
Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis


Abstract
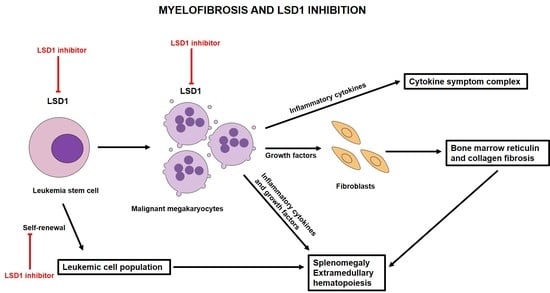
:
1. Introduction
2. Mutations in Epigenetic Regulators in Myelofibrosis
3. The Functional Role of LSD1 in Hematopoiesis
4. LSD1 as an Epigenetic Regulator
5. The Biological Role of LSD1 and the Effect of LSD1 Inhibitors in Murine Models of MPNs
6. Clinical Data of LSD1 Inhibitors in Myelofibrosis
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spivak, J.L. Myeloproliferative Neoplasms. N. Engl. J. Med. 2017, 376, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Moulard, O.; Mehta, J.; Fryzek, J.; Olivares, R.; Iqbal, U.; Mesa, R.A. Epidemiology of myelofibrosis, essential thrombocythemia, and polycythemia vera in the European Union. Eur. J. Haematol. 2014, 92, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Srour, S.A.; Devesa, S.S.; Morton, L.M.; Check, D.P.; Curtis, R.E.; Linet, M.S.; Dores, G.M. Incidence and patient survival of myeloproliferative neoplasms and myelodysplastic/myeloproliferative neoplasms in the United States, 2001-12. Br. J. Haematol. 2016, 174, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Hultcrantz, M.; Kristinsson, S.Y.; Andersson, T.M.-L.; Landgren, O.; Eloranta, S.; Derolf, A.R.; Dickman, P.W.; Björkholm, M. Patterns of Survival Among Patients With Myeloproliferative Neoplasms Diagnosed in Sweden From 1973 to 2008: A Population-Based Study. J. Clin. Oncol. 2012, 30, 2995–3001. [Google Scholar] [CrossRef]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2513. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Harrison, C.N. Emerging treatments for classical myeloproliferative neoplasms. Blood 2017, 129, 693–703. [Google Scholar] [CrossRef]
- Tefferi, A. Primary myelofibrosis: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2018, 93, 1551–1560. [Google Scholar] [CrossRef]
- Tefferi, A.; Barbui, T. Polycythemia vera and essential thrombocythemia: 2019 update on diagnosis, risk-stratification and management. Am. J. Hematol. 2019, 94, 133–143. [Google Scholar] [CrossRef]
- England, J.; Gupta, V. Novel therapies vs hematopoietic cell transplantation in myelofibrosis: Who, when, how? Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 453–462. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind, Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. N. Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Harrison, C.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Waltzman, R.; Stalbovskaya, V.; McQuitty, M.; Hunter, D.S.; Levy, R.; Knoops, L.; et al. JAK Inhibition with Ruxolitinib versus Best Available Therapy for Myelofibrosis. N. Engl. J. Med. 2012, 366, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Mesa, R.A.; Gotlib, J.; Gupta, V.; Catalano, J.V.; Deininger, M.W.; Shields, A.L.; Miller, C.B.; Silver, R.T.; Talpaz, M.; Winton, E.F.; et al. Effect of Ruxolitinib Therapy on Myelofibrosis-Related Symptoms and Other Patient-Reported Outcomes in COMFORT-I: A Randomized, Double-Blind, Placebo-Controlled Trial. J. Clin. Oncol. 2013, 31, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. The clinical benefit of ruxolitinib across patient subgroups: Analysis of a placebo-controlled, Phase III study in patients with myelofibrosis. Br. J. Haematol. 2013, 161, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Verstovsek, S.; Gotlib, J.; Mesa, R.A.; Vannucchi, A.M.; Kiladjian, J.-J.; Cervantes, F.; Harrison, C.N.; Paquette, R.; Sun, W.; Naim, A.; et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J. Hematol. Oncol. 2017, 10, 156. [Google Scholar] [CrossRef]
