, A.; Chen, Y.; Fu, R.; Deng, Y. Liraglutide Improves Water Maze Learning and Memory Performance While Reduces Hyperphosphorylation of Tau and Neurofilaments in APP/PS1/Tau Triple Transgenic Mice. Neurochem. Res. 2017, 42, 2326–2335. [Google Scholar] [CrossRef] [PubMed]
- Eakin, K.; Li, Y.; Chiang, Y.H.; Hoffer, B.J.; Rosenheim, H.; Greig, N.H.; Miller, J.P. Exendin-4 ameliorates traumatic brain injury-induced cognitive impairment in rats. PLoS ONE 2013, 8, e82016. [Google Scholar] [CrossRef] [PubMed]
- Hansen, H.H.; Barkholt, P.; Fabricius, K.; Jelsing, J.; Terwel, D.; Pyke, C.; Knudsen, L.B.; Vrang, N. The GLP-1 receptor agonist liraglutide reduces pathology-specific tau phosphorylation and improves motor function in a transgenic hTauP301L mouse model of tauopathy. Brain Res. 2016, 1634, 158–170. [Google Scholar] [CrossRef]
- Hansen, H.H.; Fabricius, K.; Barkholt, P.; Mikkelsen, J.D.; Jelsing, J.; Pyke, C.; Knudsen, L.B.; Vrang, N. Characterization of liraglutide, a glucagon-like peptide-1 (GLP-1) receptor agonist, in rat partial and full nigral 6-hydroxydopamine lesion models of Parkinson’s disease. Brain Res. 2016, 1646, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Harkavyi, A.; Abuirmeileh, A.; Lever, R.; Kingsbury, A.E.; Biggs, C.S.; Whitton, P.S. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson’s disease. J. Neuroinflamm. 2008, 5, 19. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Wang, H.; Zhao, C.; Tian, X.; Li, L.; Wang, H. Role of liraglutide in brain repair promotion through Sirt1-mediated mitochondrial improvement in stroke. J. Cell. Physiol. 2020, 235, 2986–3001. [Google Scholar] [CrossRef]
- Li, Y.; Duffy, K.B.; Ottinger, M.A.; Ray, B.; Bailey, J.A.; Holloway, H.W.; Tweedie, D.; Perry, T.; Mattson, M.P.; Kapogiannis, D.; et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2010, 19, 1205–1219. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Perry, T.; Kindy, M.S.; Harvey, B.K.; Tweedie, D.; Holloway, H.W.; Powers, K.; Shen, H.; Egan, J.M.; Sambamurti, K.; et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc. Natl. Acad. Sci. USA 2009, 106, 1285–1290. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Jalewa, J.; Sharma, M.; Li, G.; Li, L.; Hölscher, C. Neuroprotective effects of lixisenatide and liraglutide in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. Neuroscience 2015, 303, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef] [Green Version]
- Perry, T.; Holloway, H.W.; Weerasuriya, A.; Mouton, P.R.; Duffy, K.; Mattison, J.A.; Greig, N.H. Evidence of GLP-1-mediated neuroprotection in an animal model of pyridoxine-induced peripheral sensory neuropathy. Exp. Neurol. 2007, 203, 293–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, L.; Ke, L.; Liu, X.; Liao, L.; Ke, S.; Liu, X.; Wang, Y.; Lin, X.; Zhou, Y.; Wu, L.; et al. Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced alzheimer disease mouse model. Eur. J. Pharm. 2016, 783, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Rachmany, L.; Tweedie, D.; Li, Y.; Rubovitch, V.; Holloway, H.W.; Miller, J.; Hoffer, B.J.; Greig, N.H.; Pick, C.G. Exendin-4 induced glucagon-like peptide-1 receptor activation reverses behavioral impairments of mild traumatic brain injury in mice. Age 2013, 35, 1621–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, J.; Xie, Y.; Ren, L.; Qi, L.; Wu, L.; Pan, X.; Zhou, J.; Chen, Z.; Liu, L. GLP-1 improves the supportive ability of astrocytes to neurons by promoting aerobic glycolysis in Alzheimer’s disease. Mol. Metab. 2021, 47, 101180. [Google Scholar] [CrossRef]
- Han, W.N.; Holscher, C.; Yuan, L.; Yang, W.; Wang, X.H.; Wu, M.N.; Qi, J.S. Liraglutide protects against amyloid-beta protein-induced impairment of spatial learning and memory in rats. Neurobiol. Aging 2013, 34, 576–588. [Google Scholar] [CrossRef]
- McClean, P.L.; Parthsarathy, V.; Faivre, E.; Hölscher, C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 6587–6594. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.-Y.; Yang, J.-T.; Wang, Z.-J.; Zhang, J.; Yang, W.; Wu, M.-N.; Qi, J.-S. Lixisenatide reduces amyloid plaques, neurofibrillary tangles and neuroinflammation in an APP/PS1/tau mouse model of Alzheimer’s disease. Biochem. Biophys. Res. Commun. 2018, 495, 1034–1040. [Google Scholar] [CrossRef]
- Zhou, M.; Chen, S.; Peng, P.; Gu, Z.; Yu, J.; Zhao, G.; Deng, Y. Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3β. Biochem. Biophys. Res. Commun. 2019, 511, 154–160. [Google Scholar] [CrossRef]
- Batista, A.F.; Forny-Germano, L.; Clarke, J.R.; Lyra e Silva, N.M.; Brito-Moreira, J.; Boehnke, S.E.; Winterborn, A.; Coe, B.C.; Lablans, A.; Vital, J.F.; et al. The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J. Pathol. 2018, 245, 85–100. [Google Scholar] [CrossRef]
- Sharma, M.K.; Jalewa, J.; Holscher, C. Neuroprotective and anti-apoptotic effects of liraglutide on SH-SY5Y cells exposed to methylglyoxal stress. J. Neurochem. 2014, 128, 459–471. [Google Scholar] [CrossRef]
- McClean, P.L.; Hölscher, C. Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 2014, 86, 241–258. [Google Scholar] [CrossRef] [PubMed]
- McClean, P.L.; Jalewa, J.; Hölscher, C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav. Brain Res. 2015, 293, 96–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paladugu, L.; Gharaibeh, A.; Kolli, N.; Learman, C.; Hall, T.C.; Li, L.; Rossignol, J.; Maiti, P.; Dunbar, G.L. Liraglutide Has Anti-Inflammatory and Anti-Amyloid Properties in Streptozotocin-Induced and 5xFAD Mouse Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 860. [Google Scholar] [CrossRef] [PubMed]
- Carranza-Naval, M.J.; del Marco, A.; Hierro-Bujalance, C.; Alves-Martinez, P.; Infante-Garcia, C.; Vargas-Soria, M.; Herrera, M.; Barba-Cordoba, B.; Atienza-Navarro, I.; Lubian-Lopez, S.; et al. Liraglutide Reduces Vascular Damage, Neuronal Loss, and Cognitive Impairment in a Mixed Murine Model of Alzheimer’s Disease and Type 2 Diabetes. Front. Aging Neurosci. 2021, 13, 741923. [Google Scholar] [CrossRef]
- Cai, H.Y.; Hölscher, C.; Yue, X.H.; Zhang, S.X.; Wang, X.H.; Qiao, F.; Yang, W.; Qi, J.S. Lixisenatide rescues spatial memory and synaptic plasticity from amyloid β protein-induced impairments in rats. Neuroscience 2014, 277, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Bergkvist, L.; Johnson, M.E.; Mercado, G.; Steiner, J.A.; Meyerdirk, L.; Schulz, E.; Madaj, Z.; Ma, J.; Becker, K.; Li, Y.; et al. An extended release GLP-1 analogue increases alpha-synuclein accumulation in a mouse model of prodromal Parkinson’s disease. Exp. Neurol. 2021, 341, 113693. [Google Scholar] [CrossRef]
- Athauda, D.; Foltynie, T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: Mechanisms of action. Drug Discov. Today 2016, 21, 802–818. [Google Scholar] [CrossRef] [Green Version]
