
Ribosomes and Ribosomal Proteins Promote Plasticity and Stemness Induction in Glioma Cells via Reprogramming


Abstract
:1. Introduction
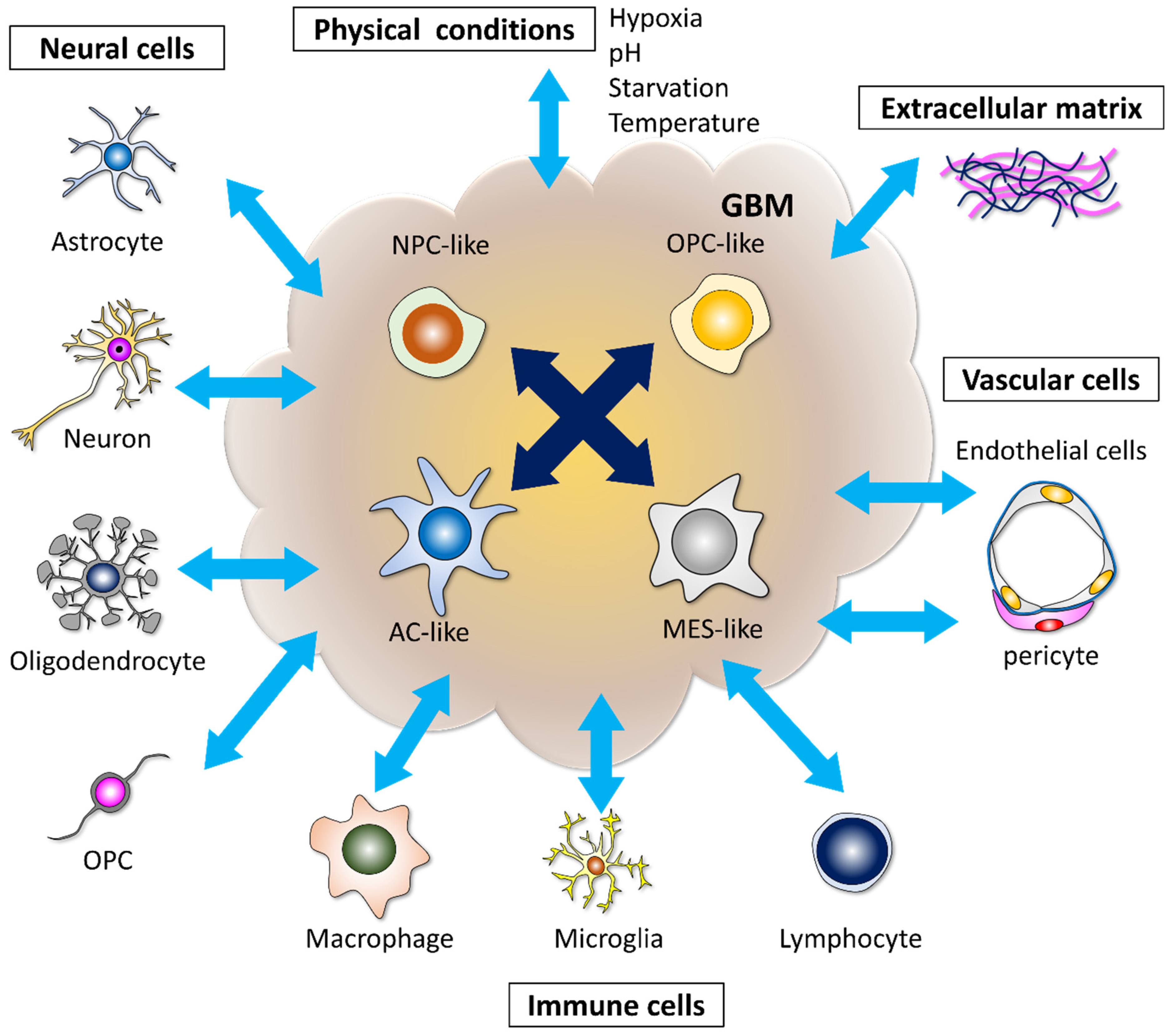
2. Microenvironments Induce Heterogeneity and Therapeutic Resistance in GBM
3. Reprogramming Potential of Glioma Cells
3.1. Indirect Reprogramming and Direct Reprogramming of Non-Cancer Cells
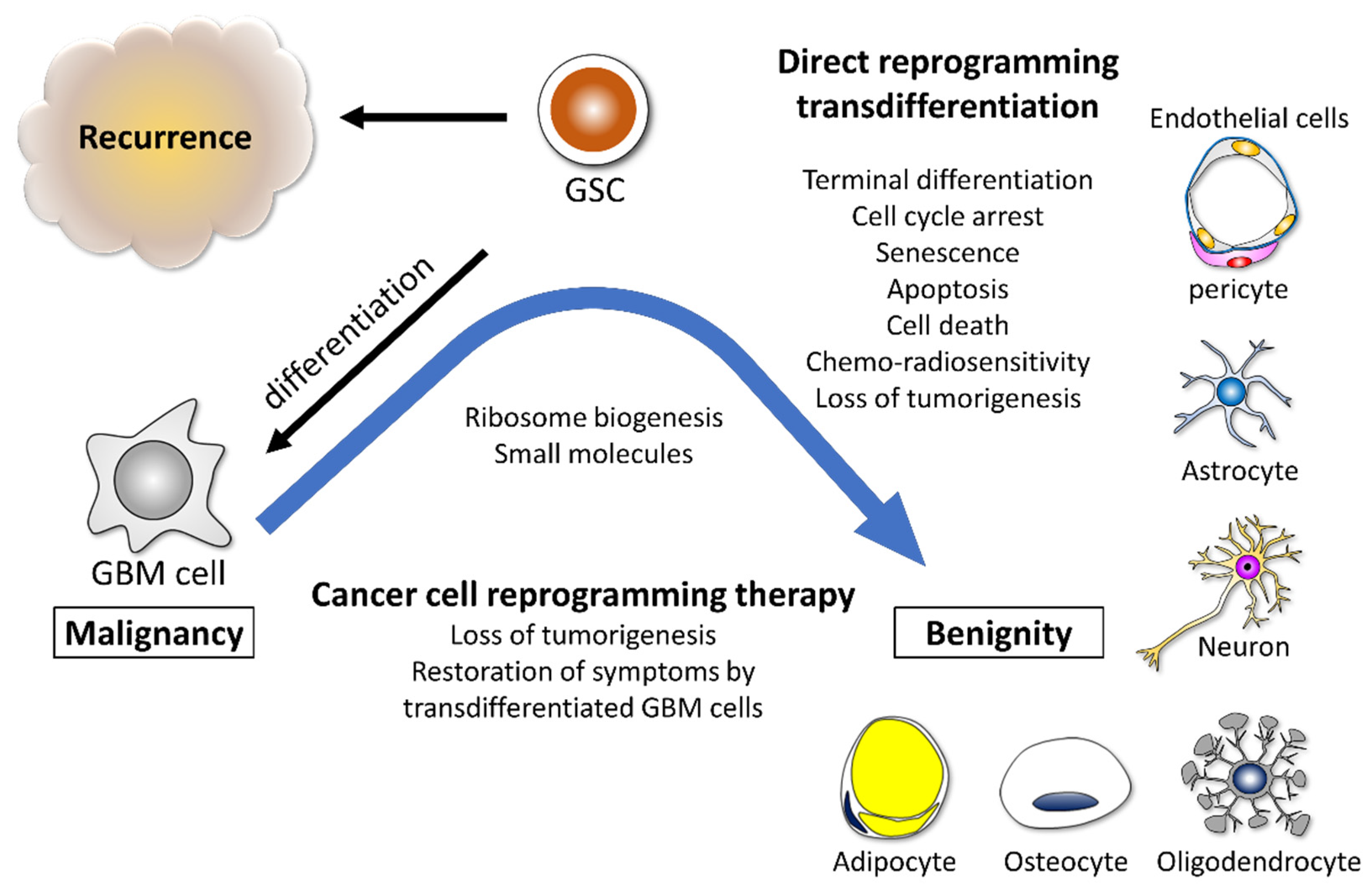
3.2. Glioma Cells Possess Potential for Reprogramming and Transdifferentiation
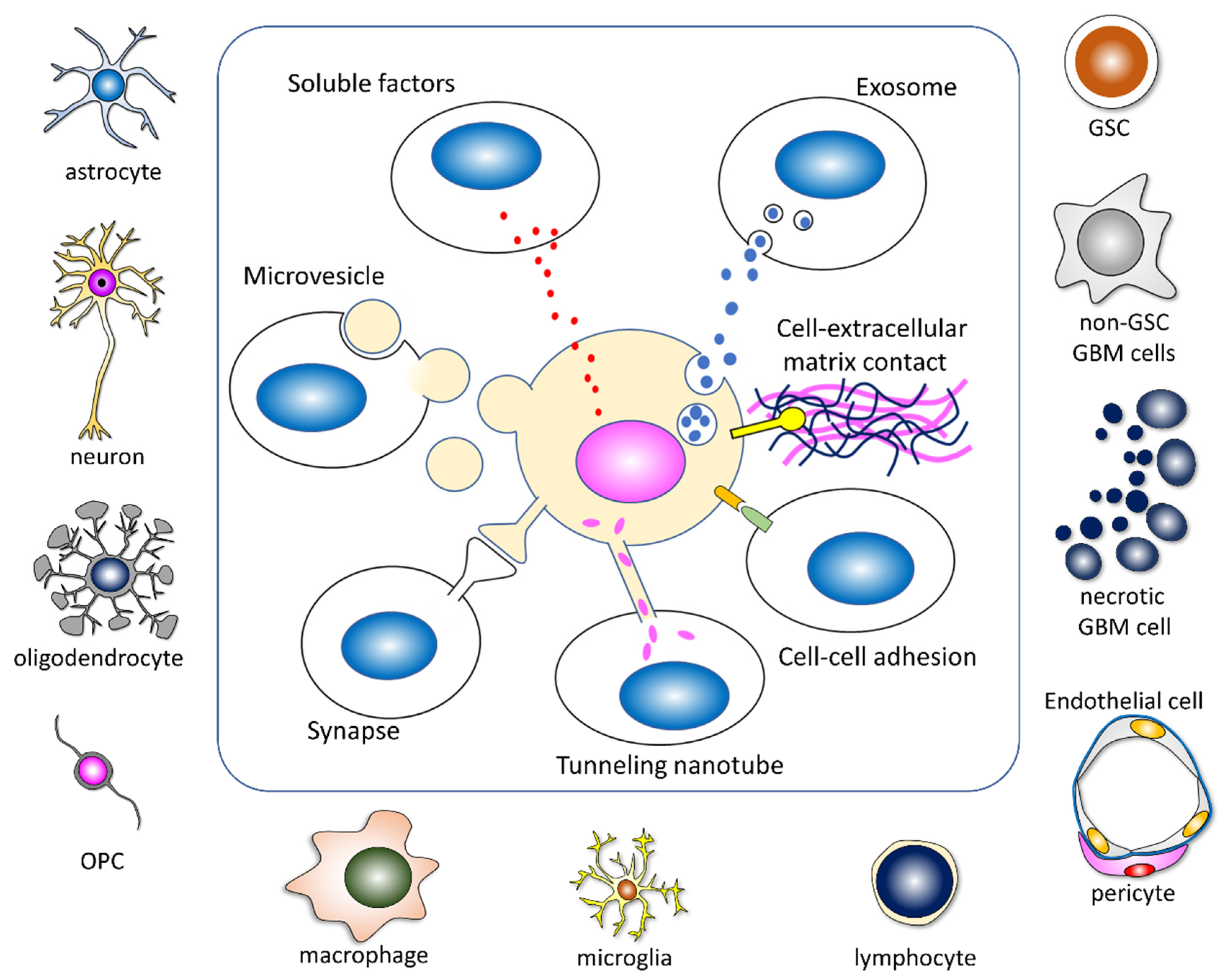
3.3. GBM Cells Acquire an Aggressive Phenotype by Reprogramming through Intercellular Communication
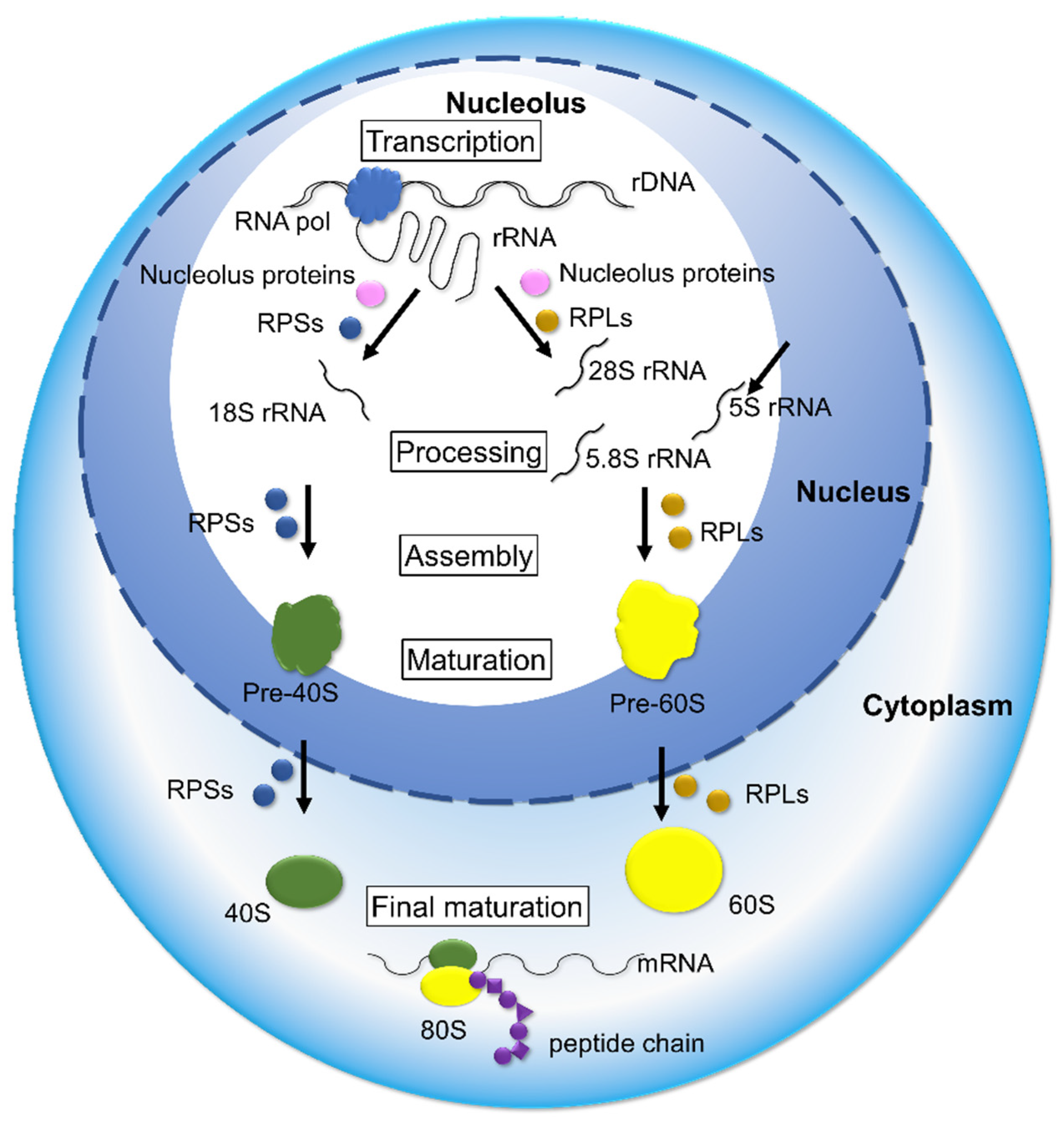
4. Deregulation of Ribosome Biogenesis in Cancer
5. Ribosome Incorporation Induces Reprogramming in Somatic Cells
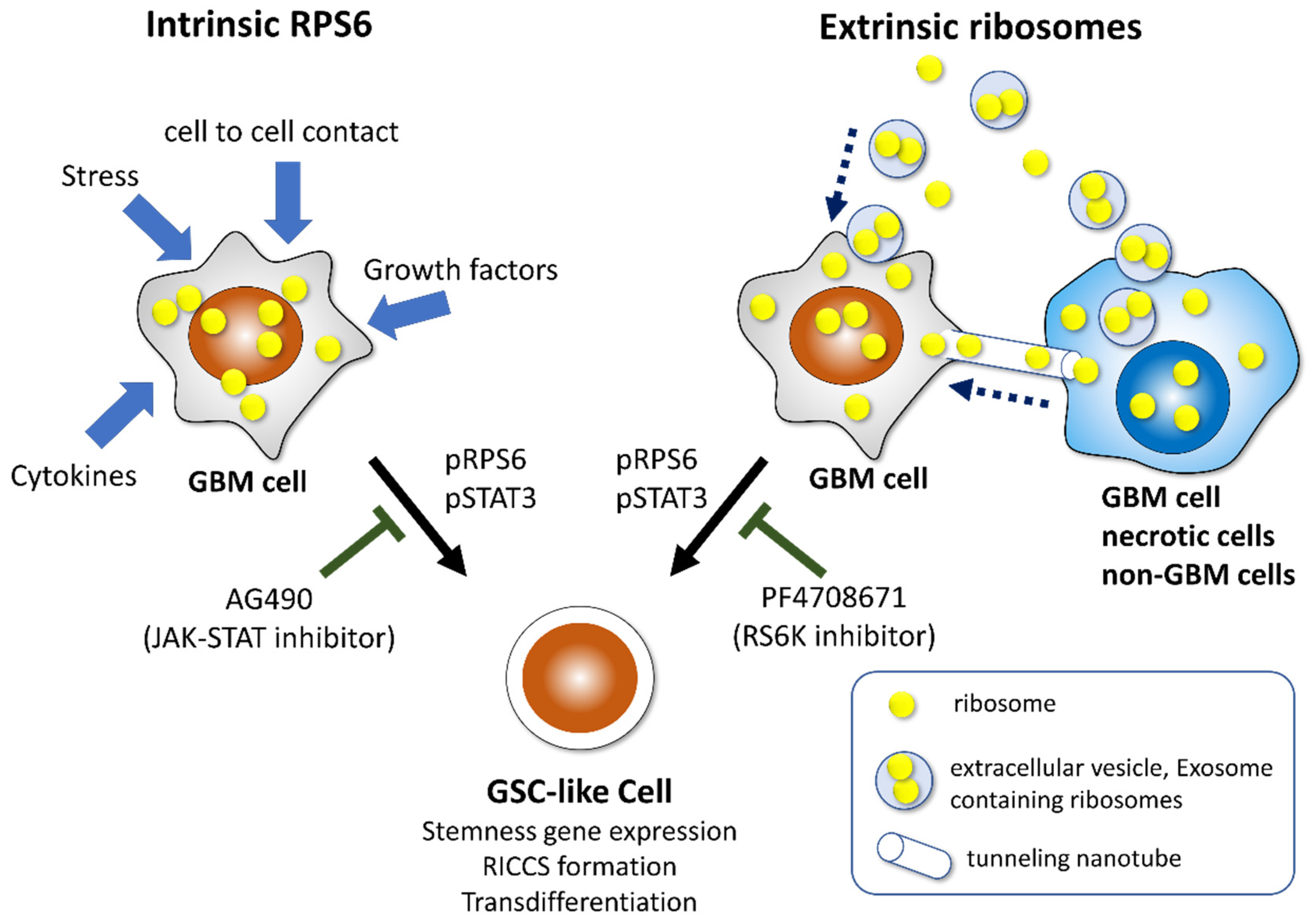
6. Incorporation of Ribosomal Proteins S6 Induces Reprogramming in Glioma Cells
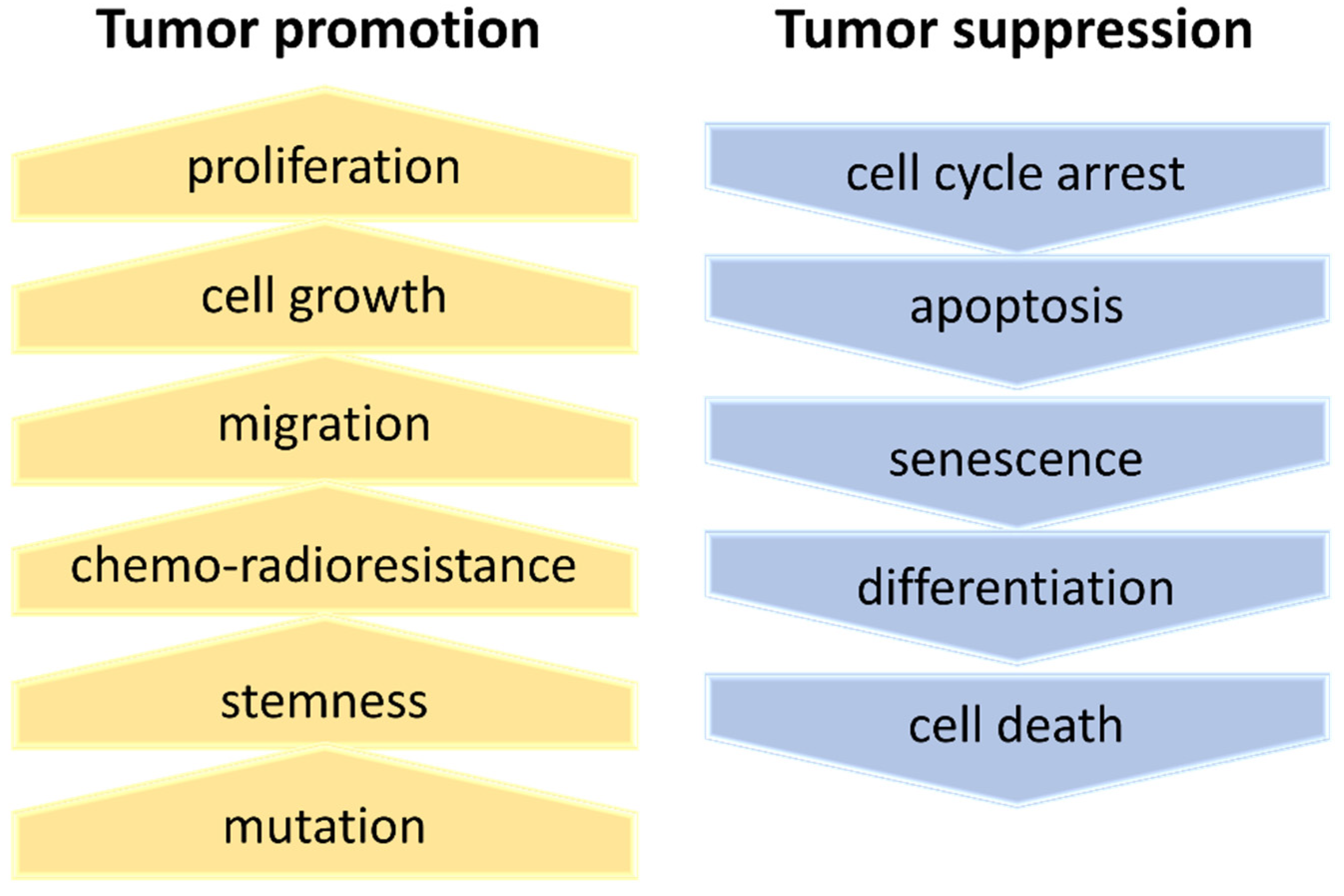
7. Deregulation of Ribosome Biogenesis Modulates Aggressiveness in GBM
8. Incorporation of Ribosome Induces Reprogramming and Transdifferentiation Potential in Cancer Cells
9. Ribosome Biogenesis as the Therapeutic Target for GBM
10. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro. Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Hide, T.; Takezaki, T.; Nakatani, Y.; Nakamura, H.; Kuratsu, J.; Kondo, T. Sox11 prevents tumorigenesis of glioma-initiating cells by inducing neuronal differentiation. Cancer Res. 2009, 69, 7953–7959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hide, T.; Takezaki, T.; Nakatani, Y.; Nakamura, H.; Kuratsu, J.; Kondo, T. Combination of a ptgs2 inhibitor and an epidermal growth factor receptor-signaling inhibitor prevents tumorigenesis of oligodendrocyte lineage-derived glioma-initiating cells. Stem Cells 2011, 29, 590–599. [Google Scholar] [CrossRef]
- Agnihotri, S.; Burrell, K.E.; Wolf, A.; Jalali, S.; Hawkins, C.; Rutka, J.T.; Zadeh, G. Glioblastoma, a brief review of history, molecular genetics, animal models and novel therapeutic strategies. Arch. Immunol. Ther. Exp. 2013, 61, 25–41. [Google Scholar] [CrossRef]
- Miyai, M.; Tomita, H.; Soeda, A.; Yano, H.; Iwama, T.; Hara, A. Current trends in mouse models of glioblastoma. J. Neurooncol. 2017, 135, 423–432. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e122. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Mooney, J.H.; Ilyas, A.; Chagoya, G.; Estevez-Ordonez, D.; Ibrahim, A.; Nakano, I. Molecular and cellular intratumoral heterogeneity in primary glioblastoma: Clinical and translational implications. J. Neurosurg. 2019, 133, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, I.H.; Cho, H.J.; Park, C.K.; Jung, Y.S.; Kim, Y.; Nam, S.H.; Kim, B.S.; Johnson, M.D.; Kong, D.S.; et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer Cell 2015, 28, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Wang, J.; Sa, J.K.; Ladewig, E.; Lee, H.O.; Lee, I.H.; Kang, H.J.; Rosenbloom, D.S.; Camara, P.G.; Liu, Z.; et al. Spatiotemporal genomic architecture informs precision oncology in glioblastoma. Nat. Genet. 2017, 49, 594–599. [Google Scholar] [CrossRef]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F.; et al. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavare, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849.e821. [Google Scholar] [CrossRef]
- Chuang, D.F.; Lin, X. Targeted Therapies for the Treatment of Glioblastoma in Adults. Curr. Oncol. Rep. 2019, 21, 61. [Google Scholar] [CrossRef]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef]
- Hide, T.; Komohara, Y.; Miyasato, Y.; Nakamura, H.; Makino, K.; Takeya, M.; Kuratsu, J.I.; Mukasa, A.; Yano, S. Oligodendrocyte Progenitor Cells and Macrophages/Microglia Produce Glioma Stem Cell Niches at the Tumor Border. EBioMedicine 2018, 30, 94–104. [Google Scholar] [CrossRef] [Green Version]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef] [PubMed]
- Papale, M.; Buccarelli, M.; Mollinari, C.; Russo, M.A.; Pallini, R.; Ricci-Vitiani, L.; Tafani, M. Hypoxia, Inflammation and Necrosis as Determinants of Glioblastoma Cancer Stem Cells Progression. Int. J. Mol. Sci. 2020, 21, 2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colwell, N.; Larion, M.; Giles, A.J.; Seldomridge, A.N.; Sizdahkhani, S.; Gilbert, M.R.; Park, D.M. Hypoxia in the glioblastoma microenvironment: Shaping the phenotype of cancer stem-like cells. Neuro Oncol. 2017, 19, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Shang, Z.; Zhou, Y.; Hu, X.; Chen, Y.; Fan, Y.; Wei, X.; Wu, L.; Liang, Q.; Zhang, J.; et al. Autophagy mediates glucose starvation-induced glioblastoma cell quiescence and chemoresistance through coordinating cell metabolism, cell cycle, and survival. Cell Death Dis. 2018, 9, 213. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Fei, X.; Sun, G.; Wang, Z.; Wan, Y.; Zeng, Y.; Guo, J. Hypothermia stimulates glioma stem spheres to spontaneously dedifferentiate adjacent non-stem glioma cells. Cell Mol. Neurobiol. 2015, 35, 217–230. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef]