- Guglielmelli, P.; Ghirardi, A.; Carobbio, A.; Masciulli, A.; Maccari, C.; Mora, B.; Rumi, E.; Triguero, A.; Finazzi, M.C.; Pettersson, H.; et al. Impact of ruxolitinib on survival of patients with myelofibrosis in the real world: Update of the ERNEST Study. Blood Adv. 2022, 6, 373–375. [Google Scholar] [CrossRef]
- Harrison, C.N.; on behalf of the COMFORT-II Investigators; Vannucchi, A.M.; Kiladjian, J.-J.; Al-Ali, H.K.; Gisslinger, H.; Knoops, L.; Cervantes, F.; Jones, M.M.; Sun, K.; et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016, 30, 1701–1707. [Google Scholar] [CrossRef]
- Harrison, C.N.; Schaap, N.; Vannucchi, A.M.; Kiladjian, J.-J.; Tiu, R.V.; Zachee, P.; Jourdan, E.; Winton, E.; Silver, R.T.; Schouten, H.C.; et al. Janus kinase-2 inhibitor fedratinib in patients with myelofibrosis previously treated with ruxolitinib (JAKARTA-2): A single-arm, open-label, non-randomised, phase 2, multicentre study. Lancet Haematol. 2017, 4, e317–e324. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Hoffman, R.; Talpaz, M.; Gerds, A.T.; Stein, B.; Gupta, V.; Szoke, A.; Drummond, M.; Pristupa, A.; Granston, T.; et al. Pacritinib vs Best Available Therapy, Including Ruxolitinib, in Patients With Myelofibrosis: A Randomized Clinical Trial. JAMA Oncol. 2018, 4, 652–659. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Bose, P.; Verstovsek, S. Momelotinib: An emerging treatment for myelofibrosis patients with anemia. J. Hematol. Oncol. 2022, 15, 1–17. [Google Scholar] [CrossRef]
- Vachhani, P.; Verstovsek, S.; Bose, P. Disease Modification in Myelofibrosis: An Elusive Goal? J. Clin. Oncol. 2022, 40, 1147–1154. [Google Scholar] [CrossRef]
- Tremblay, D.; Hoffman, R. Emerging drugs for the treatment of myelofibrosis: Phase II & III clinical trials. Expert Opin. Emerg. Drugs 2021, 26, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood 2017, 129, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Leung, A.Y.; Kwong, Y.-L. Molecular and Cellular Mechanisms of Myelodysplastic Syndrome: Implications on Targeted Therapy. Int. J. Mol. Sci. 2016, 17, 440. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Leung, A.Y.; Kwong, Y.-L. Molecularly targeted therapy in acute myeloid leukemia. Futur. Oncol. 2016, 12, 827–838. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation inTET2in Myeloid Cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Tefferi, A.; Pardanani, A.; Lim, K.-H.; Abdel-Wahab, O.; Lasho, T.L.; Patel, J.; Gangat, N.; Finke, C.M.; Schwager, S.; Mullally, A.; et al. TET2 mutations and their clinical correlates in polycythemia vera, essential thrombocythemia and myelofibrosis. Leukemia 2009, 23, 905–911. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 601–612. [Google Scholar] [CrossRef]
- Kent, D.G.; Ortmann, C.A.; Green, A.R.; Delhommeau, F. Effect of Mutation Order on Myeloproliferative Neoplasms. N. Engl. J. Med. 2015, 372, 1865–1866. [Google Scholar] [CrossRef]
- Stegelmann, F.; Bullinger, L.; Schlenk, R.F.; Paschka, P.; Griesshammer, M.; Blersch, C.; Kuhn, S.; Schauer, S.; Dohner, H.; Dohner, K. DNMT3A mutations in myeloproliferative neoplasms. Leukemia 2011, 25, 1217–1219. [Google Scholar] [CrossRef]
- Challen, G.A.; Sun, D.; Jeong, M.; Luo, M.; Jelinek, J.; Berg, J.S.; Bock, C.; Vasanthakumar, A.; Gu, H.; Xi, Y.; et al. Dnmt3a is essential for hematopoietic stem cell differentiation. Nat. Genet. 2011, 44, 23–31. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Lu, C.; Wang, J.; McLellan, M.D.; Johnson, K.J.; Wendl, M.C.; McMichael, J.F.; Schmidt, H.K.; Yellapantula, V.; Miller, C.A.; et al. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014, 20, 1472–1478. [Google Scholar] [CrossRef] [PubMed]