- Vadini, F.; Simeone, P.G.; Boccatonda, A.; Guagnano, M.T.; Liani, R.; Tripaldi, R.; Di Castelnuovo, A.; Cipollone, F.; Consoli, A.; Santilli, F. Liraglutide improves memory in obese patients with prediabetes or early type 2 diabetes: A randomized, controlled study. Int. J. Obes. 2020, 44, 1254–1263. [Google Scholar] [CrossRef]
- Watson, K.T.; Wroolie, T.E.; Tong, G.; Foland-Ross, L.C.; Frangou, S.; Singh, M.; McIntyre, R.S.; Roat-Shumway, S.; Myoraku, A.; Reiss, A.L.; et al. Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav. Brain Res. 2019, 356, 271–278. [Google Scholar] [CrossRef]
- Egefjord, L.; Gejl, M.; Møller, A.; Brændgaard, H.; Gottrup, H.; Antropova, O.; Møller, N.; Poulsen, H.E.; Gjedde, A.; Brock, B.; et al. Effects of liraglutide on neurodegeneration, blood flow and cognition in Alzheimer´s disease—Protocol for a controlled, randomized double-blinded trial. Dan. Med. J. 2012, 59, A4519. [Google Scholar]
- Gejl, M.; Gjedde, A.; Egefjord, L.; Møller, A.; Hansen, S.B.; Vang, K.; Rodell, A.; Brændgaard, H.; Gottrup, H.; Schacht, A.; et al. In Alzheimer’s Disease, 6-Month Treatment with GLP-1 Analog Prevents Decline of Brain Glucose Metabolism: Randomized, Placebo-Controlled, Double-Blind Clinical Trial. Front. Aging Neurosci. 2016, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Femminella, G.D.; Frangou, E.; Love, S.B.; Busza, G.; Holmes, C.; Ritchie, C.; Lawrence, R.; McFarlane, B.; Tadros, G.; Ridha, B.H.; et al. Evaluating the effects of the novel GLP-1 analogue liraglutide in Alzheimer’s disease: Study protocol for a randomised controlled trial (ELAD study). Trials 2019, 20, 191. [Google Scholar] [CrossRef] [Green Version]
- Mullins, J.R.; Mustapic, M.; Chia, W.C.; Carlson, O.; Gulyani, S.; Tran, J.; Li, Y.; Mattson, P.M.; Resnick, S.; Egan, M.J.; et al. A Pilot Study of Exenatide Actions in Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Athauda, D.; Maclagan, K.; Skene, S.S.; Bajwa-Joseph, M.; Letchford, D.; Chowdhury, K.; Hibbert, S.; Budnik, N.; Zampedri, L.; Dickson, J.; et al. Exenatide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1664–1675. [Google Scholar] [CrossRef]
- Aviles-Olmos, I.; Dickson, J.; Kefalopoulou, Z.; Djamshidian, A.; Ell, P.; Soderlund, T.; Whitton, P.; Wyse, R.; Isaacs, T.; Lees, A.; et al. Exenatide and the treatment of patients with Parkinson’s disease. J. Clin. Investig. 2013, 123, 2730–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulvaney, C.A.; Duarte, G.S.; Handley, J.; Evans, D.J.; Menon, S.; Wyse, R.; Emsley, H.C. GLP-1 receptor agonists for Parkinson’s disease. Cochrane Database Syst. Rev. 2020, 7, CD012990. [Google Scholar] [CrossRef] [PubMed]
- Martin, B.; Golden, E.; Carlson, O.D.; Pistell, P.; Zhou, J.; Kim, W.; Frank, B.P.; Thomas, S.; Chadwick, W.A.; Greig, N.H.; et al. Exendin-4 improves glycemic control, ameliorates brain and pancreatic pathologies, and extends survival in a mouse model of Huntington’s disease. Diabetes 2009, 58, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Seppa, K.; Toots, M.; Reimets, R.; Jagomae, T.; Koppel, T.; Pallase, M.; Hasselholt, S.; Krogsbaek Mikkelsen, M.; Randel Nyengaard, J.; Vasar, E.; et al. GLP-1 receptor agonist liraglutide has a neuroprotective effect on an aged rat model of Wolfram syndrome. Sci. Rep. 2019, 9, 15742. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Magistretti, P.J.; Pellerin, L. Cellular bases of brain energy metabolism and their relevance to functional brain imaging: Evidence for a prominent role of astrocytes. Cereb. Cortex 1996, 6, 50–61. [Google Scholar] [CrossRef]
- Barros, L.F.; San Martín, A.; Ruminot, I.; Sandoval, P.Y.; Baeza-Lehnert, F.; Arce-Molina, R.; Rauseo, D.; Contreras-Baeza, Y.; Galaz, A.; Valdivia, S. Fluid Brain Glycolysis: Limits, Speed, Location, Moonlighting, and the Fates of Glycogen and Lactate. Neurochem. Res. 2020, 45, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- El Hayek, L.; Khalifeh, M.; Zibara, V.; Abi Assaad, R.; Emmanuel, N.; Karnib, N.; El-Ghandour, R.; Nasrallah, P.; Bilen, M.; Ibrahim, P.; et al. Lactate Mediates the Effects of Exercise on Learning and Memory through SIRT1-Dependent Activation of Hippocampal Brain-Derived Neurotrophic Factor (BDNF). J. Neurosci. Off. J. Soc. Neurosci. 2019, 39, 2369–2382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beard, E.; Lengacher, S.; Dias, S.; Magistretti, P.J.; Finsterwald, C. Astrocytes as Key Regulators of Brain Energy Metabolism: New Therapeutic Perspectives. Front. Physiol. 2021, 12, 825816. [Google Scholar] [CrossRef]
- Patching, S.G. Glucose Transporters at the Blood-Brain Barrier: Function, Regulation and Gateways for Drug Delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef] [PubMed]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Figley, C.R.; Stroman, P.W. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur. J. Neurosci. 2011, 33, 577–588. [Google Scholar] [CrossRef]
- Raichle, M.E.; Mintun, M.A. Brain work and brain imaging. Annu. Rev. Neurosci. 2006, 29, 449–476. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Kisler, K.; Montagne, A.; Toga, A.W.; Zlokovic, B.V. The role of brain vasculature in neurodegenerative disorders. Nat. Neurosci. 2018, 21, 1318–1331. [Google Scholar] [CrossRef]
- Tarasoff-Conway, J.M.; Carare, R.O.; Osorio, R.S.; Glodzik, L.; Butler, T.; Fieremans, E.; Axel, L.; Rusinek, H.; Nicholson, C.; Zlokovic, B.V.; et al. Clearance systems in the brain-implications for Alzheimer disease. Nat. Rev. Neurol. 2015, 11, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Ackerman, J.J.; Sukstanskii, A.L.; Yablonskiy, D.A. How the body controls brain temperature: The temperature shielding effect of cerebral blood flow. J. Appl. Physiol. 2006, 101, 1481–1488. [Google Scholar] [CrossRef] [Green Version]
- Newman, L.A.; Korol, D.L.; Gold, P.E. Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS ONE 2011, 6, e28427. [Google Scholar] [CrossRef]
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738. [Google Scholar] [CrossRef]
- Nippert, A.R.; Biesecker, K.R.; Newman, E.A. Mechanisms Mediating Functional Hyperemia in the Brain. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2018, 24, 73–83. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef] [Green Version]
- Zonta, M.; Angulo, M.C.; Gobbo, S.; Rosengarten, B.; Hossmann, K.A.; Pozzan, T.; Carmignoto, G. Neuron-to-astrocyte signaling is central to the dynamic control of brain microcirculation. Nat. Neurosci. 2003, 6, 43–50. [Google Scholar] [CrossRef]
- Biesecker, K.R.; Srienc, A.I.; Shimoda, A.M.; Agarwal, A.; Bergles, D.E.; Kofuji, P.; Newman, E.A. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 9435–9445. [Google Scholar] [CrossRef] [Green Version]
- Ko, K.R.; Ngai, A.C.; Winn, H.R. Role of adenosine in regulation of regional cerebral blood flow in sensory cortex. Am. J. Physiol. 1990, 259, H1703–H1708. [Google Scholar] [CrossRef]
- Kur, J.; Newman, E.A. Purinergic control of vascular tone in the retina. J. Physiol. 2014, 592, 491–504. [Google Scholar] [CrossRef]
