
Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
3. Method Details
3.1. Materials
3.2. Staining Methods
3.3. Detailed DNA/mRNA/Antibody Staining
- Samples were fixed and prepared as described above (antigen retrieval and elimination of autofluorescence).
- The sections were deparaffinized as described above using sequential alcohols and xylenes.
- Afterward, either antigen retrieval or endogenous biotin blocking was performed as described above. The brain generally has low to undetectable levels of endogenous biotin. However, liver, spleen, and lymph node tissues had significant endogenous biotin amounts.
- After HIV DNA and mRNA staining, tissue sections or cells were blocked overnight using a blocking solution containing 50 mM EDTA, 1% horse serum, 1% Ig-free BSA, 5% human serum, and 1% fish gelatin in PBS.
- Samples were incubated overnight in the primary antibody at 4 °C (HIV and cell marker antibodies). A critical point was determining how many antibodies can be used concomitantly based on antibody species and isotypes. Several combinations, including species, isotypes, labeling, and secondary antibodies, can be used to avoid repeating the same host.
- F.
- After incubation with the primary antibodies, at least 5 washes with PBS every 10 min were required to eliminate the unbounded antibodies.
- G.
- To detect the antibodies conjugated to biotin, incubation with streptavidin-conjugated to a fluorophore was necessary. An additional control was required here: infected tissues without primary biotinylated antibodies but with streptavidin conjugated to a fluorophore, to examine whether the endogenous biotin was properly eliminated. The detection of low levels of HIV proteins required at least 3 h of incubation with the labeled streptavidin or secondary antibody.
- H.
- After incubation with the secondary antibody or conjugated streptavidin, at least 5 washes with PBS were required to eliminate the unbounded streptavidin or secondary labeling antibodies. Detection was achieved using confocal equipment equipped with unmixing and spectrum-detection systems that enabled the separation of extremely narrow wavelengths (up to 2.5 nm) to identify and isolate multiple colors (wavelengths) without overlay or unspecific signals (see examples in [31,33,43,44,48]).
- I.
- Then, samples were mounted using Prolong Gold/Diamond anti-fade reagent with DAPI or Vector Shield (preferred).
- J.
- After staining, samples were kept in the dark before confocal analysis to enable the mounting media to penetrate the tissue and allow for the media to cure.
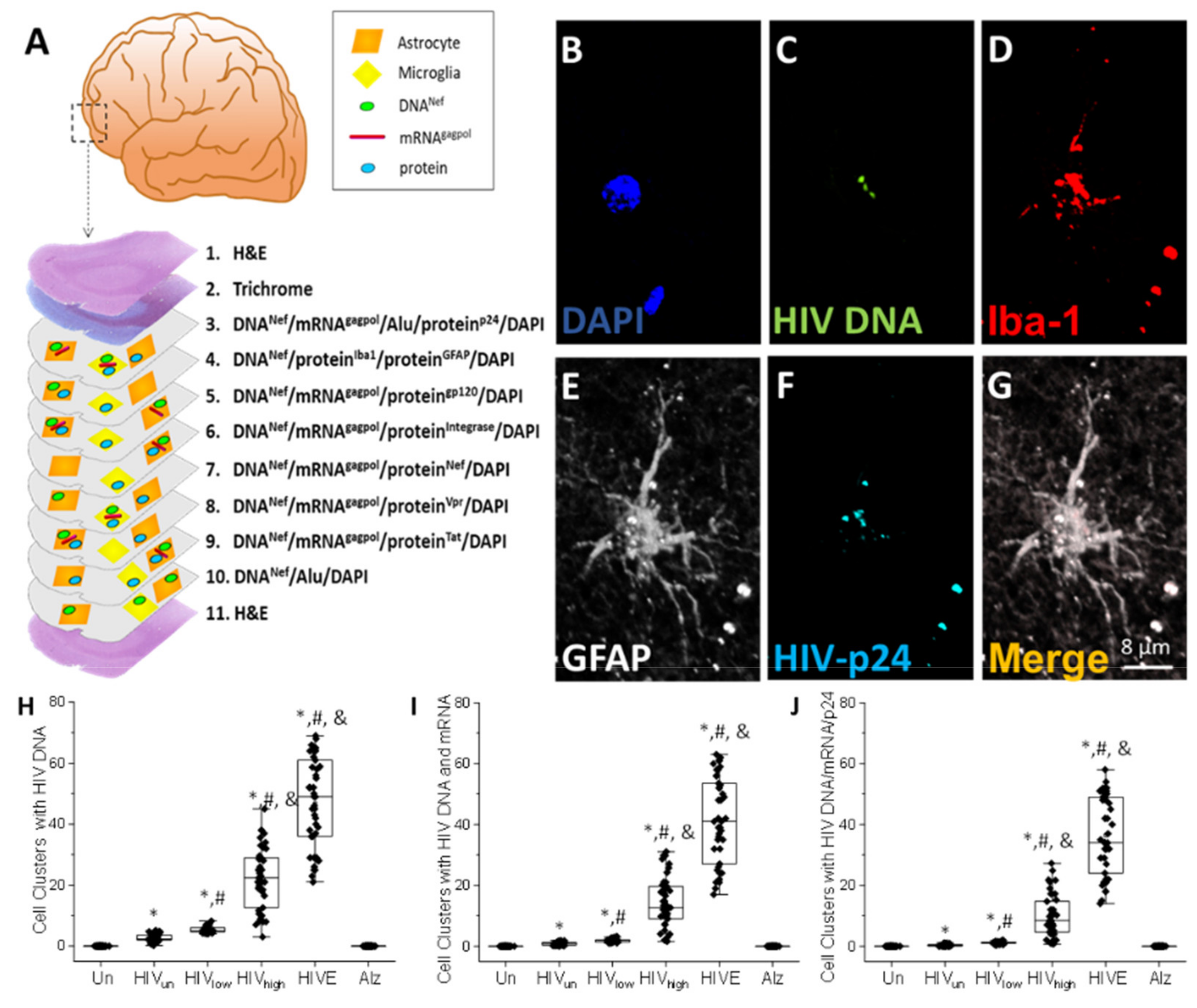
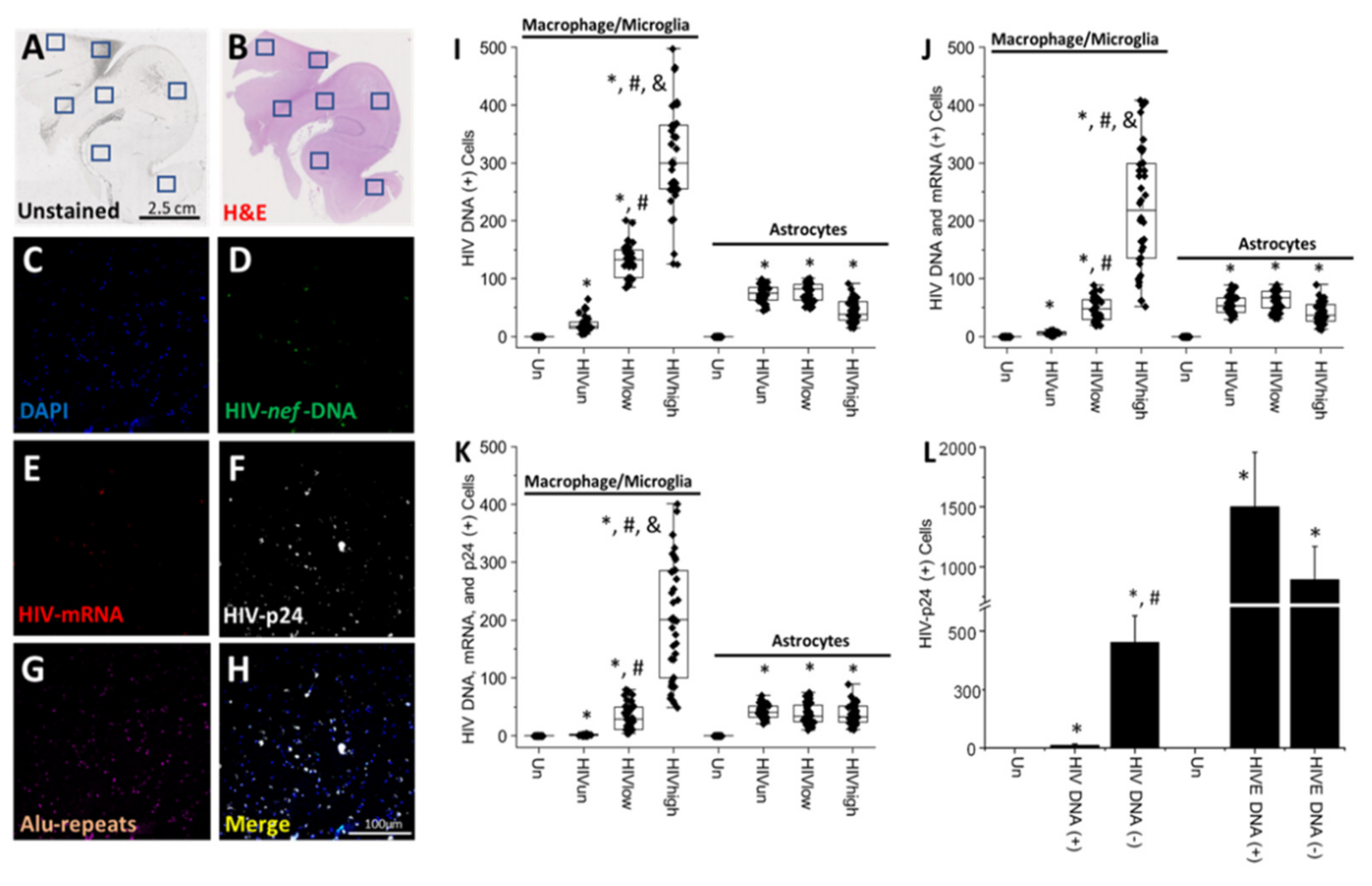
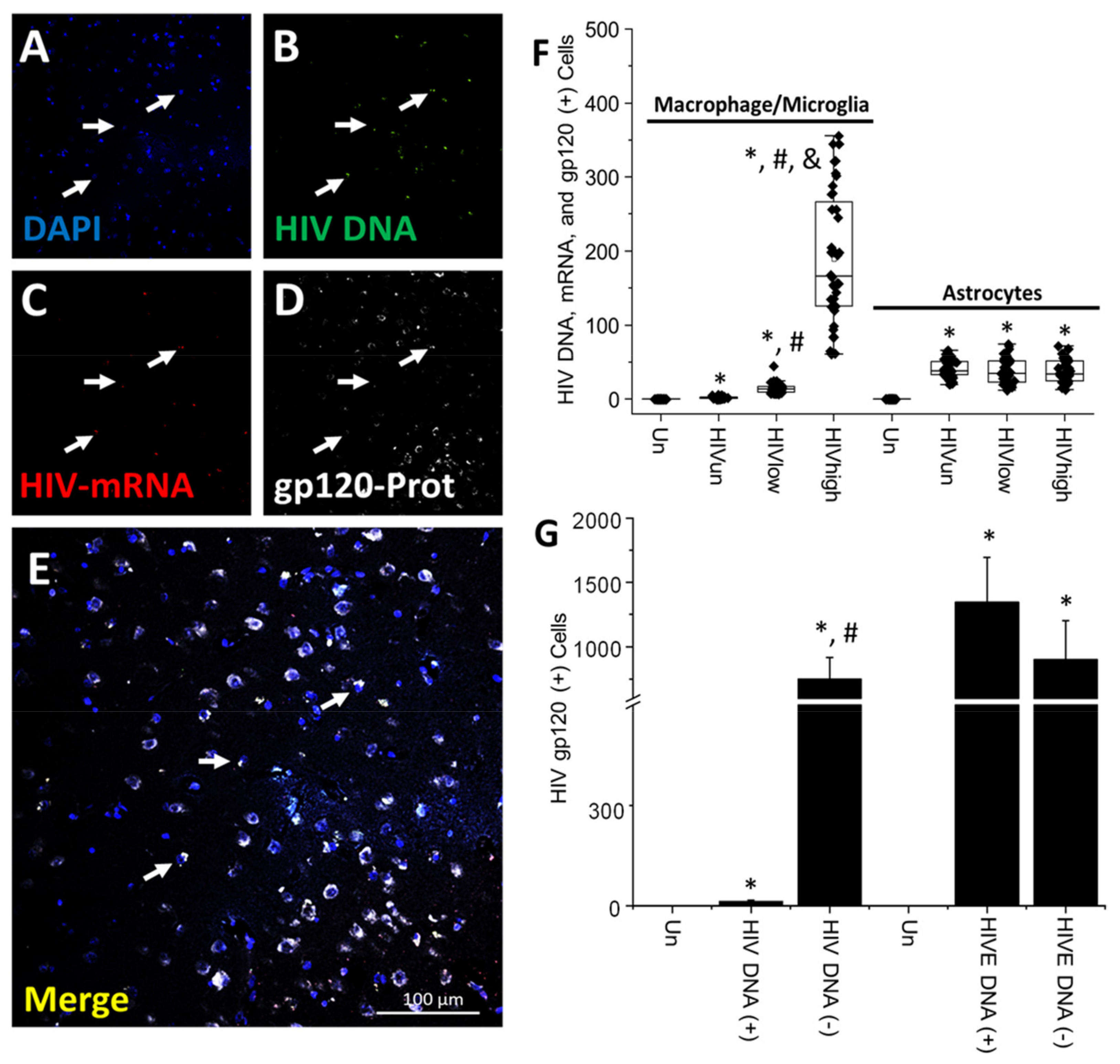
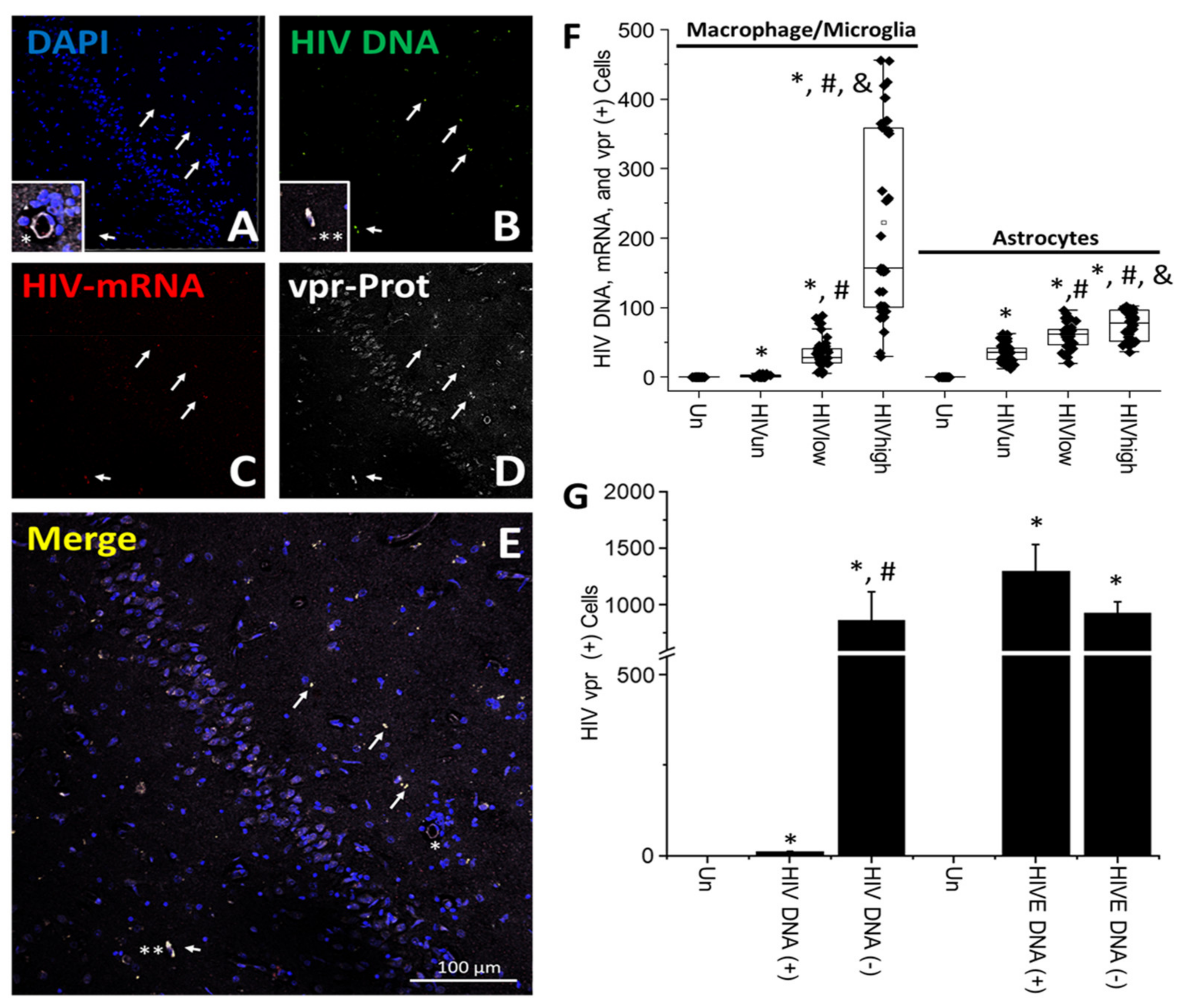
4. Results
5. Discussion
6. Conclusions
- HIV-infected microglia/macrophages and a small population of astrocytes are the main brain cell types infected in the pre- and current ART era.