- Mega, A.; Hartmark Nilsen, M.; Leiss, L.W.; Tobin, N.P.; Miletic, H.; Sleire, L.; Strell, C.; Nelander, S.; Krona, C.; Hagerstrand, D.; et al. Astrocytes enhance glioblastoma growth. Glia 2020, 68, 316–327. [Google Scholar] [CrossRef]
- Ito, N.; Katoh, K.; Kushige, H.; Saito, Y.; Umemoto, T.; Matsuzaki, Y.; Kiyonari, H.; Kobayashi, D.; Soga, M.; Era, T.; et al. Ribosome Incorporation into Somatic Cells Promotes Lineage Transdifferentiation towards Multipotency. Sci. Rep. 2018, 8, 1634. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kressler, D.; Hurt, E.; Bassler, J. Driving ribosome assembly. Biochim. Biophys. Acta 2010, 1803, 673–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Liao, W.J.; Liao, J.M.; Liao, P.; Lu, H. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 2015, 7, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, J.R.; McIntosh, K.B. How common are extraribosomal functions of ribosomal proteins? Mol. Cell 2009, 34, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Khoury, W.; Nasr, Z. Deregulation of ribosomal proteins in human cancers. Biosci. Rep. 2021, 41, BSR20211577. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, Y.; Hide, T.; Yamaoka, M.; Ito, Y.; Ito, N.; Ohta, K.; Shinojima, N.; Mukasa, A.; Saito, H.; Jono, H. Ribosomal protein S6 promotes stem-like characters in glioma cells. Cancer Sci. 2020, 111, 2041–2051. [Google Scholar] [CrossRef]
- Chow, S.; Minden, M.D.; Hedley, D.W. Constitutive phosphorylation of the S6 ribosomal protein via mTOR and ERK signaling in the peripheral blasts of acute leukemia patients. Exp. Hematol. 2006, 34, 1183–1191. [Google Scholar] [CrossRef]
- Khalaileh, A.; Dreazen, A.; Khatib, A.; Apel, R.; Swisa, A.; Kidess-Bassir, N.; Maitra, A.; Meyuhas, O.; Dor, Y.; Zamir, G. Phosphorylation of ribosomal protein S6 attenuates DNA damage and tumor suppression during development of pancreatic cancer. Cancer Res. 2013, 73, 1811–1820. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Tan, Z.; Gao, J.; Wu, W.; Liu, L.; Jin, W.; Cao, Y.; Zhao, S.; Zhang, W.; Qiu, Z.; et al. Hyperphosphorylation of ribosomal protein S6 predicts unfavorable clinical survival in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2015, 34, 126. [Google Scholar] [CrossRef] [Green Version]
- Shirakawa, Y.; Ohta, K.; Miyake, S.; Kanemaru, A.; Kuwano, A.; Yonemaru, K.; Uchino, S.; Yamaoka, M.; Ito, Y.; Ito, N.; et al. Glioma Cells Acquire Stem-like Characters by Extrinsic Ribosome Stimuli. Cells 2021, 10, 2970. [Google Scholar] [CrossRef]
- Wilson, C.B. Glioblastoma: The past, the present, and the future. Clin. Neurosurg. 1992, 38, 32–48. [Google Scholar] [PubMed]
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tonjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, L.; Yan, Q.; Zhang, Y.; Fang, X.; Liu, B.; Guan, X. Cancer cell reprogramming: A promising therapy converting malignancy to benignity. Cancer Commun. 2019, 39, 48. [Google Scholar] [CrossRef] [Green Version]
- Reik, W.; Dean, W.; Walter, J. Epigenetic reprogramming in mammalian development. Science 2001, 293, 1089–1093. [Google Scholar] [CrossRef] [Green Version]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Grath, A.; Dai, G. Direct cell reprogramming for tissue engineering and regenerative medicine. J. Biol. Eng. 2019, 13, 14. [Google Scholar] [CrossRef]
- Sell, S. Cancer stem cells and differentiation therapy. Tumour Biol. 2006, 27, 59–70. [Google Scholar] [CrossRef]
- Massard, C.; Deutsch, E.; Soria, J.C. Tumour stem cell-targeted treatment: Elimination or differentiation. Ann. Oncol. 2006, 17, 1620–1624. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Chen, A.T.; Gao, X.; Himes, B.T.; Zhang, H.; Chen, Z.; Wang, J.; Sheu, W.C.; Deng, G.; et al. ZNF117 regulates glioblastoma stem cell differentiation towards oligodendroglial lineage. Nat. Commun. 2022, 13, 2196. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Zhang, F.; Hallahan, D.; Zhang, Z.; He, L.; Wu, L.G.; You, M.; Yang, Q. Reprogramming glioblastoma multiforme cells into neurons by protein kinase inhibitors. J. Exp. Clin. Cancer Res. 2018, 37, 181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Robinson, M.; Willerth, S.M. Direct Reprogramming of Glioblastoma Cells into Neurons Using Small Molecules. ACS Chem. Neurosci. 2018, 9, 3175–3185. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Huang, S.; Zhang, H.; Hua, W.; Xin, S.; Cheng, L.; Guan, W.; Yu, Y.; Mao, Y.; Pei, G. Suppression of glioblastoma by a drug cocktail reprogramming tumor cells into neuronal like cells. Sci. Rep. 2019, 9, 3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef]