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood-brain barrier. Nat. Aging 2021, 1, 243–254. [Google Scholar] [CrossRef]
- Watkins, S.; Robel, S.; Kimbrough, I.F.; Robert, S.M.; Ellis-Davies, G.; Sontheimer, H. Disruption of astrocyte-vascular coupling and the blood-brain barrier by invading glioma cells. Nat. Commun. 2014, 5, 4196. [Google Scholar] [CrossRef] [Green Version]
- Iadecola, C.; Nedergaard, M. Glial regulation of the cerebral microvasculature. Nat. Neurosci. 2007, 10, 1369–1376. [Google Scholar] [CrossRef]
- Cunnane, S.; Nugent, S.; Roy, M.; Courchesne-Loyer, A.; Croteau, E.; Tremblay, S.; Castellano, A.; Pifferi, F.; Bocti, C.; Paquet, N.; et al. Brain fuel metabolism, aging, and Alzheimer’s disease. Nutrition 2011, 27, 3–20. [Google Scholar] [CrossRef] [Green Version]
- Sonnen, J.A.; Santa Cruz, K.; Hemmy, L.S.; Woltjer, R.; Leverenz, J.B.; Montine, K.S.; Jack, C.R.; Kaye, J.; Lim, K.; Larson, E.B.; et al. Ecology of the aging human brain. Arch. Neurol. 2011, 68, 1049–1056. [Google Scholar] [CrossRef] [Green Version]
- Mattay, V.S.; Fera, F.; Tessitore, A.; Hariri, A.R.; Berman, K.F.; Das, S.; Meyer-Lindenberg, A.; Goldberg, T.E.; Callicott, J.H.; Weinberger, D.R. Neurophysiological correlates of age-related changes in working memory capacity. Neurosci. Lett. 2006, 392, 32–37. [Google Scholar] [CrossRef]
- Glisky, E.L. Frontiers in Neuroscience. Changes in Cognitive Function in Human Aging. In Brain Aging: Models, Methods, and Mechanisms; Riddle, D.R., Ed.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group, LLC.: Boca Raton, FL, USA, 2007. [Google Scholar]
- Masliah, E.; Mallory, M.; Hansen, L.; DeTeresa, R.; Terry, R.D. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology 1993, 43, 192–197. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [Green Version]
- Nelson, A.R.; Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Neurovascular dysfunction and neurodegeneration in dementia and Alzheimer’s disease. Biochim. Biophys. Acta 2016, 1862, 887–900. [Google Scholar] [CrossRef]
- Bender, A.R.; Prindle, J.J.; Brandmaier, A.M.; Raz, N. White matter and memory in healthy adults: Coupled changes over two years. NeuroImage 2016, 131, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Bi, Q.; Wang, W.; Niu, N.; Li, H.; Wang, Y.; Huang, W.; Chen, K.; Xu, K.; Zhang, J.; Chen, Y.; et al. Relationship between the disrupted topological efficiency of the structural brain connectome and glucose hypometabolism in normal aging. NeuroImage 2021, 226, 117591. [Google Scholar] [CrossRef]
- Fox, N.C.; Schott, J.M. Imaging cerebral atrophy: Normal ageing to Alzheimer’s disease. Lancet 2004, 363, 392–394. [Google Scholar] [CrossRef]
- Resnick, S.M.; Pham, D.L.; Kraut, M.A.; Zonderman, A.B.; Davatzikos, C. Longitudinal magnetic resonance imaging studies of older adults: A shrinking brain. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 3295–3301. [Google Scholar] [CrossRef] [Green Version]
- Salat, D.H.; Buckner, R.L.; Snyder, A.Z.; Greve, D.N.; Desikan, R.S.; Busa, E.; Morris, J.C.; Dale, A.M.; Fischl, B. Thinning of the cerebral cortex in aging. Cereb. Cortex 2004, 14, 721–730. [Google Scholar] [CrossRef] [Green Version]
- Wrigglesworth, J.; Ward, P.; Harding, I.H.; Nilaweera, D.; Wu, Z.; Woods, R.L.; Ryan, J. Factors associated with brain ageing—A systematic review. BMC Neurol. 2021, 21, 312. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef]
- Rudow, G.; O’Brien, R.; Savonenko, A.V.; Resnick, S.M.; Zonderman, A.B.; Pletnikova, O.; Marsh, L.; Dawson, T.M.; Crain, B.J.; West, M.J.; et al. Morphometry of the human substantia nigra in ageing and Parkinson’s disease. Acta Neuropathol. 2008, 115, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Cabello, C.R.; Thune, J.J.; Pakkenberg, H.; Pakkenberg, B. Ageing of substantia nigra in humans: Cell loss may be compensated by hypertrophy. Neuropathol. Appl. Neurobiol. 2002, 28, 283–291. [Google Scholar] [CrossRef]
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. alpha-Synucleinopathy phenotypes. Parkinsonism Relat. Disord. 2014, 20 (Suppl. S1), S62–S67. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.S.; Gertler, T.S.; Surmeier, D.J. A molecular basis for the increased vulnerability of substantia nigra dopamine neurons in aging and Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. S1), S63–S70. [Google Scholar] [CrossRef]
- Wilson, R.S.; Nag, S.; Boyle, P.A.; Hizel, L.P.; Yu, L.; Buchman, A.S.; Schneider, J.A.; Bennett, D.A. Neural reserve, neuronal density in the locus ceruleus, and cognitive decline. Neurology 2013, 80, 1202–1208. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Hu, Y.; Wang, B.; Wang, S.; Zhang, X. Metabolic Dysregulation Contributes to the Progression of Alzheimer’s Disease. Front. Neurosci. 2020, 14, 530219. [Google Scholar] [CrossRef]
- Kivipelto, M.; Ngandu, T.; Laatikainen, T.; Winblad, B.; Soininen, H.; Tuomilehto, J. Risk score for the prediction of dementia risk in 20 years among middle aged people: A longitudinal, population-based study. Lancet. Neurol. 2006, 5, 735–741. [Google Scholar] [CrossRef]
- Rabin, J.S.; Schultz, A.P.; Hedden, T.; Viswanathan, A.; Marshall, G.A.; Kilpatrick, E.; Klein, H.; Buckley, R.F.; Yang, H.S.; Properzi, M.; et al. Interactive Associations of Vascular Risk and β-Amyloid Burden with Cognitive Decline in Clinically Normal Elderly Individuals: Findings from the Harvard Aging Brain Study. JAMA Neurol. 2018, 75, 1124–1131. [Google Scholar] [CrossRef]
- Saito, S.; Ihara, M. Interaction between cerebrovascular disease and Alzheimer pathology. Curr. Opin. Psychiatry 2016, 29, 168–173. [Google Scholar] [CrossRef]
- Brundel, M.; Heringa, S.M.; de Bresser, J.; Koek, H.L.; Zwanenburg, J.J.; Jaap Kappelle, L.; Luijten, P.R.; Biessels, G.J. High prevalence of cerebral microbleeds at 7Tesla MRI in patients with early Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2012, 31, 259–263. [Google Scholar] [CrossRef]
- Heringa, S.M.; Reijmer, Y.D.; Leemans, A.; Koek, H.L.; Kappelle, L.J.; Biessels, G.J. Multiple microbleeds are related to cerebral network disruptions in patients with early Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2014, 38, 211–221. [Google Scholar] [CrossRef]
- Shams, S.; Wahlund, L.O. Cerebral microbleeds as a biomarker in Alzheimer’s disease? A review in the field. Biomark. Med. 2016, 10, 9–18. [Google Scholar] [CrossRef]
- Uetani, H.; Hirai, T.; Hashimoto, M.; Ikeda, M.; Kitajima, M.; Sakamoto, F.; Utsunomiya, D.; Oda, S.; Sugiyama, S.; Matsubara, J.; et al. Prevalence and topography of small hypointense foci suggesting microbleeds on 3T susceptibility-weighted imaging in various types of dementia. AJNR. Am. J. Neuroradiol. 2013, 34, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Poliakova, T.; Levin, O.; Arablinskiy, A.; Vasenina, E.; Zerr, I. Cerebral microbleeds in early Alzheimer’s disease. J. Neurol. 2016, 263, 1961–1968. [Google Scholar] [CrossRef]