- HIV brain reservoirs still produce residual viral mRNAs and protein expression despite ART.
- HIV-p24, gp120, nef, vpr, and tat proteins, but not integrase, are expressed in HIV-infected cells, released, and taken up by neighboring uninfected cells, even in ART conditions.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Scheerder, M.A.; Vrancken, B.; Dellicour, S.; Schlub, T.; Lee, E.; Shao, W.; Rutsaert, S.; Verhofstede, C.; Kerre, T.; Malfait, T.; et al. HIV Rebound Is Predominantly Fueled by Genetically Identical Viral Expansions from Diverse Reservoirs. Cell Host Microbe. 2019, 26, 347–358.e347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Scheerder, M.A.; Depelseneer, B.; Vandekerckhove, L.; Trypsteen, W. Evolution of Experimental Design and Research Techniques in HIV-1 Reservoir Studies: A Systematic Review. AIDS Rev. 2020, 22, 16–24. [Google Scholar] [CrossRef]
- Kimata, J.T.; Rice, A.P.; Wang, J. Challenges and strategies for the eradication of the HIV reservoir. Curr. Opin. Immunol. 2016, 42, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujinaga, K.; Cary, D.C. Experimental Systems for Measuring HIV Latency and Reactivation. Viruses 2020, 12, 1279. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.K.; Yukl, S.A. Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 2016, 11, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Bender, A.M.; Hackman, J.; Kwon, K.J.; Lynch, B.A.; Bruno, D.; Martens, C.; Beg, S.; Florman, S.S.; Desai, N.; et al. Similar frequency and inducibility of intact HIV-1 proviruses in blood and lymph nodes. J. Infect. Dis 2020. [Google Scholar] [CrossRef]
- Denton, P.W.; Sogaard, O.S.; Tolstrup, M. Impacts of HIV Cure Interventions on Viral Reservoirs in Tissues. Front. Microbiol. 2019, 10, 1956. [Google Scholar] [CrossRef] [PubMed]
- Fernando Real, C.C.; Sennepin, A.; Arrigucci, R.; Zhu, A.; Sannier, G.; Zheng, J.; Xu, L.; Massé, J.-M.; Greffe, S.; Cazabat, M.; et al. Platelets from cART-suppressed HIV-infected patients with poor CD4+T cell recovery carry infectious HIV. Sci. Transl. Med. 2020. [Google Scholar]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.; Shirk, E.N.; Queen, S.E.; Mankowski, J.L.; Gama, L.; Clements, J.E. A Quantitative Approach to SIV Functional Latency in Brain Macrophages. J. Neuroimmune Pharm. 2019, 14, 23–32. [Google Scholar] [CrossRef]
- Abdel-Mohsen, M.; Richman, D.; Siliciano, R.F.; Nussenzweig, M.C.; Howell, B.J.; Martinez-Picado, J.; Chomont, N.; Bar, K.J.; Yu, X.G.; Lichterfeld, M.; et al. Recommendations for measuring HIV reservoir size in cure-directed clinical trials. Nat. Med. 2020, 26, 1339–1350. [Google Scholar] [CrossRef]
- Wang, Z.; Simonetti, F.R.; Siliciano, R.F.; Laird, G.M. Measuring replication competent HIV-1: Advances and challenges in defining the latent reservoir. Retrovirology 2018, 15, 21. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. Assays to Measure Latency, Reservoirs, and Reactivation. Curr. Top. Microbiol. Immunol. 2018, 417, 23–41. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. HIV reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2016, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Bruner, K.M.; Hosmane, N.N.; Siliciano, R.F. Towards an HIV-1 cure: Measuring the latent reservoir. Trends Microbiol. 2015, 23, 192–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, E.H.; O’Doherty, U. Quantitation of integrated proviral DNA in viral reservoirs. Curr. Opin. HIV AIDS 2013, 8, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Kreider, E.F.; Bar, K.J. HIV-1 Reservoir Persistence and Decay: Implications for Cure Strategies. Curr. HIV/AIDS Rep. 2022, 19, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Salantes, D.B.; Zheng, Y.; Mampe, F.; Srivastava, T.; Beg, S.; Lai, J.; Li, J.Z.; Tressler, R.L.; Koup, R.A.; Hoxie, J.; et al. HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Invest. 2018, 128, 3102–3115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eriksson, S.; Graf, E.H.; Dahl, V.; Strain, M.C.; Yukl, S.A.; Lysenko, E.S.; Bosch, R.J.; Lai, J.; Chioma, S.; Emad, F.; et al. Comparative analysis of measures of viral reservoirs in HIV-1 eradication studies. PLoS Pathog 2013, 9, e1003174. [Google Scholar] [CrossRef]
- Strain, M.C.; Richman, D.D. New assays for monitoring residual HIV burden in effectively treated individuals. Curr. Opin. HIV AIDS 2013, 8, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Deere, J.D.; Kauffman, R.C.; Cannavo, E.; Higgins, J.; Villalobos, A.; Adamson, L.; Schinazi, R.F.; Luciw, P.A.; North, T.W. Analysis of multiply spliced transcripts in lymphoid tissue reservoirs of rhesus macaques infected with RT-SHIV during HAART. PLoS ONE 2014, 9, e87914. [Google Scholar] [CrossRef] [Green Version]
- Cole, B.; Lambrechts, L.; Boyer, Z.; Noppe, Y.; De Scheerder, M.A.; Eden, J.S.; Vrancken, B.; Schlub, T.E.; McLaughlin, S.; Frenkel, L.M.; et al. Extensive characterization of HIV-1 reservoirs reveals links to plasma viremia before and during analytical treatment interruption. Cell Rep. 2022, 39, 110739. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Munoz, O.; Perreau, M. HIV persistence in lymph nodes. Curr. Opin. HIV AIDS 2021, 16, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo-Redondo, R.; Fryer, H.R.; Bedford, T.; Kim, E.Y.; Archer, J.; Pond, S.L.K.; Chung, Y.S.; Penugonda, S.; Chipman, J.; Fletcher, C.V.; et al. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 2016, 530, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaillon, A.; Gianella, S.; Dellicour, S.; Rawlings, S.A.; Schlub, T.E.; De Oliveira, M.F.; Ignacio, C.; Porrachia, M.; Vrancken, B.; Smith, D.M. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Investig. 2020, 130, 1699–1712. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, M.A.; Chun, T.W.; Justement, S.J.; Motola, I.; Spinelli, M.A.; Adelsberger, J.; Ehler, L.A.; Mizell, S.B.; Hallahan, C.W.; Fauci, A.S. Both memory and CD45RA+/CD62L+ naive CD4(+) T cells are infected in human immunodeficiency virus type 1-infected individuals. J. Virol. 1999, 73, 6430–6435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, L.J.; Reoma, L.B.; Kovacs, J.A.; Nath, A. Advances toward Curing HIV-1 Infection in Tissue Reservoirs. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Churchill, M.J.; Gorry, P.R.; Cowley, D.; Lal, L.; Sonza, S.; Purcell, D.F.; Thompson, K.A.; Gabuzda, D.; McArthur, J.C.; Pardo, C.A.; et al. Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 2006, 12, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci. Rep. 2017, 7, 12866. [Google Scholar] [CrossRef] [Green Version]