- Soda, Y.; Marumoto, T.; Friedmann-Morvinski, D.; Soda, M.; Liu, F.; Michiue, H.; Pastorino, S.; Yang, M.; Hoffman, R.M.; Kesari, S.; et al. Transdifferentiation of glioblastoma cells into vascular endothelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 4274–4280. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Christianson, H.C.; Welch, J.E.; Svensson, K.J.; Fredlund, E.; Ringner, M.; Morgelin, M.; Bourseau-Guilmain, E.; Bengzon, J.; Belting, M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc. Natl. Acad. Sci. USA 2013, 110, 7312–7317. [Google Scholar] [CrossRef] [Green Version]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074.e4. [Google Scholar] [CrossRef] [Green Version]
- Domenech, M.; Hernandez, A.; Plaja, A.; Martinez-Balibrea, E.; Balana, C. Hypoxia: The Cornerstone of Glioblastoma. Int. J. Mol. Sci. 2021, 22, 12608. [Google Scholar] [CrossRef]
- Chedeville, A.L.; Madureira, P.A. The Role of Hypoxia in Glioblastoma Radiotherapy Resistance. Cancers 2021, 13, 542. [Google Scholar] [CrossRef] [PubMed]
- Kolenda, J.; Jensen, S.S.; Aaberg-Jessen, C.; Christensen, K.; Andersen, C.; Brunner, N.; Kristensen, B.W. Effects of hypoxia on expression of a panel of stem cell and chemoresistance markers in glioblastoma-derived spheroids. J. Neurooncol. 2011, 103, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma Stem Cells Microenvironment: The Paracrine Roles of the Niche in Drug and Radioresistance. Stem Cells Int. 2016, 2016, 6809105. [Google Scholar] [CrossRef] [Green Version]
- Bhushan, A.; Kumari, R.; Srivastava, T. Scouting for common genes in the heterogenous hypoxic tumor microenvironment and their validation in glioblastoma. 3 Biotech 2021, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Martinez Gala, J.; Delmas, C.; Monferran, S.; Malric, L.; Zentkowski, D.; Lubrano, V.; Toulas, C.; Cohen-Jonathan Moyal, E.; Lemarie, A. Ionizing radiations sustain glioblastoma cell dedifferentiation to a stem-like phenotype through survivin: Possible involvement in radioresistance. Cell Death Dis. 2014, 5, e1543. [Google Scholar] [CrossRef] [Green Version]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Korber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.; Robitaille, R.; Volterra, A. Gliotransmitters travel in time and space. Neuron 2014, 81, 728–739. [Google Scholar]
- Allen, N.J.; Lyons, D.A. Glia as architects of central nervous system formation and function. Science 2018, 362, 181–185. [Google Scholar] [CrossRef] [Green Version]
- Bergles, D.E.; Roberts, J.D.; Somogyi, P.; Jahr, C.E. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 2000, 405, 187–191. [Google Scholar] [CrossRef]
- Habermacher, C.; Angulo, M.C.; Benamer, N. Glutamate versus GABA in neuron-oligodendroglia communication. Glia 2019, 67, 2092–2106. [Google Scholar] [CrossRef] [PubMed]
- Kula, B.; Chen, T.J.; Kukley, M. Glutamatergic signaling between neurons and oligodendrocyte lineage cells: Is it synaptic or non-synaptic? Glia 2019, 67, 2071–2091. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [Green Version]
- Hide, T.; Shibahara, I.; Kumabe, T. Novel concept of the border niche: Glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol. 2019, 36, 63–73. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the rising stars of the glioblastoma microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Guan, X.; Hasan, M.N.; Maniar, S.; Jia, W.; Sun, D. Reactive Astrocytes in Glioblastoma Multiforme. Mol. Neurobiol. 2018, 55, 6927–6938. [Google Scholar] [CrossRef]
- Lois, C.; Alvarez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl. Acad. Sci. USA 1993, 90, 2074–2077. [Google Scholar] [CrossRef] [Green Version]
- Li, G.; Chen, Z.; Hu, Y.D.; Wei, H.; Li, D.; Ji, H.; Wang, D.L. Autocrine factors sustain glioblastoma stem cell self-renewal. Oncol. Rep. 2009, 21, 419–424. [Google Scholar]
- Kuratsu, J.; Leonard, E.J.; Yoshimura, T. Production and characterization of human glioma cell-derived monocyte chemotactic factor. J. Natl. Cancer Inst. 1989, 81, 347–351. [Google Scholar] [CrossRef]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shono, K.; Yamaguchi, I.; Mizobuchi, Y.; Kagusa, H.; Sumi, A.; Fujihara, T.; Nakajima, K.; Kitazato, K.T.; Matsuzaki, K.; Saya, H.; et al. Downregulation of the CCL2/CCR2 and CXCL10/CXCR3 axes contributes to antitumor effects in a mouse model of malignant glioma. Sci. Rep. 2020, 10, 15286. [Google Scholar] [CrossRef] [PubMed]
- Zeis, T.; Enz, L.; Schaeren-Wiemers, N. The immunomodulatory oligodendrocyte. Brain Res. 2016, 1641, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbantat, R.M.; Vajkoczy, P.; Brandenburg, S. Advances in Chemokine Signaling Pathways as Therapeutic Targets in Glioblastoma. Cancers 2021, 13, 2983. [Google Scholar] [CrossRef] [PubMed]