- Yates, P.A.; Desmond, P.M.; Phal, P.M.; Steward, C.; Szoeke, C.; Salvado, O.; Ellis, K.A.; Martins, R.N.; Masters, C.L.; Ames, D.; et al. Incidence of cerebral microbleeds in preclinical Alzheimer disease. Neurology 2014, 82, 1266–1273. [Google Scholar] [CrossRef] [Green Version]
- Vernooij, M.W.; van der Lugt, A.; Ikram, M.A.; Wielopolski, P.A.; Niessen, W.J.; Hofman, A.; Krestin, G.P.; Breteler, M.M. Prevalence and risk factors of cerebral microbleeds: The Rotterdam Scan Study. Neurology 2008, 70, 1208–1214. [Google Scholar] [CrossRef]
- De la Torre, J.C. Pathophysiology of neuronal energy crisis in Alzheimer’s disease. Neuro-Degener. Dis. 2008, 5, 126–132. [Google Scholar] [CrossRef]
- Love, S.; Miners, J.S. Cerebrovascular disease in ageing and Alzheimer’s disease. Acta Neuropathol. 2016, 131, 645–658. [Google Scholar] [CrossRef] [Green Version]
- Roher, A.E.; Esh, C.; Rahman, A.; Kokjohn, T.A.; Beach, T.G. Atherosclerosis of cerebral arteries in Alzheimer disease. Stroke 2004, 35, 2623–2627. [Google Scholar] [CrossRef] [Green Version]
- Faucheux, B.A.; Bonnet, A.M.; Agid, Y.; Hirsch, E.C. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Guan, J.; Pavlovic, D.; Dalkie, N.; Waldvogel, H.J.; O’Carroll, S.J.; Green, C.R.; Nicholson, L.F. Vascular degeneration in Parkinson’s disease. Brain Pathol. 2013, 23, 154–164. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.C.; Parkes, L.M. Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotypes. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2017, 37, 3409–3421. [Google Scholar] [CrossRef]
- Borghammer, P.; Chakravarty, M.; Jonsdottir, K.Y.; Sato, N.; Matsuda, H.; Ito, K.; Arahata, Y.; Kato, T.; Gjedde, A. Cortical hypometabolism and hypoperfusion in Parkinson’s disease is extensive: Probably even at early disease stages. Brain Struct. Funct. 2010, 214, 303–317. [Google Scholar] [CrossRef]
- Fernandez-Seara, M.A.; Mengual, E.; Vidorreta, M.; Aznarez-Sanado, M.; Loayza, F.R.; Villagra, F.; Irigoyen, J.; Pastor, M.A. Cortical hypoperfusion in Parkinson’s disease assessed using arterial spin labeled perfusion MRI. NeuroImage 2012, 59, 2743–2750. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, P.; Sorg, C.; Förschler, A.; Grimmer, T.; Skokou, M.; Wohlschläger, A.; Perneczky, R.; Zimmer, C.; Kurz, A.; Preibisch, C. Perfusion abnormalities in mild cognitive impairment and mild dementia in Alzheimer’s disease measured by pulsed arterial spin labeling MRI. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Lopez, O.L.; Carmichael, O.T.; Becker, J.T.; Kuller, L.H.; Gach, H.M. Mild cognitive impairment and alzheimer disease: Patterns of altered cerebral blood flow at MR imaging. Radiology 2009, 250, 856–866. [Google Scholar] [CrossRef]
- den Abeelen, A.S.; Lagro, J.; van Beek, A.H.; Claassen, J.A. Impaired cerebral autoregulation and vasomotor reactivity in sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2014, 11, 11–17. [Google Scholar] [CrossRef]
- Hays, C.C.; Zlatar, Z.Z.; Wierenga, C.E. The Utility of Cerebral Blood Flow as a Biomarker of Preclinical Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Ruitenberg, A.; den Heijer, T.; Bakker, S.L.; van Swieten, J.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam Study. Ann. Neurol. 2005, 57, 789–794. [Google Scholar] [CrossRef]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9 (Suppl. S2), S16. [Google Scholar] [CrossRef] [Green Version]
- Goyal, M.S.; Vlassenko, A.G.; Blazey, T.M.; Su, Y.; Couture, L.E.; Durbin, T.J.; Bateman, R.J.; Benzinger, T.L.; Morris, J.C.; Raichle, M.E. Loss of Brain Aerobic Glycolysis in Normal Human Aging. Cell Metab. 2017, 26, 353–360. [Google Scholar] [CrossRef]
- Jagust, W.J.; Seab, J.P.; Huesman, R.H.; Valk, P.E.; Mathis, C.A.; Reed, B.R.; Coxson, P.G.; Budinger, T.F. Diminished glucose transport in Alzheimer’s disease: Dynamic PET studies. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 1991, 11, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Bailly, M.; Destrieux, C.; Hommet, C.; Mondon, K.; Cottier, J.P.; Beaufils, E.; Vierron, E.; Vercouillie, J.; Ibazizene, M.; Voisin, T.; et al. Precuneus and Cingulate Cortex Atrophy and Hypometabolism in Patients with Alzheimer’s Disease and Mild Cognitive Impairment: MRI and (18)F-FDG PET Quantitative Analysis Using FreeSurfer. BioMed Res. Int. 2015, 2015, 583931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoshima, S.; Giordani, B.; Berent, S.; Frey, K.A.; Foster, N.L.; Kuhl, D.E. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Ann. Neurol. 1997, 42, 85–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, L.; Chiotis, K.; Alongi, P.; Almkvist, O.; Wall, A.; Cerami, C.; Bettinardi, V.; Gianolli, L.; Nordberg, A.; Perani, D. A Cross-Validation of FDG- and Amyloid-PET Biomarkers in Mild Cognitive Impairment for the Risk Prediction to Dementia due to Alzheimer’s Disease in a Clinical Setting. J. Alzheimer’s Dis. JAD 2017, 59, 603–614. [Google Scholar] [CrossRef] [Green Version]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Weiner, M.W.; Jagust, W.J. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- De la Monte, S.M. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009, 42, 475–481. [Google Scholar] [CrossRef]
- Hoyer, S. Glucose metabolism and insulin receptor signal transduction in Alzheimer disease. Eur. J. Pharmacol. 2004, 490, 115–125. [Google Scholar] [CrossRef]
- Kellar, D.; Craft, S. Brain insulin resistance in Alzheimer’s disease and related disorders: Mechanisms and therapeutic approaches. Lancet. Neurol. 2020, 19, 758–766. [Google Scholar] [CrossRef]
- Liang, W.S.; Reiman, E.M.; Valla, J.; Dunckley, T.; Beach, T.G.; Grover, A.; Niedzielko, T.L.; Schneider, L.E.; Mastroeni, D.; Caselli, R.; et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 4441–4446. [Google Scholar] [CrossRef] [Green Version]
- Marcus, D.L.; Freedman, M.L. Decreased brain glucose metabolism in microvessels from patients with Alzheimer’s disease. Ann. N. Y. Acad. Sci. 1997, 826, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Neth, B.J.; Craft, S. Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front. Aging Neurosci. 2017, 9, 345. [Google Scholar] [CrossRef] [PubMed]
- Rivera, E.J.; Goldin, A.; Fulmer, N.; Tavares, R.; Wands, J.R.; de la Monte, S.M. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J. Alzheimer’s Dis. JAD 2005, 8, 247–268. [Google Scholar] [CrossRef]
- Chiu, S.L.; Chen, C.M.; Cline, H.T. Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 2008, 58, 708–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Felice, F.G.; Vieira, M.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Abeta oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 1971–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholerton, B.; Baker, L.D.; Craft, S. Insulin resistance and pathological brain ageing. Diabet. Med. 2011, 28, 1463–1475. [Google Scholar] [CrossRef]