- Guglielmelli, P.; Biamonte, F.; Score, J.; Hidalgo-Curtis, C.; Cervantes, F.; Maffioli, M.; Fanelli, T.; Ernst, T.; Winkelman, N.; Jones, A.V.; et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood 2011, 118, 5227–5234. [Google Scholar] [CrossRef]
- Yang, Y.; Akada, H.; Nath, D.; Hutchison, R.E.; Mohi, G. Loss of Ezh2 cooperates with Jak2V617F in the development of myelofibrosis in a mouse model of myeloproliferative neoplasm. Blood 2016, 127, 3410–3423. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Kubovcakova, L.; Nienhold, R.; Zmajkovic, J.; Meyer, S.C.; Hao-Shen, H.; Geier, F.; Dirnhofer, S.; Guglielmelli, P.; Vannucchi, A.M.; et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J. Exp. Med. 2016, 213, 1479–1496. [Google Scholar] [CrossRef]
- Sashida, G.; Wang, C.; Tomioka, T.; Oshima, M.; Aoyama, K.; Kanai, A.; Mochizuki-Kashio, M.; Harada, H.; Shimoda, K.; Iwama, A. The loss of Ezh2 drives the pathogenesis of myelofibrosis and sensitizes tumor-initiating cells to bromodomain inhibition. J. Exp. Med. 2016, 213, 1459–1477. [Google Scholar] [CrossRef]
- Carbuccia, N.; Murati, A.; Trouplin, V.; Brecqueville, M.; Adelaide, J.; Rey, J.; Vainchenker, W.; Bernard, O.A.; Chaffanet, M.; Vey, N.; et al. Mutations of ASXL1 gene in myeloproliferative neoplasms. Leukemia 2009, 23, 2183–2186. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Lasho, T.L.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C.; et al. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869. [Google Scholar] [CrossRef]
- Gelsi-Boyer, V.; Trouplin, V.; Adélaïde, J.; Bonansea, J.; Cervera, N.; Carbuccia, N.; Lagarde, A.; Prebet, T.; Nezri, M.; Sainty, D.; et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br. J. Haematol. 2009, 145, 788–800. [Google Scholar] [CrossRef]
- Abdel-Wahab, O.; Gao, J.; Adli, M.; Dey, A.; Trimarchi, T.; Chung, Y.R.; Kuscu, C.; Hricik, T.; Ndiaye-Lobry, D.; LaFave, L.M.; et al. Deletion of Asxl1 results in myelodysplasia and severe developmental defects in vivo. J. Exp. Med. 2013, 210, 2641–2659. [Google Scholar] [CrossRef]
- Inoue, D.; Kitaura, J.; Togami, K.; Nishimura, K.; Enomoto, Y.; Uchida, T.; Kagiyama, Y.; Kawabata, K.C.; Nakahara, F.; Izawa, K.; et al. Myelodysplastic syndromes are induced by histone methylation–altering ASXL1 mutations. J. Clin. Investig. 2013, 123, 4627–4640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-J.; Rampal, R.; Manshouri, T.; Patel, J.; Mensah, N.; Kayserian, A.; Hricik, T.; Heguy, A.; Hedvat, C.; Gönen, M.; et al. Genetic analysis of patients with leukemic transformation of myeloproliferative neoplasms shows recurrent SRSF2 mutations that are associated with adverse outcome. Blood 2012, 119, 4480–4485. [Google Scholar] [CrossRef] [PubMed]
- Harutyunyan, A.; Klampfl, T.; Cazzola, M.; Kralovics, R. p53 Lesions in Leukemic Transformation. N. Engl. J. Med. 2011, 364, 488–490. [Google Scholar] [CrossRef]
- Wong, T.N.; Ramsingh, G.; Young, A.L.; Miller, C.A.; Touma, W.; Welch, J.S.; Lamprecht, T.L.; Shen, D.; Hundal, J.; Fulton, R.S.; et al. Role of TP53 mutations in the origin and evolution of therapy-related acute myeloid leukaemia. Nature 2015, 518, 552–555. [Google Scholar] [CrossRef]