- Yeo, E.C.F.; Brown, M.P.; Gargett, T.; Ebert, L.M. The Role of Cytokines and Chemokines in Shaping the Immune Microenvironment of Glioblastoma: Implications for Immunotherapy. Cells 2021, 10, 607. [Google Scholar] [CrossRef]
- Sil, S.; Periyasamy, P.; Thangaraj, A.; Chivero, E.T.; Buch, S. PDGF/PDGFR axis in the neural systems. Mol. Aspects Med. 2018, 62, 63–74. [Google Scholar] [CrossRef]
- Almiron Bonnin, D.A.; Havrda, M.C.; Lee, M.C.; Liu, H.; Zhang, Z.; Nguyen, L.N.; Harrington, L.X.; Hassanpour, S.; Cheng, C.; Israel, M.A. Secretion-mediated STAT3 activation promotes self-renewal of glioma stem-like cells during hypoxia. Oncogene 2018, 37, 1107–1118. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, S.M.B.; Staicu, G.A.; Sevastre, A.S.; Baloi, C.; Ciubotaru, V.; Dricu, A.; Tataranu, L.G. Glioblastoma Stem Cells-Useful Tools in the Battle against Cancer. Int. J. Mol. Sci. 2022, 23, 4602. [Google Scholar] [CrossRef]
- Quezada, C.; Torres, A.; Niechi, I.; Uribe, D.; Contreras-Duarte, S.; Toledo, F.; San Martin, R.; Gutierrez, J.; Sobrevia, L. Role of extracellular vesicles in glioma progression. Mol. Aspects Med. 2018, 60, 38–51. [Google Scholar] [CrossRef]
- Matarredona, E.R.; Pastor, A.M. Extracellular Vesicle-Mediated Communication between the Glioblastoma and Its Microenvironment. Cells 2019, 9, 96. [Google Scholar] [CrossRef] [Green Version]
- Simon, T.; Jackson, E.; Giamas, G. Breaking through the glioblastoma micro-environment via extracellular vesicles. Oncogene 2020, 39, 4477–4490. [Google Scholar] [CrossRef] [PubMed]
- Maas, S.L.N.; Breakefield, X.O.; Weaver, A.M. Extracellular Vesicles: Unique Intercellular Delivery Vehicles. Trends Cell Biol. 2017, 27, 172–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skog, J.; Wurdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, J.; Sun, G.; Meng, H.; Wang, J.; Guan, Y.; Yin, Y.; Zhao, Z.; Dong, X.; Yin, S.; et al. Glioblastoma extracellular vesicles induce the tumour-promoting transformation of neural stem cells. Cancer Lett. 2019, 466, 1–12. [Google Scholar] [CrossRef]
- Sun, Z.; Wang, L.; Zhou, Y.; Dong, L.; Ma, W.; Lv, L.; Zhang, J.; Wang, X. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell Mol. Neurobiol. 2020, 40, 767–784. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, Z.; Mashimo, T.; Shen, B.; Nyagilo, J.; Wang, H.; Wang, Y.; Liu, Z.; Mulgaonkar, A.; Hu, X.L.; et al. Gliomas Interact with Non-glioma Brain Cells via Extracellular Vesicles. Cell Rep. 2020, 30, 2489–2500.e5. [Google Scholar] [CrossRef] [Green Version]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [Green Version]
- Oushy, S.; Hellwinkel, J.E.; Wang, M.; Nguyen, G.J.; Gunaydin, D.; Harland, T.A.; Anchordoquy, T.J.; Graner, M.W. Glioblastoma multiforme-derived extracellular vesicles drive normal astrocytes towards a tumour-enhancing phenotype. Philos. Trans. R Soc. Lond B Biol. Sci. 2018, 373, 20160477. [Google Scholar] [CrossRef] [Green Version]
- Zeng, A.; Wei, Z.; Rabinovsky, R.; Jun, H.J.; El Fatimy, R.; Deforzh, E.; Arora, R.; Yao, Y.; Yao, S.; Yan, W.; et al. Glioblastoma-Derived Extracellular Vesicles Facilitate Transformation of Astrocytes via Reprogramming Oncogenic Metabolism. iScience 2020, 23, 101420. [Google Scholar] [CrossRef]
- Piazzi, M.; Bavelloni, A.; Gallo, A.; Faenza, I.; Blalock, W.L. Signal Transduction in Ribosome Biogenesis: A Recipe to Avoid Disaster. Int. J. Mol. Sci. 2019, 20, 2718. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Brajanovski, N.; Chan, K.T.; Xuan, J.; Pearson, R.B.; Sanij, E. Ribosomal proteins and human diseases: Molecular mechanisms and targeted therapy. Signal Transduct. Target. Ther. 2021, 6, 323. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Yoshihama, M.; Kenmochi, N. RPG: The Ribosomal Protein Gene database. Nucleic Acids Res. 2004, 32, D168–D170. [Google Scholar] [CrossRef] [PubMed]
- Gaviraghi, M.; Vivori, C.; Tonon, G. How Cancer Exploits Ribosomal RNA Biogenesis: A Journey beyond the Boundaries of rRNA Transcription. Cells 2019, 8, 1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drygin, D.; Rice, W.G.; Grummt, I. The RNA polymerase I transcription machinery: An emerging target for the treatment of cancer. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 131–156. [Google Scholar] [CrossRef]
- Stefanovsky, V.Y.; Pelletier, G.; Hannan, R.; Gagnon-Kugler, T.; Rothblum, L.I.; Moss, T. An immediate response of ribosomal transcription to growth factor stimulation in mammals is mediated by ERK phosphorylation of UBF. Mol. Cell 2001, 8, 1063–1073. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef]