- Holscher, C. Diabetes as a risk factor for Alzheimer’s disease: Insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem. Soc. Trans. 2011, 39, 891–897. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Wilson, R.S.; Bennett, D.A. Diabetes mellitus, dementia, and cognitive function in older persons. J. Nutr. Health Aging 2006, 10, 287–291. [Google Scholar]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Willette, A.A.; Modanlo, N.; Kapogiannis, D.; Alzheimer’s Disease Neuroimaging, I. Insulin resistance predicts medial temporal hypermetabolism in mild cognitive impairment conversion to Alzheimer disease. Diabetes 2015, 64, 1933–1940. [Google Scholar] [CrossRef] [Green Version]
- Harr, S.D.; Simonian, N.A.; Hyman, B.T. Functional alterations in Alzheimer’s disease: Decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J. Neuropathol. Exp. Neurol. 1995, 54, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Mooradian, A.D.; Chung, H.C.; Shah, G.N. GLUT-1 expression in the cerebra of patients with Alzheimer’s disease. Neurobiol. Aging 1997, 18, 469–474. [Google Scholar] [CrossRef]
- Simpson, I.A.; Chundu, K.R.; Davies-Hill, T.; Honer, W.G.; Davies, P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol. 1994, 35, 546–551. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Sheu, K.F.; Blass, J.P.; Baker, A.; Carlson, K.C.; Harding, B.; Perrino, P. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch. Neurol. 1988, 45, 836–840. [Google Scholar] [CrossRef]
- Kish, S.J.; Bergeron, C.; Rajput, A.; Dozic, S.; Mastrogiacomo, F.; Chang, L.J.; Wilson, J.M.; DiStefano, L.M.; Nobrega, J.N. Brain cytochrome oxidase in Alzheimer’s disease. J. Neurochem. 1992, 59, 776–779. [Google Scholar] [CrossRef]
- Bosco, D.; Plastino, M.; Cristiano, D.; Colica, C.; Ermio, C.; De Bartolo, M.; Mungari, P.; Fonte, G.; Consoli, D.; Consoli, A.; et al. Dementia is associated with Insulin Resistance in patients with Parkinson’s Disease. J. Neurol. Sci. 2012, 315, 39–43. [Google Scholar] [CrossRef]
- Santiago, J.A.; Potashkin, J.A. Shared dysregulated pathways lead to Parkinson’s disease and diabetes. Trends Mol. Med. 2013, 19, 176–186. [Google Scholar] [CrossRef]
- Pagano, G.; Polychronis, S.; Wilson, H.; Giordano, B.; Ferrara, N.; Niccolini, F.; Politis, M. Diabetes mellitus and Parkinson disease. Neurology 2018, 90, e1654–e1662. [Google Scholar] [CrossRef]
- Giuntini, M.; Baldacci, F.; Del Prete, E.; Bonuccelli, U.; Ceravolo, R. Diabetes is associated with postural and cognitive domains in Parkinson’s disease. Results from a single-center study. Parkinsonism Relat. Disord. 2014, 20, 671–672. [Google Scholar] [CrossRef]
- Hu, G.; Jousilahti, P.; Bidel, S.; Antikainen, R.; Tuomilehto, J. Type 2 diabetes and the risk of Parkinson’s disease. Diabetes Care 2007, 30, 842–847. [Google Scholar] [CrossRef] [Green Version]
- Dunn, L.; Allen, G.F.; Mamais, A.; Ling, H.; Li, A.; Duberley, K.E.; Hargreaves, I.P.; Pope, S.; Holton, J.L.; Lees, A.; et al. Dysregulation of glucose metabolism is an early event in sporadic Parkinson’s disease. Neurobiol. Aging 2014, 35, 1111–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaidelli, A.; Jan, A.; Herms, J.; Sorensen, P.H. Translational control in brain pathologies: Biological significance and therapeutic opportunities. Acta Neuropathol. 2019, 137, 535–555. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Alafuzoff, I.; Soininen, H.; Winblad, B.; Pei, J.J. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005, 272, 4211–4220. [Google Scholar] [CrossRef] [PubMed]
- Vingtdeux, V.; Davies, P.; Dickson, D.W.; Marambaud, P. AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol. 2011, 121, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jan, A.; Jansonius, B.; Delaidelli, A.; Bhanshali, F.; An, Y.A.; Ferreira, N.; Smits, L.M.; Negri, G.L.; Schwamborn, J.C.; Jensen, P.H.; et al. Activity of translation regulator eukaryotic elongation factor-2 kinase is increased in Parkinson disease brain and its inhibition reduces alpha synuclein toxicity. Acta Neuropathol. Commun. 2018, 6, 54. [Google Scholar] [CrossRef] [Green Version]
- Jan, A.; Jansonius, B.; Delaidelli, A.; Somasekharan, S.P.; Bhanshali, F.; Vandal, M.; Negri, G.L.; Moerman, D.; MacKenzie, I.; Calon, F.; et al. eEF2K inhibition blocks Abeta42 neurotoxicity by promoting an NRF2 antioxidant response. Acta Neuropathol. 2017, 133, 101–119. [Google Scholar] [CrossRef]
- Chang, R.C.; Wong, A.K.; Ng, H.K.; Hugon, J. Phosphorylation of eukaryotic initiation factor-2alpha (eIF2alpha) is associated with neuronal degeneration in Alzheimer’s disease. Neuroreport 2002, 13, 2429–2432. [Google Scholar] [CrossRef]
- Wang, Y.; Parlevliet, E.T.; Geerling, J.J.; van der Tuin, S.J.; Zhang, H.; Bieghs, V.; Jawad, A.H.; Shiri-Sverdlov, R.; Bot, I.; de Jager, S.C.; et al. Exendin-4 decreases liver inflammation and atherosclerosis development simultaneously by reducing macrophage infiltration. Br. J. Pharmacol. 2014, 171, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Helmstadter, J.; Keppeler, K.; Aust, F.; Kuster, L.; Frenis, K.; Filippou, K.; Vujacic-Mirski, K.; Tsohataridis, S.; Kalinovic, S.; Kroller-Schon, S.; et al. GLP-1 Analog Liraglutide Improves Vascular Function in Polymicrobial Sepsis by Reduction of Oxidative Stress and Inflammation. Antioxidants 2021, 10, 1175. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Ferrannini, E.; Groop, L.; Henry, R.R.; Herman, W.H.; Holst, J.J.; Hu, F.B.; Kahn, C.R.; Raz, I.; Shulman, G.I.; et al. Type 2 diabetes mellitus. Nat. Rev. Dis. Primers 2015, 1, 15019. [Google Scholar] [CrossRef]
- Kitada, M.; Zhang, Z.; Mima, A.; King, G.L. Molecular mechanisms of diabetic vascular complications. J. Diabetes Investig. 2010, 1, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R.; et al. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Srikanth, V.; Maczurek, A.; Phan, T.; Steele, M.; Westcott, B.; Juskiw, D.; Munch, G. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol. Aging 2011, 32, 763–777. [Google Scholar] [CrossRef]
- Luth, H.J.; Ogunlade, V.; Kuhla, B.; Kientsch-Engel, R.; Stahl, P.; Webster, J.; Arendt, T.; Munch, G. Age- and stage-dependent accumulation of advanced glycation end products in intracellular deposits in normal and Alzheimer’s disease brains. Cereb. Cortex 2005, 15, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Jana, A.K.; Batkulwar, K.B.; Kulkarni, M.J.; Sengupta, N. Glycation induces conformational changes in the amyloid-beta peptide and enhances its aggregation propensity: Molecular insights. Phys. Chem. Chem. Phys. 2016, 18, 31446–31458. [Google Scholar] [CrossRef]
- Craft, S.; Newcomer, J.; Kanne, S.; Dagogo-Jack, S.; Cryer, P.; Sheline, Y.; Luby, J.; Dagogo-Jack, A.; Alderson, A. Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 123–130. [Google Scholar] [CrossRef]
- Sartorius, T.; Peter, A.; Heni, M.; Maetzler, W.; Fritsche, A.; Haring, H.U.; Hennige, A.M. The brain response to peripheral insulin declines with age: A contribution of the blood-brain barrier? PLoS ONE 2015, 10, e0126804. [Google Scholar] [CrossRef] [Green Version]