- Valdebenito, S.; Castellano, P.; Ajasin, D.; Eugenin, E.A. Astrocytes are HIV reservoirs in the brain: A cell type with poor HIV infectivity and replication but efficient cell-to-cell viral transfer. J. Neurochem. 2021, 158, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Valdebenito, S.; D’Amico, D.; Prideaux, B.; Eugenin, E.A. HIV infection of astrocytes compromises inter-organelle interactions and inositol phosphate metabolism: A potential mechanism of bystander damage and viral reservoir survival. Prog. Neurobiol. 2021, 206, 102157. [Google Scholar] [CrossRef] [PubMed]
- Lutgen, V.; Narasipura, S.D.; Barbian, H.J.; Richards, M.; Wallace, J.; Razmpour, R.; Buzhdygan, T.; Ramirez, S.H.; Prevedel, L.; Eugenin, E.A.; et al. HIV infects astrocytes in vivo and egresses from the brain to the periphery. PLoS Pathog. 2020, 16, e1008381. [Google Scholar] [CrossRef] [PubMed]
- Barat, C.; Proust, A.; Deshiere, A.; Leboeuf, M.; Drouin, J.; Tremblay, M.J. Astrocytes sustain long-term productive HIV-1 infection without establishment of reactivable viral latency. Glia 2018, 66, 1363–1381. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A. Enigma of HIV-1 latent infection in astrocytes: An in-vitro study using protein kinase C agonist as a latency reversing agent. Microbes Infect. 2015, 17, 651–659. [Google Scholar] [CrossRef] [Green Version]
- Kasparov, S. Are Astrocytes the Pressure-Reservoirs of Lactate in the Brain? Cell Metab. 2016, 23, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Ko, A.; Kang, G.; Hattler, J.B.; Galadima, H.I.; Zhang, J.; Li, Q.; Kim, W.K. Macrophages but not Astrocytes Harbor HIV DNA in the Brains of HIV-1-Infected Aviremic Individuals on Suppressive Antiretroviral Therapy. J. Neuroimmune Pharm. 2019, 14, 110–119. [Google Scholar] [CrossRef] [Green Version]
- Marban, C.; Forouzanfar, F.; Ait-Ammar, A.; Fahmi, F.; El Mekdad, H.; Daouad, F.; Rohr, O.; Schwartz, C. Targeting the Brain Reservoirs: Toward an HIV Cure. Front. Immunol. 2016, 7, 397. [Google Scholar] [CrossRef] [Green Version]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr HIV Res. 2016, 14, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.A.; Chojnacki, J.; Jones, D.M.; Johnson, E.; Do, T.; Eggeling, C.; Padilla-Parra, S.; Sattentau, Q.J. Astrocytes Resist HIV-1 Fusion but Engulf Infected Macrophage Material. Cell Rep. 2017, 18, 1473–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon-Rivera, R.; Veenstra, M.; Donoso, M.; Tell, E.; Eugenin, E.A.; Morgello, S.; Berman, J.W. Central Nervous System (CNS) Viral Seeding by Mature Monocytes and Potential Therapies To Reduce CNS Viral Reservoirs in the cART Era. mBio 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Capron, C.; Sennepin, A.; Arrigucci, R.; Zhu, A.; Sannier, G.; Zheng, J.; Xu, L.; Masse, J.M.; Greffe, S.; et al. Platelets from HIV-infected individuals on antiretroviral drug therapy with poor CD4(+) T cell recovery can harbor replication-competent HIV despite viral suppression. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Prevedel, L.; Ruel, N.; Castellano, P.; Smith, C.; Malik, S.; Villeux, C.; Bomsel, M.; Morgello, S.; Eugenin, E.A. Identification, Localization, and Quantification of HIV Reservoirs Using Microscopy. Curr. Protoc. Cell Biol. 2019, 82, e64. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723-742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butera, S.T.; Roberts, B.D.; Leung, K.; Nabel, G.J.; Folks, T.M. Tumor necrosis factor receptor expression and signal transduction in HIV-1-infected cells. AIDS 1993, 7, 911–918. [Google Scholar] [CrossRef]
- Firestein, G.S.; Reifler, D.; Richman, D.; Gruber, H.E. Rapid and reversible modulation of T4 (CD4) on monocytoid cells by phorbol myristate acetate: Effect on HIV susceptibility. Cell Immunol. 1988, 113, 63–69. [Google Scholar] [CrossRef]
- Luu, R.; Valdebenito, S.; Scemes, E.; Cibelli, A.; Spray, D.C.; Rovegno, M.; Tichauer, J.; Cottignies-Calamarte, A.; Rosenberg, A.; Capron, C.; et al. Pannexin-1 channel opening is critical for COVID-19 pathogenesis. iScience 2021, 24, 103478. [Google Scholar] [CrossRef] [PubMed]
- Deyl, Z.; Horakova, M.; Vancikova, O. Changes in pyridinoline content of elastin during ontogeny. Mech Ageing Dev. 1981, 17, 321–325. [Google Scholar] [CrossRef]
- Deyl, Z.; Macek, K.; Adam, M.; Vancikova, O. Studies on the chemical nature of elastin fluorescence. Biochim. Biophys. Acta 1980, 625, 248–254. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Clements, J.E.; Zink, M.C.; Berman, J.W. Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 2011, 31, 9456–9465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jernigan, T.L.; Archibald, S.L.; Fennema-Notestine, C.; Taylor, M.J.; Theilmann, R.J.; Julaton, M.D.; Notestine, R.J.; Wolfson, T.; Letendre, S.L.; Ellis, R.J.; et al. Clinical factors related to brain structure in HIV: The CHARTER study. J. Neurovirol. 2011, 17, 248–257. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, S.P.; Rippeth, J.D.; Frol, A.B.; Levy, J.K.; Ryan, E.; Soukup, V.M.; Hinkin, C.H.; Lazzaretto, D.; Cherner, M.; Marcotte, T.D.; et al. Interrater reliability of clinical ratings and neurocognitive diagnoses in HIV. J. Clin. Exp. Neuropsychol. 2004, 26, 759–778. [Google Scholar] [CrossRef] [PubMed]