- Courtier, F.; Carbuccia, N.; Garnier, S.; Guille, A.; Adélaïde, J.; Cervera, N.; Gelsi-Boyer, V.; Mozziconacci, M.-J.; Rey, J.; Vey, N.; et al. Genomic analysis of myeloproliferative neoplasms in chronic and acute phases. Haematologica 2016, 102, e11–e14. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, J.D.; Puda, A.; Malcovati, L.; Berg, T.; Hofbauer, M.; Stukalov, A.; Klampfl, T.; Harutyunyan, A.S.; Gisslinger, H.; Gisslinger, B.; et al. Clinical significance of genetic aberrations in secondary acute myeloid leukemia. Am. J. Hematol. 2012, 87, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Lasho, T.L.; Gangat, N.; Ketterling, R.P.; Pardanani, A.; Vannucchi, A.M. MIPSS70+ Version 2.0: Mutation and Karyotype-Enhanced International Prognostic Scoring System for Primary Myelofibrosis. J. Clin. Oncol. 2018, 36, 1769–1770. [Google Scholar] [CrossRef] [PubMed]
- Tefferi, A.; Guglielmelli, P.; Nicolosi, M.; Mannelli, F.; Mudireddy, M.; Bartalucci, N.; Finke, C.M.; Lasho, T.L.; Hanson, C.A.; Ketterling, R.P.; et al. GIPSS: Genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia 2018, 32, 1631–1642. [Google Scholar] [CrossRef]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The lysine demethylase LSD1 (KDM1) is required for maintenance of global DNA methylation. Nat. Genet. 2009, 41, 125–129. [Google Scholar] [CrossRef]
- Foster, C.T.; Dovey, O.M.; Lezina, L.; Luo, J.L.; Gant, T.W.; Barlev, N.; Bradley, A.; Cowley, S.M. Lysine-Specific Demethylase 1 Regulates the Embryonic Transcriptome and CoREST Stability. Mol. Cell. Biol. 2010, 30, 4851–4863. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Scully, K.; Zhu, X.; Cai, L.; Zhang, J.; Prefontaine, G.G.; Krones, A.; Ohgi, K.A.; Zhu, P.; Garcia-Bassets, I.; et al. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 2007, 446, 882–887. [Google Scholar] [CrossRef]
- Sprüssel, A.; Schulte, J.H.; Weber, S.; Necke, M.; Händschke, K.; Thor, T.; Pajtler, K.W.; Schramm, A.; König, K.; Diehl, L.; et al. Lysine-specific demethylase 1 restricts hematopoietic progenitor proliferation and is essential for terminal differentiation. Leukemia 2012, 26, 2039–2051. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Bilodeau, S.; Orlando, D.A.; Hoke, H.A.; Frampton, G.M.; Foster, C.T.; Cowley, S.M.; Young, R.A. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 2012, 482, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef]
- Kaniskan, H.U.; Martini, M.L.; Jin, J. Inhibitors of Protein Methyltransferases and Demethylases. Chem. Rev. 2018, 118, 989–1068. [Google Scholar] [CrossRef]
- Hoffmann, I.; Roatsch, M.; Schmitt, M.L.; Carlino, L.; Pippel, M.; Sippl, W.; Jung, M. The role of histone demethylases in cancer therapy. Mol. Oncol. 2012, 6, 683–703. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A.; Shi, Y. Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Beisel, C.; Paro, R. Silencing chromatin: Comparing modes and mechanisms. Nat. Rev. Genet. 2011, 12, 123–135. [Google Scholar] [CrossRef]
- Kornberg, R.D. Chromatin Structure: A Repeating Unit of Histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Karakaidos, P.; Verigos, J.; Magklara, A. LSD1/KDM1A, a Gate-Keeper of Cancer Stemness and a Promising Therapeutic Target. Cancers 2019, 11, 1821. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Liao, G.; Yu, B. LSD1/KDM1A inhibitors in clinical trials: Advances and prospects. J. Hematol. Oncol. 2019, 12, 1–14. [Google Scholar] [CrossRef]