- Thoms, H.C.; Stark, L.A. The NF-kappaB Nucleolar Stress Response Pathway. Biomedicines 2021, 9, 1082. [Google Scholar] [CrossRef]
- Dameshek, W. Riddle: What do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and hypoplastic leukemia have in common? Blood 1967, 30, 251–254. [Google Scholar] [CrossRef]
- Kampen, K.R.; Sulima, S.O.; Vereecke, S.; De Keersmaecker, K. Hallmarks of ribosomopathies. Nucleic Acids Res. 2020, 48, 1013–1028. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; You, K.S.; Park, J.S.; Lee, S.G.; Seong, Y.S. Ribosomal Protein S6: A Potential Therapeutic Target against Cancer? Int. J. Mol. Sci. 2021, 23, 48. [Google Scholar] [CrossRef] [PubMed]
- Hide, T.; Shibahara, I.; Inukai, M.; Shigeeda, R.; Shirakawa, Y.; Jono, H.; Shinojima, N.; Mukasa, A.; Kumabe, T. Ribosomal proteins induce stem cell-like characteristics in glioma cells as an extra-ribosomal function. Brain Tumor Pathol. 2022, 39, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Penzo, M.; Montanaro, L.; Trere, D.; Derenzini, M. The Ribosome Biogenesis-Cancer Connection. Cells 2019, 8, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecoraro, A.; Pagano, M.; Russo, G.; Russo, A. Ribosome Biogenesis and Cancer: Overview on Ribosomal Proteins. Int. J. Mol. Sci. 2021, 22, 5496. [Google Scholar] [CrossRef]
- Pestov, D.G.; Strezoska, Z.; Lau, L.F. Evidence of p53-dependent cross-talk between ribosome biogenesis and the cell cycle: Effects of nucleolar protein Bop1 on G(1)/S transition. Mol. Cell Biol. 2001, 21, 4246–4255. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Deisenroth, C.; Zhang, Y. RP-MDM2-p53 Pathway: Linking Ribosomal Biogenesis and Tumor Surveillance. Trends Cancer 2016, 2, 191–204. [Google Scholar] [CrossRef] [Green Version]
- Lo, S.J.; Fan, L.C.; Tsai, Y.F.; Lin, K.Y.; Huang, H.L.; Wang, T.H.; Liu, H.; Chen, T.C.; Huang, S.F.; Chang, C.J.; et al. A novel interaction of nucleophosmin with BCL2-associated X protein regulating death evasion and drug sensitivity in human hepatoma cells. Hepatology 2013, 57, 1893–1905. [Google Scholar] [CrossRef]
- Eymin, B.; Claverie, P.; Salon, C.; Leduc, C.; Col, E.; Brambilla, E.; Khochbin, S.; Gazzeri, S. p14ARF activates a Tip60-dependent and p53-independent ATM/ATR/CHK pathway in response to genotoxic stress. Mol. Cell Biol. 2006, 26, 4339–4350. [Google Scholar] [CrossRef] [Green Version]
- James, A.; Wang, Y.; Raje, H.; Rosby, R.; DiMario, P. Nucleolar stress with and without p53. Nucleus 2014, 5, 402–426. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Russo, G. Ribosomal Proteins Control or Bypass p53 during Nucleolar Stress. Int. J. Mol. Sci. 2017, 18, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correll, C.C.; Bartek, J.; Dundr, M. The Nucleolus: A Multiphase Condensate Balancing Ribosome Synthesis and Translational Capacity in Health, Aging and Ribosomopathies. Cells 2019, 8, 869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aspesi, A.; Ellis, S.R. Rare ribosomopathies: Insights into mechanisms of cancer. Nat. Rev. Cancer 2019, 19, 228–238. [Google Scholar] [CrossRef] [PubMed]
- Orgebin, E.; Lamoureux, F.; Isidor, B.; Charrier, C.; Ory, B.; Lezot, F.; Baud’huin, M. Ribosomopathies: New Therapeutic Perspectives. Cells 2020, 9, 2080. [Google Scholar]
- Ohta, K.; Kawano, R.; Ito, N. Lactic acid bacteria convert human fibroblasts to multipotent cells. PLoS ONE 2012, 7, e51866. [Google Scholar] [CrossRef] [Green Version]
- Istiaq, A.; Ohta, K. Ribosome-Induced Cellular Multipotency, an Emerging Avenue in Cell Fate Reversal. Cells 2021, 10, 2276. [Google Scholar] [CrossRef]
- Grundy, M.; Jones, T.; Elmi, L.; Hall, M.; Graham, A.; Russell, N.; Pallis, M. Early changes in rpS6 phosphorylation and BH3 profiling predict response to chemotherapy in AML cells. PLoS ONE 2018, 13, e0196805. [Google Scholar]
- Hagner, P.R.; Mazan-Mamczarz, K.; Dai, B.; Balzer, E.M.; Corl, S.; Martin, S.S.; Zhao, X.F.; Gartenhaus, R.B. Ribosomal protein S6 is highly expressed in non-Hodgkin lymphoma and associates with mRNA containing a 5’ terminal oligopyrimidine tract. Oncogene 2011, 30, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- Chaisuparat, R.; Rojanawatsirivej, S.; Yodsanga, S. Ribosomal protein S6 phosphorylation is associated with epithelial dysplasia and squamous cell carcinoma of the oral cavity. Pathol. Oncol. Res. 2013, 19, 189–193. [Google Scholar] [CrossRef]
- Yanai, A.; Inoue, N.; Yagi, T.; Nishimukai, A.; Miyagawa, Y.; Murase, K.; Imamura, M.; Enomoto, Y.; Takatsuka, Y.; Watanabe, T.; et al. Activation of mTOR/S6K But Not MAPK Pathways Might Be Associated With High Ki-67, ER(+), and HER2(−) Breast Cancer. Clin. Breast Cancer 2015, 15, 197–203. [Google Scholar] [CrossRef]