- Chompre, G.; Cruz, E.; Maldonado, L.; Rivera-Amill, V.; Porter, J.T.; Noel, R.J., Jr. Astrocytic expression of HIV-1 Nef impairs spatial and recognition memory. Neurobiol. Dis. 2013, 49, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarandi, S.S.; Robinson, J.A.; Vakili, S.; Donadoni, M.; Burdo, T.H.; Sariyer, I.K. Characterization of Nef expression in different brain regions of SIV-infected macaques. PLoS ONE 2020, 15, e0241667. [Google Scholar] [CrossRef] [PubMed]
- Sami Saribas, A.; Cicalese, S.; Ahooyi, T.M.; Khalili, K.; Amini, S.; Sariyer, I.K. HIV-1 Nef is released in extracellular vesicles derived from astrocytes: Evidence for Nef-mediated neurotoxicity. Cell Death Dis 2017, 8, e2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, P.K.; Deshmane, S.; Khalili, K.; Merali, S.; Gordon, J.C.; Fecchio, C.; Barrero, C.A. Glutamate metabolism in HIV-1 infected macrophages: Role of HIV-1 Vpr. Cell Cycle 2016, 15, 2288–2298. [Google Scholar] [CrossRef] [Green Version]
- Kehn-Hall, K.; Guendel, I.; Carpio, L.; Skaltsounis, L.; Meijer, L.; Al-Harthi, L.; Steiner, J.P.; Nath, A.; Kutsch, O.; Kashanchi, F. Inhibition of Tat-mediated HIV-1 replication and neurotoxicity by novel GSK3-beta inhibitors. Virology 2011, 415, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, E.D.; Bachis, A.; Avdoshina, V.; Taraballi, F.; Tasciotti, E.; Mocchetti, I. Endocytic Trafficking of HIV gp120 is Mediated by Dynamin and Plays a Role in gp120 Neurotoxicity. J. Neuroimmune Pharm. 2017, 12, 492–503. [Google Scholar] [CrossRef]
- Bachis, A.; Aden, S.A.; Nosheny, R.L.; Andrews, P.M.; Mocchetti, I. Axonal transport of human immunodeficiency virus type 1 envelope protein glycoprotein 120 is found in association with neuronal apoptosis. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 6771–6780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, D.T.; Silvestri, G. Nonhuman primate models in AIDS research. Curr. Opin. HIV AIDS 2013, 8, 255–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, H.; Cheng, Y.; Sravanam, S.; Mathews, S.; Gorantla, S.; Poluektova, L.Y.; Dash, P.K.; Gendelman, H.E. Immune Activations and Viral Tissue Compartmentalization During Progressive HIV-1 Infection of Humanized Mice. Front. Immunol. 2019, 10, 340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar, K.J.; Coronado, E.; Hensley-McBain, T.; O’Connor, M.A.; Osborn, J.M.; Miller, C.; Gott, T.M.; Wangari, S.; Iwayama, N.; Ahrens, C.Y.; et al. Simian-Human Immunodeficiency Virus SHIV.CH505 Infection of Rhesus Macaques Results in Persistent Viral Replication and Induces Intestinal Immunopathology. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, F.; Ma, X.; Zhao, X.; Li, W.; Zhang, Z.; Zhang, J.; Zhang, X.E.; Cui, Z. Identification of interaction between HIV-1 glycoprotein 41 and integrase. Virol. Sin. 2016, 31, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Koneru, P.C.; Francis, A.C.; Deng, N.; Rebensburg, S.V.; Hoyte, A.C.; Lindenberger, J.; Adu-Ampratwum, D.; Larue, R.C.; Wempe, M.F.; Engelman, A.N.; et al. HIV-1 integrase tetramers are the antiviral target of pyridine-based allosteric integrase inhibitors. Elife 2019, 8. [Google Scholar] [CrossRef]
- Wadhwa, P.; Jain, P.; Rudrawar, S.; Jadhav, H.R.A. Quinoline, Coumarin and Other Heterocyclic Analogs Based HIV-1 Integrase Inhibitors. Curr. Drug Discov. Technol. 2018, 15, 2–19. [Google Scholar] [CrossRef]
- Gill, M.S.A.; Hassan, S.S.; Ahemad, N. Evolution of HIV-1 reverse transcriptase and integrase dual inhibitors: Recent advances and developments. Eur. J. Med. Chem. 2019, 179, 423–448. [Google Scholar] [CrossRef] [PubMed]
- Craigie, R.; Bushman, F.D. HIV DNA integration. Cold Spring Harb Perspect. Med. 2012, 2, a006890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheney, L.; Guzik, H.; Macaluso, F.P.; Macian, F.; Cuervo, A.M.; Berman, J.W. HIV Nef and Antiretroviral Therapy Have an Inhibitory Effect on Autophagy in Human Astrocytes that May Contribute to HIV-Associated Neurocognitive Disorders. Cells 2020, 9, 1426. [Google Scholar] [CrossRef]
- Fields, J.; Dumaop, W.; Eleuteri, S.; Campos, S.; Serger, E.; Trejo, M.; Kosberg, K.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV-1 Tat alters neuronal autophagy by modulating autophagosome fusion to the lysosome: Implications for HIV-associated neurocognitive disorders. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 1921–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarandi, S.S.; Duggan, M.R.; Sariyer, I.K. Emerging Role of Nef in the Development of HIV Associated Neurological Disorders. J. Neuroimmune Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kogan, M.; Rappaport, J. HIV-1 accessory protein Vpr: Relevance in the pathogenesis of HIV and potential for therapeutic intervention. Retrovirology 2011, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Spector, C.; Mele, A.R.; Wigdahl, B.; Nonnemacher, M.R. Genetic variation and function of the HIV-1 Tat protein. Med. Microbiol Immunol 2019, 208, 131–169. [Google Scholar] [CrossRef]
- Hategan, A.; Masliah, E.; Nath, A. HIV and Alzheimer’s disease: Complex interactions of HIV-Tat with amyloid beta peptide and Tau protein. J. Neurovirol. 2019, 25, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.J.; Johnson, T.P.; Smith, B.R.; Reoma, L.B.; Santamaria, U.A.; Bachani, M.; Demarino, C.; Barclay, R.A.; Snow, J.; Sacktor, N.; et al. Presence of Tat and transactivation response element in spinal fluid despite antiretroviral therapy. AIDS 2019, 33 (Suppl. S2), S145–S157. [Google Scholar] [CrossRef]