- Steen, E.; Terry, B.M.; Rivera, E.J.; Cannon, J.L.; Neely, T.R.; Tavares, R.; Xu, X.J.; Wands, J.R.; de la Monte, S.M. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease—Is this type 3 diabetes? J. Alzheimer’s Dis. JAD 2005, 7, 63–80. [Google Scholar] [CrossRef] [Green Version]
- Horwood, N.; Davies, D.C. Immunolabelling of hippocampal microvessel glucose transporter protein is reduced in Alzheimer’s disease. Virchows Arch. Int. J. Pathol. 1994, 425, 69–72. [Google Scholar] [CrossRef]
- Winkler, E.A.; Nishida, Y.; Sagare, A.P.; Rege, S.V.; Bell, R.D.; Perlmutter, D.; Sengillo, J.D.; Hillman, S.; Kong, P.; Nelson, A.R.; et al. GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat. Neurosci. 2015, 18, 521–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.; Yan, B.; Yoo, K.Y.; Choi, J.H.; Kwon, S.H.; Her, S.; Sohn, Y.; Hwang, I.K.; Cho, J.H.; Kim, Y.M.; et al. Ischemia-induced changes in glucagon-like peptide-1 receptor and neuroprotective effect of its agonist, exendin-4, in experimental transient cerebral ischemia. J. Neurosci. Res. 2011, 89, 1103–1113. [Google Scholar] [CrossRef] [PubMed]
- Reiner, D.J.; Mietlicki-Baase, E.G.; McGrath, L.E.; Zimmer, D.J.; Bence, K.K.; Sousa, G.L.; Konanur, V.R.; Krawczyk, J.; Burk, D.H.; Kanoski, S.E.; et al. Astrocytes Regulate GLP-1 Receptor-Mediated Effects on Energy Balance. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 3531–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timper, K.; Del Rio-Martin, A.; Cremer, A.L.; Bremser, S.; Alber, J.; Giavalisco, P.; Varela, L.; Heilinger, C.; Nolte, H.; Trifunovic, A.; et al. GLP-1 Receptor Signaling in Astrocytes Regulates Fatty Acid Oxidation, Mitochondrial Integrity, and Function. Cell Metab. 2020, 31, 1189–1205.e13. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Supplie, L.M.; Duking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Gotz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.A. Respiration-Deficient Astrocytes Survive As Glycolytic Cells In Vivo. J. Neurosci. Off. J. Soc. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef] [Green Version]
- Karagiannis, A.; Gallopin, T.; Lacroix, A.; Plaisier, F.; Piquet, J.; Geoffroy, H.; Hepp, R.; Naudé, J.; Le Gac, B.; Egger, R.; et al. Lactate is an energy substrate for rodent cortical neurons and enhances their firing activity. eLife 2021, 10, e71424. [Google Scholar] [CrossRef]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [Green Version]
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef]
- Suzuki, A.; Stern, S.A.; Bozdagi, O.; Huntley, G.W.; Walker, R.H.; Magistretti, P.J.; Alberini, C.M. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 2011, 144, 810–823. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Zhang, Z.; Zhao, S.; Zhu, Y.; Guo, D.; Yang, B.; Zhuo, L.; Han, J.; Wang, R.; Fang, Z.; et al. Dynamic Variations in Brain Glycogen are Involved in Modulating Isoflurane Anesthesia in Mice. Neurosci. Bull. 2020, 36, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Kajihara, H.; Tsutsumi, E.; Kinoshita, A.; Nakano, J.; Takagi, K.; Takeo, S. Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: Immunohistochemical and electron microscopic studies. Brain Res. 2001, 909, 92–101. [Google Scholar] [CrossRef]
- Alcantara, A.I.; Morales, M.; Delgado, E.; Lopez-Delgado, M.I.; Clemente, F.; Luque, M.A.; Malaisse, W.J.; Valverde, I.; Villanueva-Penacarrillo, M.L. Exendin-4 agonist and exendin(9-39)amide antagonist of the GLP-1(7-36)amide effects in liver and muscle. Arch. Biochem. Biophys. 1997, 341, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Redondo, A.; Trigo, M.V.; Acitores, A.; Valverde, I.; Villanueva-Penacarrillo, M.L. Cell signalling of the GLP-1 action in rat liver. Mol. Cell Endocrinol. 2003, 204, 43–50. [Google Scholar] [CrossRef]
- Ambrosi, G.; Cerri, S.; Blandini, F. A further update on the role of excitotoxicity in the pathogenesis of Parkinson’s disease. J. Neural. Transm. 2014, 121, 849–859. [Google Scholar] [CrossRef]
- Garcia, J.D.; Gookin, S.E.; Crosby, K.C.; Schwartz, S.L.; Tiemeier, E.; Kennedy, M.J.; Dell’Acqua, M.L.; Herson, P.S.; Quillinan, N.; Smith, K.R. Stepwise disassembly of GABAergic synapses during pathogenic excitotoxicity. Cell Rep. 2021, 37, 110142. [Google Scholar] [CrossRef]
- Garry, D.J.; Appel, N.M.; Garry, M.G.; Sorenson, R.L. Cellular and subcellular immunolocalization of L-glutamate decarboxylase in rat pancreatic islets. J. Histochem. Cytochem. 1988, 36, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Sorenson, R.L.; Garry, D.G.; Brelje, T.C. Structural and functional considerations of GABA in islets of Langerhans. Beta-cells and nerves. Diabetes 1991, 40, 1365–1374. [Google Scholar] [CrossRef]
- Rorsman, P.; Berggren, P.O.; Bokvist, K.; Ericson, H.; Mohler, H.; Ostenson, C.G.; Smith, P.A. Glucose-inhibition of glucagon secretion involves activation of GABAA-receptor chloride channels. Nature 1989, 341, 233–236. [Google Scholar] [CrossRef]
- Wang, C.; Mao, R.; Van de Casteele, M.; Pipeleers, D.; Ling, Z. Glucagon-like peptide-1 stimulates GABA formation by pancreatic beta-cells at the level of glutamate decarboxylase. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1201–E1206. [Google Scholar] [CrossRef]
- Wendt, A.; Birnir, B.; Buschard, K.; Gromada, J.; Salehi, A.; Sewing, S.; Rorsman, P.; Braun, M. Glucose inhibition of glucagon secretion from rat alpha-cells is mediated by GABA released from neighboring beta-cells. Diabetes 2004, 53, 1038–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Liu, L.; Pei, L.; Ju, W.; Ahmadian, G.; Lu, J.; Wang, Y.; Liu, F.; Wang, Y.T. Control of synaptic strength, a novel function of Akt. Neuron 2003, 38, 915–928. [Google Scholar] [CrossRef] [Green Version]
- Fortin, S.M.; Lipsky, R.K.; Lhamo, R.; Chen, J.; Kim, E.; Borner, T.; Schmidt, H.D.; Hayes, M.R. GABA neurons in the nucleus tractus solitarius express GLP-1 receptors and mediate anorectic effects of liraglutide in rats. Sci. Transl. Med. 2020, 12, eaay8071. [Google Scholar] [CrossRef] [PubMed]
- Kabahizi, A.; Wallace, B.; Lieu, L.; Chau, D.; Dong, Y.; Hwang, E.S.; Williams, K.W. Glucagon-like peptide-1 (GLP-1) signalling in the brain: From neural circuits and metabolism to therapeutics. Br. J. Pharmacol. 2022, 179, 600–624. [Google Scholar] [CrossRef]
- Laurindo, L.F.; Barbalho, S.M.; Guiguer, E.L.; da Silva Soares de Souza, M.; de Souza, G.A.; Fidalgo, T.M.; Araujo, A.C.; de Souza Gonzaga, H.F.; de Bortoli Teixeira, D.; de Oliveira Silva Ullmann, T.; et al. GLP-1a: Going Beyond Traditional Use. Int. J. Mol. Sci. 2022, 23, 739. [Google Scholar] [CrossRef]
- Göke, R.; Larsen, P.J.; Mikkelsen, J.D.; Sheikh, S.P. Distribution of GLP-1 Binding Sites in the Rat Brain: Evidence that Exendin-4 is a Ligand of Brain GLP-1 Binding Sites. Eur. J. Neurosci. 1995, 7, 2294–2300. [Google Scholar] [CrossRef]