- Zheng, Z.; Zheng, Y.; Zhang, M.; Wang, J.; Yu, G.; Fang, W. Reciprocal expression of p-AMPKa and p-S6 is strongly associated with the prognosis of gastric cancer. Tumour Biol. 2016, 37, 4803–4811. [Google Scholar] [CrossRef] [PubMed]
- Knoll, M.; Macher-Goeppinger, S.; Kopitz, J.; Duensing, S.; Pahernik, S.; Hohenfellner, M.; Schirmacher, P.; Roth, W. The ribosomal protein S6 in renal cell carcinoma: Functional relevance and potential as biomarker. Oncotarget 2016, 7, 418–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Yun, R.; Yu, X.; Hu, H.; Huang, G.; Tan, B.; Chen, T. Overexpression of Notch3 and pS6 Is Associated with Poor Prognosis in Human Ovarian Epithelial Cancer. Mediators Inflamm. 2016, 2016, 5953498. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Rothenberg, S.M.; Hata, A.N.; Faber, A.C.; Piris, A.; Nazarian, R.M.; Brown, R.D.; Godfrey, J.T.; Winokur, D.; Walsh, J.; et al. TORC1 suppression predicts responsiveness to RAF and MEK inhibition in BRAF-mutant melanoma. Sci. Transl. Med. 2013, 5, 196ra198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; Chen, H.P.; Duan, H.F.; Gao, L.H.; Shao, Y.; Chen, K.Y.; Wang, Y.L.; Lan, F.H.; Hu, X.W. Aggregation of Ribosomal Protein S6 at Nucleolus Is Cell Cycle-Controlled and Its Function in Pre-rRNA Processing Is Phosphorylation Dependent. J. Cell Biochem. 2016, 117, 1649–1657. [Google Scholar] [CrossRef]
- Puchalski, R.B.; Shah, N.; Miller, J.; Dalley, R.; Nomura, S.R.; Yoon, J.G.; Smith, K.A.; Lankerovich, M.; Bertagnolli, D.; Bickley, K.; et al. An anatomic transcriptional atlas of human glioblastoma. Science 2018, 360, 660–663. [Google Scholar] [CrossRef] [Green Version]
- Ganger, D.R.; Hamilton, P.D.; Fletcher, J.W.; Fernandez-Pol, J.A. Metallopanstimulin is overexpressed in a patient with colonic carcinoma. Anticancer Res. 1997, 17, 1993–1999. [Google Scholar]
- Fernandez-Pol, J.A.; Fletcher, J.W.; Hamilton, P.D.; Klos, D.J. Expression of metallopanstimulin and oncogenesis in human prostatic carcinoma. Anticancer Res. 1997, 17, 1519–1530. [Google Scholar]
- Atsuta, Y.; Aoki, N.; Sato, K.; Oikawa, K.; Nochi, H.; Miyokawa, N.; Hirata, S.; Kimura, S.; Sasajima, T.; Katagiri, M. Identification of metallopanstimulin-1 as a member of a tumor associated antigen in patients with breast cancer. Cancer Lett. 2002, 182, 101–107. [Google Scholar] [CrossRef]
- Wang, Y.W.; Qu, Y.; Li, J.F.; Chen, X.H.; Liu, B.Y.; Gu, Q.L.; Zhu, Z.G. In vitro and in vivo evidence of metallopanstimulin-1 in gastric cancer progression and tumorigenicity. Clin. Cancer Res. 2006, 12, 4965–4973. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Pol, J.A. Increased serum level of RPMPS-1/S27 protein in patients with various types of cancer is useful for the early detection, prevention and therapy. Cancer Genom. Proteom. 2012, 9, 203–256. [Google Scholar]
- Feldheim, J.; Kessler, A.F.; Schmitt, D.; Salvador, E.; Monoranu, C.M.; Feldheim, J.J.; Ernestus, R.I.; Lohr, M.; Hagemann, C. Ribosomal Protein S27/Metallopanstimulin-1 (RPS27) in Glioma-A New Disease Biomarker? Cancers 2020, 12, 1085. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Fu, J.; Xue, F.; Ryu, B.; Zhang, T.; Zhang, S.; Sun, J.; Xu, X.; Shen, Z.; Zheng, L.; et al. Knockdown of ribosomal protein S15A induces human glioblastoma cell apoptosis. World J. Surg. Oncol. 2016, 14, 129. [Google Scholar] [CrossRef] [Green Version]
- Yao, Y.; Liu, Y.; Lv, X.; Dong, B.; Wang, F.; Li, J.; Zhang, Q.; Xu, R.; Xu, Y. Down-regulation of ribosomal protein S15A inhibits proliferation of human glioblastoma cells in vivo and in vitro via AKT pathway. Tumour Biol. 2016, 37, 4979–4990. [Google Scholar] [CrossRef] [PubMed]
- Nissan, T.A.; Bassler, J.; Petfalski, E.; Tollervey, D.; Hurt, E. 60S pre-ribosome formation viewed from assembly in the nucleolus until export to the cytoplasm. EMBO J. 2002, 21, 5539–5547. [Google Scholar] [CrossRef] [PubMed]
- Fancello, L.; Kampen, K.R.; Hofman, I.J.; Verbeeck, J.; De Keersmaecker, K. The ribosomal protein gene RPL5 is a haploinsufficient tumor suppressor in multiple cancer types. Oncotarget 2017, 8, 14462–14478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awah, C.U.; Chen, L.; Bansal, M.; Mahajan, A.; Winter, J.; Lad, M.; Warnke, L.; Gonzalez-Buendia, E.; Park, C.; Zhang, D.; et al. Ribosomal protein S11 influences glioma response to TOP2 poisons. Oncogene 2020, 39, 5068–5081. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Kim, I.K.; Hong, S.H.; Nan, H.; Kim, H.J.; Lee, H.J.; Masuda, E.S.; Meyuhas, O.; Oh, B.H.; Jung, Y.K. Ribosomal protein S6 is a selective mediator of TRAIL-apoptotic signaling. Oncogene 2008, 27, 4344–4352. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Kim, H.D.; Youn, B.; Park, Y.G.; Kim, J. Ribosomal protein S3 is secreted as a homodimer in cancer cells. Biochem. Biophys. Res. Commun. 2013, 441, 805–808. [Google Scholar] [CrossRef]