- Cary, D.C.; Fujinaga, K.; Peterlin, B.M. Molecular mechanisms of HIV latency. J. Clin. Investig. 2016, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bruner, K.M.; Wang, Z.; Simonetti, F.R.; Bender, A.M.; Kwon, K.J.; Sengupta, S.; Fray, E.J.; Beg, S.A.; Antar, A.A.R.; Jenike, K.M.; et al. A quantitative approach for measuring the reservoir of latent HIV-1 proviruses. Nature 2019, 566, 120–125. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Hosmane, N.N.; Kwon, K.J.; Bruner, K.M.; Capoferri, A.A.; Beg, S.; Rosenbloom, D.I.; Keele, B.F.; Ho, Y.C.; Siliciano, J.D.; Siliciano, R.F. Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: Potential role in latent reservoir dynamics. J. Exp. Med. 2017, 214, 959–972. [Google Scholar] [CrossRef]
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 2019, 142, 502–511. [Google Scholar] [CrossRef]
- Falcinelli, S.D.; Ceriani, C.; Margolis, D.M.; Archin, N.M. New Frontiers in Measuring and Characterizing the HIV Reservoir. Front. Microbiol. 2019, 10, 2878. [Google Scholar] [CrossRef] [Green Version]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate HIV from infected patients: Elimination of latent provirus reservoirs. Cell Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef] [Green Version]
- Herzig, E.; Kim, K.C.; Packard, T.A.; Vardi, N.; Schwarzer, R.; Gramatica, A.; Deeks, S.G.; Williams, S.R.; Landgraf, K.; Killeen, N.; et al. Attacking Latent HIV with convertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell 2019, 179, 880–894.e10. [Google Scholar] [CrossRef] [PubMed]
- Strain, M.C.; Letendre, S.; Pillai, S.K.; Russell, T.; Ignacio, C.C.; Gunthard, H.F.; Good, B.; Smith, D.M.; Wolinsky, S.M.; Furtado, M.; et al. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J. Virol. 2005, 79, 1772–1788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gama, L.; Abreu, C.M.; Shirk, E.N.; Price, S.L.; Li, M.; Laird, G.M.; Pate, K.A.; Wietgrefe, S.W.; O’Connor, S.L.; Pianowski, L.; et al. Reactivation of simian immunodeficiency virus reservoirs in the brain of virally suppressed macaques. AIDS 2017, 31, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Killingsworth, L.; Spudich, S. Neuropathogenesis of HIV-1: Insights from across the spectrum of acute through long-term treated infection. Semin. Immunopathol. 2022. [Google Scholar] [CrossRef]
- Eugenin, E.A.; Berman, J.W. Cytochrome C dysregulation induced by HIV infection of astrocytes results in bystander apoptosis of uninfected astrocytes by an IP3 and calcium-dependent mechanism. J. Neurochem. 2013, 127, 644–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Valdebenito, S.; Eugenin, E.A. HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 2019, 9, 3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugenin, E.A.; Berman, J.W. Gap junctions mediate human immunodeficiency virus-bystander killing in astrocytes. J. Neurosci. 2007, 27, 12844–12850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorska, A.M.; Eugenin, E.A. The Glutamate System as a Crucial Regulator of CNS Toxicity and Survival of HIV Reservoirs. Front. Cell Infect. Microbiol. 2020, 10, 261. [Google Scholar] [CrossRef] [PubMed]
- Okafo, G.; Valdebenito, S.; Donoso, M.; Luu, R.; Ajasin, D.; Prideaux, B.; Gorantla, S.; Eugenin, E.A. Role of Tunneling Nanotube-like Structures during the Early Events of HIV Infection: Novel Features of Tissue Compartmentalization and Mechanism of HIV Spread. J. Immunol 2020. [Google Scholar] [CrossRef] [PubMed]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Ajasin, D.; Eugenin, E.A. HIV-1 Tat: Role in Bystander Toxicity. Front. Cell Infect. Microbiol. 2020, 10, 61. [Google Scholar] [CrossRef] [PubMed]
- Berman, J.W.; Carvallo, L.; Buckner, C.M.; Luers, A.; Prevedel, L.; Bennett, M.V.; Eugenin, E.A. HIV-tat alters Connexin43 expression and trafficking in human astrocytes: Role in NeuroAIDS. J. Neuroinflammation 2016, 13, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Huang, W.; Jiang, W.; Wu, X.; Ye, B.; Zhou, X. HIV-1 Tat Regulates Occludin and Abeta Transfer Receptor Expression in Brain Endothelial Cells via Rho/ROCK Signaling Pathway. Oxid Med. Cell Longev 2016, 2016, 4196572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eugenin, E.A.; King, J.E.; Nath, A.; Calderon, T.M.; Zukin, R.S.; Bennett, M.V.; Berman, J.W. HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2007, 104, 3438–3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.E.; Eugenin, E.A.; Buckner, C.M.; Berman, J.W. HIV tat and neurotoxicity. Microbes. Infect. 2006, 8, 1347–1357. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhao, L.; Jia, B.; Wu, L.; Li, Y.; Curthoys, N.; Zheng, J.C. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: Relevant to HIV-1-associated neurocognitive disorders. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 15195–15204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Xu, Q.; Qiu, J.; Zheng, H.; Ye, X.; Xue, Y.; Yin, Y.; Zhang, Z.; Liu, Y.; et al. Menin mediates Tat-induced neuronal apoptosis in brain frontal cortex of SIV-infected macaques and in Tat-treated cells. Oncotarget 2017, 8, 18082–18094. [Google Scholar] [CrossRef] [Green Version]
- Hammond, J.W.; Qiu, W.Q.; Marker, D.F.; Chamberlain, J.M.; Greaves-Tunnell, W.; Bellizzi, M.J.; Lu, S.M.; Gelbard, H.A. HIV Tat causes synapse loss in a mouse model of HIV-associated neurocognitive disorder that is independent of the classical complement cascade component C1q. Glia 2018, 66, 2563–2574. [Google Scholar] [CrossRef] [PubMed]