- Fang, Y.; Liao, G.; Yu, B. Targeting Histone Lysine Demethylase LSD1/KDM1A as a New Avenue for Cancer Therapy. Curr. Top. Med. Chem. 2019, 19, 889–891. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-C.; Ma, J.; Wang, Z.; Li, J.; Bailing, J.; Jiang, B.; Zhou, W.; Shi, X.; Wang, X.; Zhao, W.; et al. A Systematic Review of Histone Lysine-Specific Demethylase 1 and Its Inhibitors. Med. Res. Rev. 2015, 35, 1032–1071. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Müller, J.M.; Schneider, R.; Peters, A.H.; Günther, T.; Buettner, R.; Schüle, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Saleque, S.; Kim, J.; Rooke, H.M.; Orkin, S.H. Epigenetic Regulation of Hematopoietic Differentiation by Gfi-1 and Gfi-1b Is Mediated by the Cofactors CoREST and LSD1. Mol. Cell 2007, 27, 562–572. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar] [CrossRef]
- Shi, Y.-J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 Histone Demethylase Activity by Its Associated Factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Orkin, S.H.; Hochedlinger, K. Chromatin Connections to Pluripotency and Cellular Reprogramming. Cell 2011, 145, 835–850. [Google Scholar] [CrossRef] [PubMed]
- Kahl, P.; Gullotti, L.; Heukamp, L.C.; Wolf, S.; Friedrichs, N.; Vorreuther, R.; Solleder, G.; Bastian, P.J.; Ellinger, J.; Metzger, E.; et al. Androgen Receptor Coactivators Lysine-Specific Histone Demethylase 1 and Four and a Half LIM Domain Protein 2 Predict Risk of Prostate Cancer Recurrence. Cancer Res. 2006, 66, 11341–11347. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.H.; Lim, S.; Schramm, A.; Friedrichs, N.; Koster, J.; Versteeg, R.; Ora, I.; Pajtler, K.; Klein-Hitpass, L.; Kuhfittig-Kulle, S.; et al. Lysine-Specific Demethylase 1 Is Strongly Expressed in Poorly Differentiated Neuroblastoma: Implications for Therapy. Cancer Res. 2009, 69, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Niebel, D.; Kirfel, J.; Janzen, V.; Höller, T.; Majores, M.; Gütgemann, I. Lysine-specific demethylase 1 (LSD1) in hematopoietic and lymphoid neoplasms. Blood 2014, 124, 151–152. [Google Scholar] [CrossRef] [PubMed]
- Somervaille, T.C.; Cleary, M.L. Identification and characterization of leukemia stem cells in murine MLL-AF9 acute myeloid leukemia. Cancer Cell 2006, 10, 257–268. [Google Scholar] [CrossRef]
- Somervaille, T.C.; Matheny, C.J.; Spencer, G.J.; Iwasaki, M.; Rinn, J.L.; Witten, D.M.; Chang, H.Y.; Shurtleff, S.A.; Downing, J.R.; Cleary, M.L. Hierarchical Maintenance of MLL Myeloid Leukemia Stem Cells Employs a Transcriptional Program Shared with Embryonic Rather Than Adult Stem Cells. Cell Stem Cell 2009, 4, 129–140. [Google Scholar] [CrossRef]
- Harris, W.J.; Huang, X.; Lynch, J.T.; Spencer, G.J.; Hitchin, J.R.; Li, Y.; Ciceri, F.; Blaser, J.G.; Greystoke, B.F.; Jordan, A.M.; et al. The Histone Demethylase KDM1A Sustains the Oncogenic Potential of MLL-AF9 Leukemia Stem Cells. Cancer Cell 2012, 21, 473–487. [Google Scholar] [CrossRef]
- Lin, X.-M.; Zhong, W.-T.; Wang, C.-L.; Wang, S.-Q. Expression of histone demethylase lysine specific demethylase 1 in acute leukemia and its clinical significance. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2011, 19, 1348–1352. [Google Scholar]
- Rhodes, D.R.; Kalyana-Sundaram, S.; Mahavisno, V.; Varambally, R.; Yu, J.; Briggs, B.B.; Barrette, T.R.; Anstet, M.J.; Kincead-Beal, C.; Kulkarni, P.; et al. Oncomine 3.0: Genes, Pathways, and Networks in a Collection of 18,000 Cancer Gene Expression Profiles. Neoplasia 2007, 9, 166–180. [Google Scholar] [CrossRef]