- Court, F.A.; Hendriks, W.T.; MacGillavry, H.D.; Alvarez, J.; van Minnen, J. Schwann cell to axon transfer of ribosomes: Toward a novel understanding of the role of glia in the nervous system. J. Neurosci. 2008, 28, 11024–11029. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Leal, R.; Alvarez, J.; Court, F.A. Origin of axonal proteins: Is the axon-schwann cell unit a functional syncytium? Cytoskeleton 2016, 73, 629–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, G.; Saenz-de-Santa-Maria, I.; Chastagner, P.; Perthame, E.; Delmas, C.; Toulas, C.; Moyal-Jonathan-Cohen, E.; Brou, C.; Zurzolo, C. Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem. J. 2021, 478, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Kudo, M.; Anam, M.B.; Istiaq, A.; Ahmad, S.A.I.; Ito, N.; Ohta, K. Ribosome Incorporation Induces EMT-like Phenomenon with Cell Cycle Arrest in Human Breast Cancer Cell. Cells Tissues Organs 2022, 211, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Anam, M.B.; Istiaq, A.; Kariya, R.; Kudo, M.; Ishtiyaq Ahmad, S.A.; Ito, N.; Okada, S.; Ohta, K. Ribosome induces transdifferentiation of A549 and H-111-TC cancer cell lines. Biochem. Biophys. Rep. 2021, 26, 100946. [Google Scholar] [CrossRef] [PubMed]
- Setayesh-Mehr, Z.; Poorsargol, M. Toxic proteins application in cancer therapy. Mol. Biol. Rep. 2021, 48, 3827–3840. [Google Scholar] [CrossRef]
- Rotondo, R.; Ragucci, S.; Castaldo, S.; Oliva, M.A.; Landi, N.; Pedone, P.V.; Arcella, A.; Di Maro, A. Cytotoxicity Effect of Quinoin, Type 1 Ribosome-Inactivating Protein from Quinoa Seeds, on Glioblastoma Cells. Toxins 2021, 13, 684. [Google Scholar] [CrossRef]
- Lapik, Y.R.; Fernandes, C.J.; Lau, L.F.; Pestov, D.G. Physical and functional interaction between Pes1 and Bop1 in mammalian ribosome biogenesis. Mol. Cell 2004, 15, 17–29. [Google Scholar] [CrossRef]
- Holzel, M.; Rohrmoser, M.; Schlee, M.; Grimm, T.; Harasim, T.; Malamoussi, A.; Gruber-Eber, A.; Kremmer, E.; Hiddemann, W.; Bornkamm, G.W.; et al. Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J. Cell Biol. 2005, 170, 367–378. [Google Scholar] [CrossRef]
- Li, Y.Z.; Zhang, C.; Pei, J.P.; Zhang, W.C.; Zhang, C.D.; Dai, D.Q. The functional role of Pescadillo ribosomal biogenesis factor 1 in cancer. J. Cancer 2022, 13, 268–277. [Google Scholar] [CrossRef]
- Mi, L.; Qi, Q.; Ran, H.; Chen, L.; Li, D.; Xiao, D.; Wu, J.; Cai, Y.; Zhang, S.; Li, Y.; et al. Suppression of Ribosome Biogenesis by Targeting WD Repeat Domain 12 (WDR12) Inhibits Glioma Stem-Like Cell Growth. Front. Oncol. 2021, 11, 751792. [Google Scholar] [CrossRef]
- Li, J.L.; Chen, C.; Chen, W.; Zhao, L.F.; Xu, X.K.; Li, Y.; Yuan, H.Y.; Lin, J.R.; Pan, J.P.; Jin, B.L.; et al. Integrative genomic analyses identify WDR12 as a novel oncogene involved in glioblastoma. J. Cell Physiol. 2020, 235, 7344–7355. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, S.; Hirayama, A.; Eberhardt, A.O.; Kawaguchi, R.; Sugiura, Y.; Sampetrean, O.; Ikeda, Y.; Warren, M.; Sakamoto, N.; Kitahara, S.; et al. IMP dehydrogenase-2 drives aberrant nucleolar activity and promotes tumorigenesis in glioblastoma. Nat. Cell Biol. 2019, 21, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Yao, Y.; Scott, A.J.; Wilder-Romans, K.; Dresser, J.J.; Werner, C.K.; Sun, H.; Pratt, D.; Sajjakulnukit, P.; Zhao, S.G.; et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat. Commun. 2020, 11, 3811. [Google Scholar] [CrossRef] [PubMed]
- Lafita-Navarro, M.C.; Venkateswaran, N.; Kilgore, J.A.; Kanji, S.; Han, J.; Barnes, S.; Williams, N.S.; Buszczak, M.; Burma, S.; Conacci-Sorrell, M. Inhibition of the de novo pyrimidine biosynthesis pathway limits ribosomal RNA transcription causing nucleolar stress in glioblastoma cells. PLoS Genet. 2020, 16, e1009117. [Google Scholar] [CrossRef]
- Tagliaferro, M.; Rosa, P.; Bellenchi, G.C.; Bastianelli, D.; Trotta, R.; Tito, C.; Fazi, F.; Calogero, A.; Ponti, D. Nucleolar localization of the ErbB3 receptor as a new target in glioblastoma. BMC Mol. Cell Biol. 2022, 23, 13. [Google Scholar] [CrossRef]
- Park, S.J.; Kim, H.; Kim, S.H.; Joe, E.H.; Jou, I. Epigenetic downregulation of STAT6 increases HIF-1alpha expression via mTOR/S6K/S6, leading to enhanced hypoxic viability of glioma cells. Acta Neuropathol. Commun. 2019, 7, 149. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Zhang, W.; He, L.; Kong, F.; Pan, M.; Guo, J.; Xu, X.; Guo, J.; Wang, H.; Wang, Y. Jinlong capsule inhibits migration and invasion in human glioblastoma cells via the modulation of mTOR/S6 signaling pathway. Drug Des. Devel. Ther. 2019, 13, 1023–1032. [Google Scholar] [CrossRef] [Green Version]
- Udugama, M.; Sanij, E.; Voon, H.P.J.; Son, J.; Hii, L.; Henson, J.D.; Chan, F.L.; Chang, F.T.M.; Liu, Y.; Pearson, R.B.; et al. Ribosomal DNA copy loss and repeat instability in ATRX-mutated cancers. Proc. Natl. Acad. Sci. USA 2018, 115, 4737–4742. [Google Scholar] [CrossRef] [Green Version]