- Bozzelli, P.L.; Yin, T.; Avdoshina, V.; Mocchetti, I.; Conant, K.E.; Maguire-Zeiss, K.A. HIV-1 Tat promotes astrocytic release of CCL2 through MMP/PAR-1 signaling. Glia 2019, 67, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Nath, A.; Mattson, M.P. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp. Neurol. 1998, 154, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.T. HIV-1 Tat-Mediated Calcium Dysregulation and Neuronal Dysfunction in Vulnerable Brain Regions. Curr. Drug Targets 2016, 17, 4–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, J.E.; Eugenin, E.A.; Hazleton, J.E.; Morgello, S.; Berman, J.W. Mechanisms of HIV-tat-induced phosphorylation of N-methyl-D-aspartate receptor subunit 2A in human primary neurons: Implications for neuroAIDS pathogenesis. Am. J. Pathol. 2010, 176, 2819–2830. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 and Methamphetamine-Mediated Mitochondrial Damage and Neuronal Degeneration in Human Neurons. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Samikkannu, T.; Rao, K.V.K.; Salam, A.A.A.; Atluri, V.S.R.; Kaftanovskaya, E.M.; Agudelo, M.; Perez, S.; Yoo, C.; Raymond, A.D.; Ding, H.; et al. HIV Subtypes B and C gp120 and Methamphetamine Interaction: Dopaminergic System Implicates Differential Neuronal Toxicity. Sci. Rep. 2015, 5, 11130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, E.D.; Speidell, A.; Flowers, S.A.; Wu, C.; Avdoshina, V.; Mocchetti, I. Histone deacetylase 6 inhibition rescues axonal transport impairments and prevents the neurotoxicity of HIV-1 envelope protein gp120. Cell Death Dis. 2019, 10, 674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Green, M.V.; Thayer, S.A. HIV gp120-induced neuroinflammation potentiates NMDA receptors to overcome basal suppression of inhibitory synapses by p38 MAPK. J. Neurochem. 2019, 148, 499–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 gp120 and Tat Induce Mitochondrial Fragmentation and Incomplete Mitophagy in Human Neurons. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Avdoshina, V.; Fields, J.A.; Castellano, P.; Dedoni, S.; Palchik, G.; Trejo, M.; Adame, A.; Rockenstein, E.; Eugenin, E.; Masliah, E.; et al. The HIV Protein gp120 Alters Mitochondrial Dynamics in Neurons. Neurotox Res. 2016, 29, 583–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trillo-Pazos, G.; McFarlane-Abdulla, E.; Campbell, I.C.; Pilkington, G.J.; Everall, I.P. Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res. 2000, 864, 315–326. [Google Scholar] [CrossRef]
- Liu, X.; Kumar, A. Differential signaling mechanism for HIV-1 Nef-mediated production of IL-6 and IL-8 in human astrocytes. Sci. Rep. 2015, 5, 9867. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Shah, A.; Gangwani, M.R.; Silverstein, P.S.; Fu, M.; Kumar, A. HIV-1 Nef induces CCL5 production in astrocytes through p38-MAPK and PI3K/Akt pathway and utilizes NF-kB, CEBP and AP-1 transcription factors. Sci. Rep. 2014, 4, 4450. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
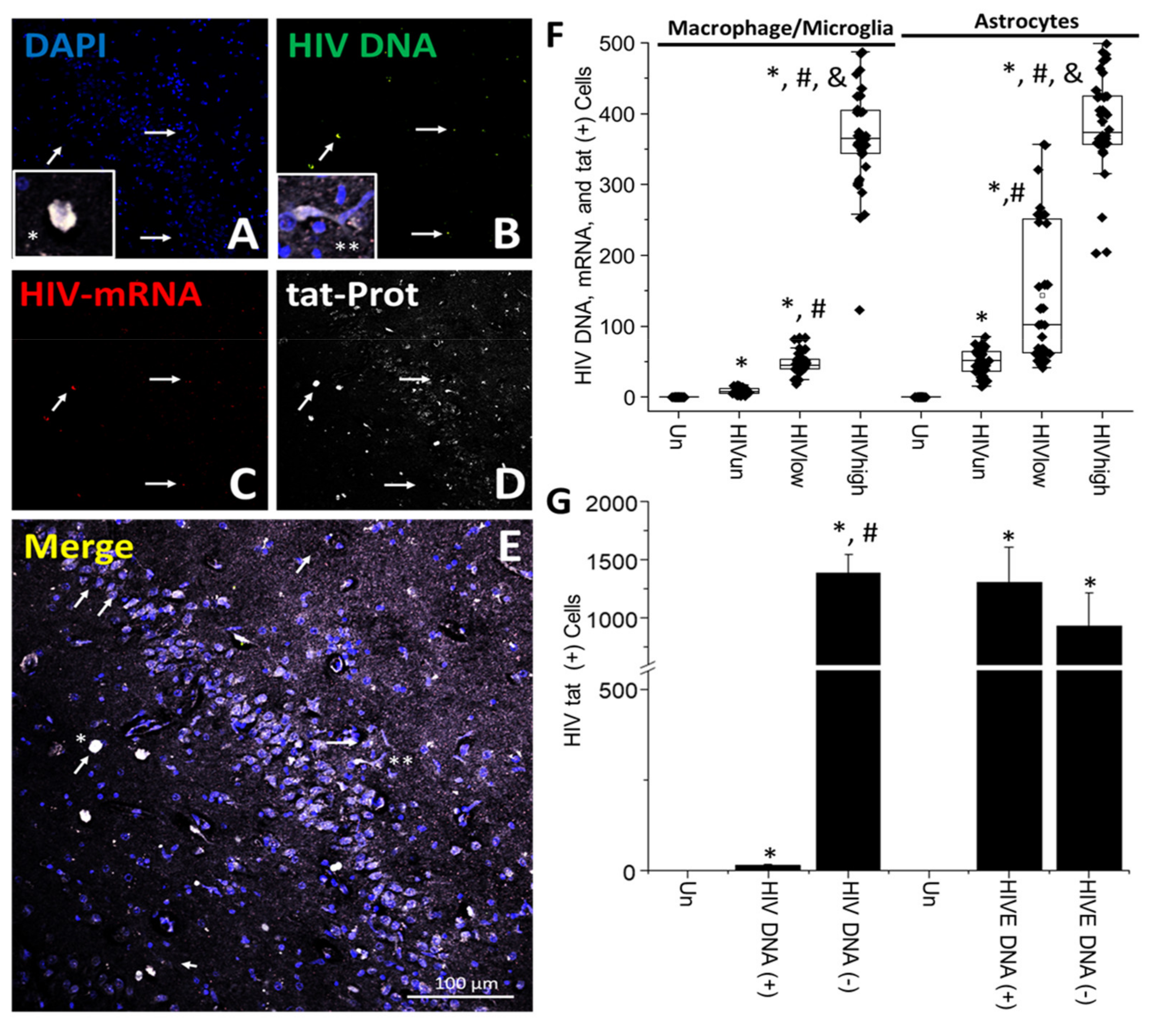{kind=link}
| No | Patient Group | HIV | Age | Sex | Cognitive Status | CD4 Cells/mm3 | CD8, Cells/mm3 | Viral Load, Copies/mL | Years with HIV | ART | Brain Area |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Un | - | 44 | M | None | - | - | - | - | - | B |
| 2 | Un | - | 50 | M | None | - | - | - | - | - | B |
| 3 | Un | - | 69 | M | None | - | - | - | - | - | B |
| 4 | Un | - | 38 | M | None | - | - | - | - | - | C |
| 5 | Un | - | 51 | F | None | - | - | - | - | - | B |
| 6 | HIVun | + | 70 | M | MCMD | 741 | N.R. | <50 | 26 | ziagen, epivir, fosamprenavir, ritonavir | B |
| 7 | HIVun | + | 69 | F | MCMD | 616 | 1190 | <50 | 22 | ziagen, epivir, fosamprenavir | C |
| 8 | HIVun | + | 61 | F | MCMD | 488 | 465 | <50 | 24 | efavirenz, ziagen, epivir | B |
| 9 | HIVun | + | 68 | F | None | 1306 | 380 | 0 | 15 | emtricitabine, tenofovir alafenamide, elvitegravir, cobicistat | B |
| 10 | HIVun | + | 48 | M | MCMD | 1658 | 50 | <50 | 13 | etravirine, ritonavir, maraviroc, darunavir | C |
| 11 | HIVun | + | 36 | M | None | 293 | 881 | <50 | 11 | zidovudine, lamivudine, nelfinavir | C |
| 12 | HIVun | + | 59 | M | None | 428 | 768 | <50 | 26 | lamivudine, darunavir, raltegravir, ritonavir, etravirine | B |
| 13 | HIVun | + | 43 | F | None | 282 | 376 | <50 | 24 | emtricitabine, tenofovir alafenamide, darunavir, cobicistat | C |
| 14 | HIVlow | + | 38 | M | None | 973 | 864 | <400 | 14 | etravirine, abacavir, zidovudine, lamivudine | B |
| 15 | HIVlow | + | 53 | M | Prob. HAD | 101 | 396 | <400 | 18 | lamivudine, nevirapine, zidovudine | C |
| 16 | HIVlow | + | 62 | F | None | 750 | 647 | 275 | 25 | darunavir, ritonavir, emtricitabine, tenofovir | C |
| 17 | HIVlow | + | 51 | M | Prob. MCMD | 126 | N.R. | 349 | 1 | lamivudine, abacavir, nelfinavir | B |
| 18 | HIVlow | + | 46 | M | Prob. MCMD | 214 | 1330 | 12,352 | 25 | darunavir, raltegravir, etravirine, emtricitabine, tenofovir | B |
| 19 | HIVlow | + | 61 | M | Prob. HAD | 500 | 1077 | <400 | 18 | atazanavir, emtricitabine, nevirapine, ritonavir | C |
| 20 | HIVhigh | + | 49 | M | Prob. MCMD | 3 | 71 | >750,000 | 21 | amprenavir, stavudine, lopinavir | C |
| 21 | HIVhigh | + | 44 | F | Prob. HAD | 1 | 139 | >750,000 | 9 | stavudine, didanosine, nevirapine, nelfinavir | C |
| 22 | HIVhigh | + | 42 | M | Prob. HAD | 37 | 1683 | 691,338 | 6 | didanosine, efavirenz, nelfinavir | B |
| 23 | HIVhigh | + | 37 | F | Prob. HAD | 1 | 1101 | >750,000 | 3 | zidovudine, didanosine, lopinavir, ritonavir | C |
| 24 | HIVhigh | + | 40 | F | Prob. HAD | 14 | N.R. | >750,000 | 4 | indinavir, zidovudine | C |
| 25 | HIVhigh | + | 34 | M | Prob. HAD | 10 | 545 | 165,862 | 6 | zidovudine, nelfinavir, indinavir, didanosine, lamivudine, abacavir, saquinavir-sgc | B |
| 26 | HIVhigh | + | 37 | M | Prob. MCMD | 91 | N.R. | >750,000 | 2 | efavirenz, lopinavir, ritonavir, nevirapine, tenofovir, abacavir, zidovudine, lamivudine | C |
| 27 | HIVE | + | 43 | M | HAD | 7 | N.R. | 119,000 | 6 | None | B |
| 28 | HIVE | + | 40 | F | HAD | 5 | N.R. | 750,000 | 8 | atazanavir | H |
| 29 | HIVE | + | 43 | M | HAD | 21 | N.R. | 172,000 | 5 | None | B |
| 30 | HIVE | + | 38 | M | HAD | 185 | N.R. | 46,284 | 3 | None | C |
| 31 | HIVE | + | 44 | F | HAD | 64 | N.R. | 750,000 | 5 | emtricitabine, tenofovir | B |
| 32 | Alz | - | 62 | F | Alz | - | - | - | - | - | C |
| 33 | Alz | - | 68 | M | Alz | - | - | - | - | - | C |
| 34 | Alz | - | 62 | M | Alz | - | - | - | - | - | C |