- Brosseron, F.; Krauthausen, M.; Kummer, M.; Heneka, M.T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 2014, 50, 534–544. [Google Scholar] [CrossRef] [Green Version]
- Domingues, C.; da Cruz, E.S.O.A.B.; Henriques, A.G. Impact of Cytokines and Chemokines on Alzheimer’s Disease Neuropathological Hallmarks. Curr. Alzheimer Res. 2017, 14, 870–882. [Google Scholar] [CrossRef] [Green Version]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The overlap between vascular disease and Alzheimer’s disease--lessons from pathology. BMC Med. 2014, 12, 206. [Google Scholar] [CrossRef] [Green Version]
- Wijdicks, E.F. Hepatic Encephalopathy. N. Engl. J. Med. 2016, 375, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- Rosner, M.H.; Husain-Syed, F.; Reis, T.; Ronco, C.; Vanholder, R. Uremic encephalopathy. Kidney Int. 2022, 101, 227–241. [Google Scholar] [CrossRef] [PubMed]
- Melendez-Flores, J.D.; Estrada-Bellmann, I. Linking chronic kidney disease and Parkinson’s disease: A literature review. Metab. Brain Dis. 2021, 36, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tryc, A.B.; Goldbecker, A.; Berding, G.; Rumke, S.; Afshar, K.; Shahrezaei, G.H.; Pflugrad, H.; Barg-Hock, H.; Strassburg, C.P.; Hecker, H.; et al. Cirrhosis-related Parkinsonism: Prevalence, mechanisms and response to treatments. J. Hepatol. 2013, 58, 698–705. [Google Scholar] [CrossRef]
- Weinstein, G.; Zelber-Sagi, S.; Preis, S.R.; Beiser, A.S.; DeCarli, C.; Speliotes, E.K.; Satizabal, C.L.; Vasan, R.S.; Seshadri, S. Association of Nonalcoholic Fatty Liver Disease with Lower Brain Volume in Healthy Middle-Aged Adults in the Framingham Study. JAMA Neurol. 2018, 75, 97–104. [Google Scholar] [CrossRef]
- Zhang, C.Y.; He, F.F.; Su, H.; Zhang, C.; Meng, X.F. Association between chronic kidney disease and Alzheimer’s disease: An update. Metab. Brain Dis. 2020, 35, 883–894. [Google Scholar] [CrossRef]
- Viggiano, D.; Wagner, C.A.; Martino, G.; Nedergaard, M.; Zoccali, C.; Unwin, R.; Capasso, G. Mechanisms of cognitive dysfunction in CKD. Nat. Rev. Nephrol. 2020, 16, 452–469. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Collaboration, G.B.D.C.K.D. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef] [Green Version]
- Cordato, D.J.; Chan, D.K. Genetics and Parkinson’s disease. J. Clin. Neurosci. 2004, 11, 119–123. [Google Scholar] [CrossRef]
- Ur Rasheed, M.S.; Mishra, A.K.; Singh, M.P. Cytochrome P450 2D6 and Parkinson’s Disease: Polymorphism, Metabolic Role, Risk and Protection. Neurochem. Res. 2017, 42, 3353–3361. [Google Scholar] [CrossRef] [PubMed]
- Wojcikowski, J.; Daniel, W.A. The brain dopaminergic system as an important center regulating liver cytochrome P450 in the rat. Expert Opin. Drug Metab. Toxicol. 2009, 5, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Csermely, A.; Lonardo, A.; Targher, G. Glucagon-like Peptide-1 Receptor Agonists for Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis: An Updated Meta-Analysis of Randomized Controlled Trials. Metabolites 2021, 11, 73. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.A.; Mells, J.; Dunham, R.M.; Grakoui, A.; Handy, J.; Saxena, N.K.; Anania, F.A. Glucagon-like peptide-1 receptor is present on human hepatocytes and has a direct role in decreasing hepatic steatosis in vitro by modulating elements of the insulin signaling pathway. Hepatology 2010, 51, 1584–1592. [Google Scholar] [CrossRef] [Green Version]
- Jin, T.; Weng, J. Hepatic functions of GLP-1 and its based drugs: Current disputes and perspectives. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E620–E627. [Google Scholar] [CrossRef] [Green Version]
- Pyke, C.; Heller, R.S.; Kirk, R.K.; Orskov, C.; Reedtz-Runge, S.; Kaastrup, P.; Hvelplund, A.; Bardram, L.; Calatayud, D.; Knudsen, L.B. GLP-1 receptor localization in monkey and human tissue: Novel distribution revealed with extensively validated monoclonal antibody. Endocrinology 2014, 155, 1280–1290. [Google Scholar] [CrossRef]
- Patel Chavez, C.; Cusi, K.; Kadiyala, S. The Emerging Role of Glucagon-like Peptide-1 Receptor Agonists for the Management of NAFLD. J. Clin. Endocrinol. Metab. 2022, 107, 29–38. [Google Scholar] [CrossRef]
- Scheen, A.J. Effects of glucose-lowering agents on surrogate endpoints and hard clinical renal outcomes in patients with type 2 diabetes. Diabetes Metab. 2019, 45, 110–121. [Google Scholar] [CrossRef]
- Cohen, S.; Sternlicht, H.; Bakris, G.L. Mineralocorticoid Receptor Antagonists in the Treatment of Diabetic Kidney Disease: Their Application in the Era of SGLT2 Inhibitors and GLP-1 Receptor Agonists. Curr. Diab. Rep. 2022, 22, 213–218. [Google Scholar] [CrossRef]
- Puglisi, S.; Rossini, A.; Poli, R.; Dughera, F.; Pia, A.; Terzolo, M.; Reimondo, G. Effects of SGLT2 Inhibitors and GLP-1 Receptor Agonists on Renin-Angiotensin-Aldosterone System. Front. Endocrinol. 2021, 12, 738848. [Google Scholar] [CrossRef]
- Wright, J.W.; Harding, J.W. The brain renin-angiotensin system: A diversity of functions and implications for CNS diseases. Pflug. Arch. 2013, 465, 133–151. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Otero-Losada, M.; Toblli, J.E.; Capani, F. Renin-angiotensin system as a potential target for new therapeutic approaches in Parkinson’s disease. Expert Opin. Investig. Drugs 2017, 26, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Fandino, J.; Vaz, A.A.; Toba, L.; Romani-Perez, M.; Gonzalez-Matias, L.; Mallo, F.; Diz-Chaves, Y. Liraglutide Enhances the Activity of the ACE-2/Ang(1-7)/Mas Receptor Pathway in Lungs of Male Pups from Food-Restricted Mothers and Prevents the Reduction of SP-A. Int. J. Endocrinol. 2018, 2018, 6920620. [Google Scholar] [CrossRef] [PubMed]
- Romani-Perez, M.; Outeirino-Iglesias, V.; Moya, C.M.; Santisteban, P.; Gonzalez-Matias, L.C.; Vigo, E.; Mallo, F. Activation of the GLP-1 Receptor by Liraglutide Increases ACE2 Expression, Reversing Right Ventricle Hypertrophy, and Improving the Production of SP-A and SP-B in the Lungs of Type 1 Diabetes Rats. Endocrinology 2015, 156, 3559–3569. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Miners, J.S.; Piva, G.; Willis, C.L.; Heard, D.M.; Kidd, E.J.; Good, M.A.; Kehoe, P.G. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020, 139, 485–502. [Google Scholar] [CrossRef] [Green Version]
- Rabie, M.A.; Abd El Fattah, M.A.; Nassar, N.N.; El-Abhar, H.S.; Abdallah, D.M. Angiotensin 1-7 ameliorates 6-hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type-1 receptor/NF-kappaB axis. Biochem. Pharm. 2018, 151, 126–134. [Google Scholar] [CrossRef]
- Regenhardt, R.W.; Desland, F.; Mecca, A.P.; Pioquinto, D.J.; Afzal, A.; Mocco, J.; Sumners, C. Anti-inflammatory effects of angiotensin-(1-7) in ischemic stroke. Neuropharmacology 2013, 71, 154–163. [Google Scholar] [CrossRef] [Green Version]
- Bairamian, D.; Sha, S.; Rolhion, N.; Sokol, H.; Dorothee, G.; Lemere, C.A.; Krantic, S. Microbiota in neuroinflammation and synaptic dysfunction: A focus on Alzheimer’s disease. Mol. Neurodegener. 2022, 17, 19. [Google Scholar] [CrossRef]