- Chen, E.; Mullally, A. How does JAK2V617F contribute to the pathogenesis of myeloproliferative neoplasms? Hematol. Am. Soc. Hematol. Educ. Program 2014, 2014, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed]
- Jutzi, J.S.; Kleppe, M.; Dias, J.; Staehle, H.F.; Shank, K.; Teruya-Feldstein, J.; Gambheer, S.M.M.; Dierks, C.; Rienhoff, H.Y., Jr.; Levine, R.L.; et al. LSD1 Inhibition Prolongs Survival in Mouse Models of MPN by Selectively Targeting the Disease Clone. HemaSphere 2018, 2, e54. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Yacoub, A.; Pettit, K.M.; Bradley, T.; Gerds, A.T.; Tatarczuch, M.; Shortt, J.; Curtin, N.J.; Rossetti, J.M.; Burbury, K.; et al. A Phase 2 Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Advanced Myelofibrosis. Blood 2021, 138, 139. [Google Scholar] [CrossRef]
- Gill, H.; Yacoub, A.; Pettit, K.; Bradley, T.; Gerds, A.; Tatarczuch, M.; Shortt, J.; Curtin, N.; Rossetti, J.; Burbury, K.; et al. A phase 2 study of IMG-7289 (bomedemstat) in patients with advanced myelofibrosis. HemaSphere 2022, 6, 1809. [Google Scholar] [CrossRef]
- Kohrogi, K.; Hino, S.; Sakamoto, A.; Anan, K.; Takase, R.; Araki, H.; Hino, Y.; Araki, K.; Sato, T.; Nakamura, K.; et al. LSD1 defines erythroleukemia metabolism by controlling the lineage-specific transcription factors GATA1 and C/EBPα. Blood Adv. 2021, 5, 2305–2318. [Google Scholar] [CrossRef]
- Rivers, A.; Vaitkus, K.; Ibanez, V.; Ruiz, M.A.; Jagadeeswaran, R.; Saunthararajah, Y.; Cui, S.; Engel, J.D.; DeSimone, J.; Lavelle, D. The LSD1 inhibitor RN-1 recapitulates the fetal pattern of hemoglobin synthesis in baboons (P. anubis). Haematologica 2016, 101, 688–697. [Google Scholar] [CrossRef]
- Palandri, F.; Vianelli, N.; Ross, D.M.; Cochrane, T.; Lane, S.W.; Larsen, S.R.; Gerds, A.T.; Halpern, A.B.; Shortt, J.; Rossetti, J.M.; et al. A Phase 2 Study of the LSD1 Inhibitor Img-7289 (bomedemstat) for the Treatment of Essential Thrombocythemia (ET). Blood 2021, 138, 386. [Google Scholar] [CrossRef]
- Kremyanskaya, M.; Mascarenhas, J.; Palandri, F.; Vannucchi, A.; Verstovsek, S.; Harrison, C.N.; Bose, P.; Schiller, G.J.; Rampal, R.; Drummond, M.W.; et al. Pelabresib (CPI-0610) Monotherapy in Patients with Myelofibrosis—Update of Clinical and Translational Data from the Ongoing Manifest Trial. Blood 2021, 138, 141. [Google Scholar] [CrossRef]
- Verstovsek, S.; Salama, M.E.; Mascarenhas, J.; Talpaz, M.; Mesa, R.A.; Vannucchi, A.; Rampal, R.; Oh, S.T.; Olteanu, H.; Chiu, A.; et al. Disease-Modifying Potential of BET Inhibitor Pelabresib (CPI-0610) As Demonstrated By Improvements in Bone Marrow Function and Clinical Activity in Patients with Myelofibrosis-Preliminary Data. Blood 2021, 138, 2568. [Google Scholar] [CrossRef]
- Mascarenhas, J.; Komrokji, R.S.; Palandri, F.; Martino, B.; Niederwieser, D.; Reiter, A.; Scott, B.L.; Baer, M.R.; Hoffman, R.; Odenike, O.; et al. Randomized, Single-Blind, Multicenter Phase II Study of Two Doses of Imetelstat in Relapsed or Refractory Myelofibrosis. J. Clin. Oncol. 2021, 39, 2881–2892. [Google Scholar] [CrossRef]
- Pemmaraju, N.; Garcia, J.S.; Potluri, J.; Holes, B.L.; Harb, J.; Jung, P.; Hutti, J.E.; Prchal, J.T.; Verstovsek, S.; Harrison, D.C. The Addition of Navitoclax to Ruxolitinib Demonstrates Efficacy within Different High-Risk Populations in Patients with Relapsed/Refractory Myelofibrosis. Blood 2020, 136, 49–50. [Google Scholar] [CrossRef]