- Burger, K.; Muhl, B.; Harasim, T.; Rohrmoser, M.; Malamoussi, A.; Orban, M.; Kellner, M.; Gruber-Eber, A.; Kremmer, E.; Holzel, M.; et al. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J. Biol. Chem. 2010, 285, 12416–12425. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Trivedi, R.; Moubarek, M.S.; Moore, H.M.; Bai, B.; Rudek, M.A.; Bieberich, C.J.; Laiho, M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, R.; Schneekloth, J.S., Jr.; Panov, K.I.; Hannan, K.M.; Hannan, R.D. Targeting the RNA Polymerase I Transcription for Cancer Therapy Comes of Age. Cells 2020, 9, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negi, S.S.; Brown, P. rRNA synthesis inhibitor, CX-5461, activates ATM/ATR pathway in acute lymphoblastic leukemia, arrests cells in G2 phase and induces apoptosis. Oncotarget 2015, 6, 18094–18104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.S.; Zeki, J.; Ornell, K.; Coburn, J.; Shimada, H.; Ikegaki, N.; Chiu, B. Down-regulation of MYCN protein by CX-5461 leads to neuroblastoma tumor growth suppression. J. Pediatr. Surg. 2019, 54, 1192–1197. [Google Scholar] [CrossRef]
- Figueiredo, V.C.; McCarthy, J.J. Targeting cancer via ribosome biogenesis: The cachexia perspective. Cell Mol. Life Sci. 2021, 78, 5775–5787. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ribosomal Protein | Function | Induced Phenotypes | Reference |
|---|---|---|---|
| RPS6 | Oncogenic | Sphere-forming ability Stemness gene expression (Nestin, Sox2) Higher expression in GBM Higher expression at GSC-dominant area | [34,38] |
| RPS27 | Oncogenic | High expression in gliomas Higher expression at GSC-dominant area No relation to survival time | [136] |
| RPS15A | Oncogenic | Higher expression in GBM Proliferation Colony formation Anti-apoptosis Tumorigenesis Poor survival | [137,138] |
| RPL34 | Oncogenic | Higher expression in GBM Proliferation Anti-apoptosis Poor survival | [139] |
| RPL5 | Tumor-suppressive | Mutation 2.5%, deletion 8.4% in GBM Poor survival time in low RPL5 expression | [140] |
| RPS11 | Tumor-suppressive | High expression levels mean high susceptibility to topoisomerase II inhibitors (etoposide and doxorubicin) | [141] |
| RPS16 | |||
| RPS18 |
| Cell Line | Ribosome | Alteration of Phenotypes | Reference |
|---|---|---|---|
| Glioblastoma U251MG | Prokaryote | RICCS formation Stemness gene expression (Nestin, Sox2) pRPS6, RPS6 expression pSTAT3 expression Transdifferentiation (adipocyte, osteocyte) | [38,107] |
| Eukaryote (U252MG) | RICCS formation Stemness gene expression (Nestin, Sox2) pRPS6, RPS6 expression pRPS6 co-expressed Nestin RPS6K inhibitor (PF4708671) suppresses RICCS formation RPS6K inhibitor (PF4708671) suppresses the expression of Nestin and Sox2 | [38,107] | |
| Breast cancer MCF7 | Prokaryote | RICCS formation Increased G0 and early G1 phase cells EMT-like phenomenon Autophagy pathway activation p53-mediated stress response | [120,147] |
| Non-small cell lung cancer A549 | Prokaryote | RICCS formation Transdifferentiation (adipocyte, osteoblast) EGFR expression was increased on day 7, then decreased on day 14 CXCR4 expression was increased on day 14 Ki67-positive cells increased gradually on day 14 cyclinD1 expression increased by day 7, then decreased by day 14 In the tumor-forming assay, direct injection of ribosomes into the tumor mass No significant difference in tumor size and volume between control and ribosome-incorporated tumor | [120,148] |
| Gastric tubular adenocarcinoma H-111-TC | Prokaryote | RICCS formation Transdifferentiation (adipocyte, osteoblast) | [120,148] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hide, T.; Shibahara, I.; Inukai, M.; Shigeeda, R.; Kumabe, T. Ribosomes and Ribosomal Proteins Promote Plasticity and Stemness Induction in Glioma Cells via Reprogramming. Cells 2022, 11, 2142. https://doi.org/10.3390/cells11142142
Hide T, Shibahara I, Inukai M, Shigeeda R, Kumabe T. Ribosomes and Ribosomal Proteins Promote Plasticity and Stemness Induction in Glioma Cells via Reprogramming. Cells. 2022; 11(14):2142. https://doi.org/10.3390/cells11142142
Chicago/Turabian StyleHide, Takuichiro, Ichiyo Shibahara, Madoka Inukai, Ryota Shigeeda, and Toshihiro Kumabe. 2022. "Ribosomes and Ribosomal Proteins Promote Plasticity and Stemness Induction in Glioma Cells via Reprogramming" Cells 11, no. 14: 2142. https://doi.org/10.3390/cells11142142
APA StyleHide, T., Shibahara, I., Inukai, M., Shigeeda, R., & Kumabe, T. (2022). Ribosomes and Ribosomal Proteins Promote Plasticity and Stemness Induction in Glioma Cells via Reprogramming. Cells, 11(14), 2142. https://doi.org/10.3390/cells11142142