- Romano, S.; Savva, G.M.; Bedarf, J.R.; Charles, I.G.; Hildebrand, F.; Narbad, A. Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis. 2021, 7, 27. [Google Scholar] [CrossRef]
- Canfora, E.E.; Meex, R.C.R.; Venema, K.; Blaak, E.E. Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 2019, 15, 261–273. [Google Scholar] [CrossRef]
- Musso, G.; Gambino, R.; Cassader, M. Obesity, diabetes, and gut microbiota: The hygiene hypothesis expanded? Diabetes Care 2010, 33, 2277–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Tredici, K.; Braak, H. Review: Sporadic Parkinson’s disease: Development and distribution of alpha-synuclein pathology. Neuropathol. Appl. Neurobiol. 2016, 42, 33–50. [Google Scholar] [CrossRef]
- Jan, A.; Goncalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The Prion-like Spreading of Alpha-Synuclein in Parkinson’s Disease: Update on Models and Hypotheses. Int. J. Mol. Sci. 2021, 22, 8338. [Google Scholar] [CrossRef]
- Ferreira, N.; Goncalves, N.P.; Jan, A.; Jensen, N.M.; van der Laan, A.; Mohseni, S.; Vaegter, C.B.; Jensen, P.H. Trans-synaptic spreading of alpha-synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31. [Google Scholar] [CrossRef]
- Ferreira, N.; Richner, M.; van der Laan, A.; Bergholdt Jul Christiansen, I.; Vaegter, C.B.; Nyengaard, J.R.; Halliday, G.M.; Weiss, J.; Giasson, B.I.; Mackenzie, I.R.; et al. Prodromal neuroinvasion of pathological alpha-synuclein in brainstem reticular nuclei and white matter lesions in a model of alpha-synucleinopathy. Brain Commun. 2021, 3, fcab104. [Google Scholar] [CrossRef]
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737. [Google Scholar] [CrossRef] [Green Version]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuthati, Y.; Rao, V.N.; Busa, P.; Wong, C.S. Teneligliptin Exerts Antinociceptive Effects in Rat Model of Partial Sciatic Nerve Transection Induced Neuropathic Pain. Antioxidants 2021, 10, 1438. [Google Scholar] [CrossRef]
- Hillman, E.M. Coupling mechanism and significance of the BOLD signal: A status report. Annu. Rev. Neurosci. 2014, 37, 161–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| GLP-1RA | Backbone Modification | Frequency of Administration | Half-Life |
|---|---|---|---|
| Exenatide (Byetta/Bydureon) | Exendin-4 (resistant to DPP-4 cleavage, largely due to the substitution of the second amino acid from alanine to glycine) | Twice daily/weekly | 3.3–4 h |
| Lixisenatide (Adlyxin, Lyxumia) | Non-acylated GLP-1 (7–37) analogue based on exendin-4, but is modified by the deletion of one proline residue and with a C-terminal hexa-lysine extension | Daily | 2.6 h |
| Oral Semaglutide (Rybelsus); Semaglutide (Ozempic) | Acylated Human GLP-1 (7–37) analogue | Once daily; weekly | 1 week |
| Liraglutide (Victoza) | Mammalian GLP-1, substitution of lysine for arginine at position 28 with the addition of C-16 fatty acid | Daily | 13 h |
| Dulaglutide (Trulicity) | Mammalian GLP-1; the GLP-1 portion of the molecule is fused to an IgG4 molecule, limiting renal clearance and prolonging activity | Weekly | 1 week |
| Albiglutide (Eperzan and Tanzeum) | Two GLP-1 (7–36) molecules fused in tandem to human serum albumin | Weekly | 1 week |
| Taspoglutide | Modifications designed to delay DPP-4 cleavage and other serine proteases, with greater receptor binding | Weekly | 1 week |
| Studies | Experiment | GLP-1RA | Observations | Publications |
|---|---|---|---|---|
| Preclinical studies | Animal model | |||
| AD features | ||||
| Plaque load | APP/PS1/tau mice 5xFAD mice APP/PS1 mice 3xTg-AD mice | Liraglutide Liraglutide Lixisenatide Exendin-4 | Reduction of plaque load Reduction of plaque load Reduction of plaque load Reduction of plaque load | [72,76,77] [78] [76] [62] |
| Tau phosphorylation | APP/PS1/tau mice hTauP301L mice Aβ injection in mice APP/PS1 x db/db mice Streptozotocin injection in mice | Liraglutide Liraglutide Liraglutide Liraglutide Dulaglutide | Reduction of neurofibrillary tangles Reduced Tau phosphorylation Reduced Tau phosphorylation Reduced Tau phosphorylation Reduced Tau phosphorylation | [56,72] [58] [67] [79] [73] |
| Cognitive and memory performance | Aβ injection in mice Aβ injection in rats Streptozotocin injection in mice | Liraglutide Lixisenatide Dulaglutide | Improved cognitive impairment Improved spatial memory Improved memory ability | [74] [80] [73] |
| Other | Aβ injection in non-human primates | Liraglutide | Reduced synaptic loss | [74] |
| PD features | ||||
| Dopaminergic neuronal loss | 6-OHDA rat model 6-OHDA rat model 6-OHDA rat model | Liraglutide Exendin-4 Exendin-4′ | No influence on dopaminergic neuronal loss Neurogenesis Reduced lesions | [59] [55] [60] |
| Motor performance | MPTP mouse model MPTP mouse model | Liraglutide Lixisenatide | Improved motor control Improved motor control | [64] [64] |
| α-synuclein aggregation | Preformed fibrils injection in striatum of human A53T α-synuclein mice Preformed fibrils injection in the olfactory bulb of C57BL/6J mice | Exendin-4 (NLY01) Exendin-4 | Reduced loss of dopaminergic neurons and improved motor performance No significant reduction of α-synuclein aggregation | [81] [82] |
| Clinical trials | Trial ID | |||
| AD | NCT02140983 | Liraglutide | Increased connectivity in the default mode network | [85] |
| NCT01469351 NCT01843075 NCT01255163 NCT04777396 NCT04777409 | Liraglutide Liraglutide Exenatide Semaglutide Semaglutide | Improved cerebral glucose uptake Improved cognition No significant changes in cognition Recruiting Recruiting | [86,87] [88] [89] | |
| PD | NCT01971242 NCT01174810 NCT02953665 NCT03439943 NCT04154072 NCT03659682 | Exenatide Exenatide Liraglutide Lixisenatide Exenatide Semaglutide | Improved motor and cognitive outcomes Improved motor and cognitive outcomes Active Active Active Not yet recruiting | [90] [91] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, G.; Gomes Moreira, D.; Richner, M.; Mutsaers, H.A.M.; Ferreira, N.; Jan, A. GLP-1 Receptor Agonists in Neurodegeneration: Neurovascular Unit in the Spotlight. Cells 2022, 11, 2023. https://doi.org/10.3390/cells11132023
Monti G, Gomes Moreira D, Richner M, Mutsaers HAM, Ferreira N, Jan A. GLP-1 Receptor Agonists in Neurodegeneration: Neurovascular Unit in the Spotlight. Cells. 2022; 11(13):2023. https://doi.org/10.3390/cells11132023
Chicago/Turabian StyleMonti, Giulia, Diana Gomes Moreira, Mette Richner, Henricus Antonius Maria Mutsaers, Nelson Ferreira, and Asad Jan. 2022. "GLP-1 Receptor Agonists in Neurodegeneration: Neurovascular Unit in the Spotlight" Cells 11, no. 13: 2023. https://doi.org/10.3390/cells11132023
APA StyleMonti, G., Gomes Moreira, D., Richner, M., Mutsaers, H. A. M., Ferreira, N., & Jan, A. (2022). GLP-1 Receptor Agonists in Neurodegeneration: Neurovascular Unit in the Spotlight. Cells, 11(13), 2023. https://doi.org/10.3390/cells11132023