- Vachani, P.; Lange, A.; Delgado, R.G.; Al-Ali, H.K.; Hernández-Rivas, J.M.; Kiladjian, J.-J.; Vannucchi, A.; Perkins, A.C.; Valmeekam, V.; Krejsa, C.M.; et al. Potential Disease-Modifying Activity of Navtemadlin (KRT-232), a First-in-Class MDM2 Inhibitor, Correlates with Clinical Benefits in Relapsed/Refractory Myelofibrosis (MF). Blood 2021, 138, 3581. [Google Scholar] [CrossRef]
- Masarova, L.; Verstovsek, S.; Hidalgo-Lopez, J.E.; Pemmaraju, N.; Bose, P.; Estrov, Z.; Jabbour, E.J.; Ravandi-Kashani, F.; Takahashi, K.; Cortes, J.; et al. A phase 2 study of ruxolitinib in combination with azacitidine in patients with myelofibrosis. Blood 2018, 132, 1664–1674. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Class | Drug | Study Population | Design | SVR35 at 24 Weeks | TSS50 at 24 Weeks | Anemia Response | VAF Reduction | BM Fibrosis Reduction |
|---|---|---|---|---|---|---|---|---|
| LSD1 inhibitor | Bomedemstat [84,85] | Ruxolitinib exposed: 83% (74/89) N = 89 | Phase 2 (ongoing) Single-agent bomedemstat | 6% (3/50) | 19% (5/26) | In TD patients: 52% (11/21) had stable or reduced transfusion burden; 14% (3/21) became TI | VAF reduction 52% (36/69), most frequently in JAK2V617F and/or ASXL1 | 31% (16/52) improved by 1 grade 50% (26/52) stable |
| BET inhibitor | Pelabresib (MANIFEST) [89,90] | Both JAKi exposed and JAKi naïve, N = 271 | Phase 1/2 (ongoing) Arm 1: JAKi exposed (pelabresib) Arm 2: JAKi exposed (pelabrasib + ruxolitinib) Arm 3: JAKi naïve (pelabresib + ruxolitinib) | Arm 1: 11% (7/64) Arm 2: 20% (16/81) Arm 3: 68% (57/84) | Arm 1: 28% (18/64) Arm 2: 37% (30/81) Arm 3: 56% (46/82) | Arm 1: In TD patients, 16% (4/25) became TI Arm 2: In TD patients, 36% (13/36) became TI Arm 3: In patients with Hb < 10 g/dL; Hb improved by 1 g/dL | Not reported | Arm 1: 23% (7/30) improved at 24 weeks Arm 2: 25% (9/36) improved at 24 weeks Arm 3: 31% (16/52) improved at 24 weeks |
| Telomerase inhibitor | Imetelstat (IMBark) [91] | JAKi exposed N = 59 | Phase 2 (complete) Single-agent imetelstat | 10.2% (6/59) | 32.2% (19/59) | In TD patients, 25% (3/12) became transfusion-independent | 42% had ≥25% reduction in VAF | 41% (15/37) had reduction in BM fibrosis |
| BH3 mimetic; Bcl-2/Bcl-XL inhibitor | Navitoclax (REFINE) [92] | Ruxolitinib exposed N = 174 | Phase 2 (ongoing) Navitoclax +/− ruxolitinib | 27% (9/34) | 30% (9/34) | In TD patients or patients with Hb < 10 g/dL; TI or ≥ 2 g/dL in 64% (7/11) | 46% (12/26) had >10% reduction in VAF | 21% (7/34) had BM fibrosis reduction at 24 weeks |
| MDM2 inhibitor | Navtemadlin (BOREAS) [93] | JAKi exposed N = 113 | Phase 2 (ongoing) Single-agent navtemadlin | Not reported | Not reported | Not reported | 34% had ≥20% reduction in VAF | 27% ≥ 1 grade reduction in BM fibrosis |
| Hypomethylating agent | Azacitidine [94] | JAKi naïve N = 60 | Phase 2 Ruxolitinib + azacitidine | NR | 54% (25/46) | In TD patients, 20% (1/5) became TI | 81% (13/16) had reduction in JAK2V617F VAF at 24 weeks | 57% (8/14) had reduction in BM fibrosis at 24 weeks |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gill, H. Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis. Cells 2022, 11, 2107. https://doi.org/10.3390/cells11132107
Gill H. Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis. Cells. 2022; 11(13):2107. https://doi.org/10.3390/cells11132107
Chicago/Turabian StyleGill, Harinder. 2022. "Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis" Cells 11, no. 13: 2107. https://doi.org/10.3390/cells11132107
APA StyleGill, H. (2022). Lysine-Specific Demethylase 1 (LSD1/KDM1A) Inhibition as a Target for Disease Modification in Myelofibrosis. Cells, 11(13), 2107. https://doi.org/10.3390/cells11132107