| % Positive for HIV DNA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| 104 | 0 | 1 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| 105 | 0 | 1 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| 109 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1010 | 0 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA + HIV-mRNA | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 104 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 105 | 2 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 109 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1010 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA + HIV-mRNA + p24 | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 0 | 0 | 0 | 0 | 0 | 0 | 2 | 0 | 0 | 0 |
| 104 | 0 | 1 | 0 | 0 | 0 | 0 | 6 | 3 | 0 | 0 | |
| 105 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 109 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1010 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 0 | 10 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 104 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 105 | 2 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 109 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| 1010 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA + HIV-mRNA | |||||||||||
| Dilution (number of HeLa cells) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| 103 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 104 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 105 | 3 | 2 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 109 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | |
| 1010 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA + HIV-mRNA + p24 | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| 104 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 105 | 1 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 106 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 107 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 108 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 109 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1010 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1011 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 1012 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| % Positive for HIV DNA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 88 | 80 | 78 | 100 | 83 | 100 | 100 | 100 | 100 | 100 |
| 104 | 89 | 94 | 93 | 100 | 83 | 100 | 100 | 98 | 100 | 95 | |
| 105 | 78 | 94 | 100 | 85 | 83 | 97 | 95 | 95 | 100 | 98 | |
| 106 | 83 | 94 | 100 | 100 | 77 | 97 | 98 | 100 | 100 | 100 | |
| 107 | 100 | 93 | 97 | 100 | 77 | 97 | 93 | 98 | 100 | 100 | |
| 108 | 100 | 85 | 100 | 90 | 70 | 95 | 83 | 98 | 100 | 100 | |
| 109 | 100 | 100 | 100 | 100 | 77 | 100 | 95 | 93 | 100 | 100 | |
| 1010 | 100 | 92 | 100 | 90 | 80 | 100 | 100 | 100 | 100 | 100 | |
| 1011 | 57 | 100 | 100 | 90 | 83 | 100 | 100 | 100 | 100 | 98 | |
| 1012 | 80 | 89 | 100 | 95 | 80 | 97 | 100 | 100 | 100 | 100 | |
| % Positive for HIV DNA + HIV-mRNA | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 100 | 70 | 84 | 100 | 83 | 100 | 100 | 100 | 100 | 100 |
| 104 | 100 | 100 | 89 | 100 | 80 | 99 | 99 | 98 | 100 | 95 | |
| 105 | 86 | 100 | 100 | 85 | 83 | 97 | 95 | 95 | 100 | 98 | |
| 106 | 100 | 100 | 100 | 100 | 75 | 96 | 98 | 100 | 100 | 99 | |
| 107 | 100 | 100 | 100 | 100 | 77 | 97 | 93 | 96 | 100 | 100 | |
| 108 | 100 | 100 | 100 | 90 | 69 | 95 | 83 | 96 | 100 | 95 | |
| 109 | 100 | 100 | 100 | 100 | 77 | 100 | 95 | 93 | 100 | 99 | |
| 1010 | 100 | 100 | 100 | 90 | 99 | 100 | 100 | 100 | 100 | 100 | |
| 1011 | 80 | 100 | 100 | 90 | 83 | 100 | 100 | 100 | 100 | 98 | |
| 1012 | 100 | 100 | 100 | 95 | 79 | 97 | 100 | 100 | 100 | 100 | |
| % Positive for HIV DNA + HIV-mRNA + p24 | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 88 | 69 | 85 | 100 | 83 | 100 | 100 | 100 | 100 | 99 |
| 104 | 100 | 100 | 88 | 100 | 83 | 100 | 99 | 98 | 100 | 96 | |
| 105 | 85 | 94 | 100 | 88 | 83 | 97 | 96 | 95 | 100 | 97 | |
| 106 | 100 | 94 | 100 | 100 | 75 | 97 | 98 | 100 | 100 | 100 | |
| 107 | 100 | 93 | 96 | 98 | 77 | 97 | 91 | 95 | 100 | 100 | |
| 108 | 100 | 85 | 100 | 90 | 69 | 95 | 81 | 98 | 100 | 96 | |
| 109 | 100 | 100 | 100 | 100 | 77 | 100 | 95 | 90 | 100 | 99 | |
| 1010 | 100 | 92 | 100 | 90 | 79 | 100 | 100 | 100 | 100 | 100 | |
| 1011 | 81 | 100 | 100 | 88 | 83 | 100 | 100 | 99 | 100 | 98 | |
| 1012 | 80 | 89 | 100 | 95 | 81 | 97 | 100 | 100 | 100 | 99 | |
| % Positive for HIV DNA | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 85 | 77 | 95 | 100 | 100 | 100 | 91 | 91 | 100 | 100 |
| 104 | 79 | 83 | 93 | 100 | 92 | 100 | 94 | 91 | 92 | 100 | |
| 105 | 100 | 81 | 93 | 100 | 88 | 100 | 86 | 94 | 89 | 95 | |
| 106 | 100 | 76 | 95 | 100 | 100 | 96 | 91 | 91 | 86 | 93 | |
| 107 | 100 | 84 | 100 | 100 | 100 | 90 | 93 | 100 | 92 | 90 | |
| 108 | 100 | 83 | 93 | 85 | 80 | 93 | 89 | 100 | 92 | 98 | |
| 109 | 100 | 83 | 87 | 90 | 84 | 100 | 86 | 88 | 89 | 100 | |
| 1010 | 100 | 81 | 88 | 95 | 88 | 87 | 75 | 75 | 95 | 100 | |
| 1011 | 100 | 83 | 93 | 89 | 100 | 90 | 87 | 87 | 89 | 100 | |
| 1012 | 100 | 83 | 89 | 81 | 84 | 87 | 87 | 87 | 100 | 100 | |
| % Positive for HIV DNA + HIV-mRNA | |||||||||||
| Dilution (number of HeLa cells) | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | |
| 103 | 84 | 76 | 91 | 100 | 100 | 100 | 90 | 92 | 100 | 100 | |
| 104 | 80 | 83 | 93 | 100 | 93 | 100 | 94 | 89 | 92 | 100 | |
| 105 | 100 | 81 | 94 | 100 | 87 | 100 | 88 | 95 | 89 | 93 | |
| 106 | 100 | 76 | 95 | 100 | 100 | 96 | 89 | 89 | 86 | 93 | |
| 107 | 100 | 84 | 100 | 100 | 92 | 91 | 94 | 100 | 92 | 91 | |
| 108 | 100 | 83 | 94 | 85 | 100 | 94 | 89 | 100 | 91 | 98 | |
| 109 | 100 | 83 | 100 | 89 | 84 | 100 | 86 | 89 | 89 | 100 | |
| 1010 | 100 | 79 | 88 | 95 | 85 | 97 | 77 | 77 | 94 | 100 | |
| 1011 | 100 | 83 | 93 | 88 | 100 | 93 | 93 | 93 | 88 | 99 | |
| 1012 | 100 | 83 | 88 | 87 | 83 | 100 | 100 | 100 | 100 | 100 | |
| % Positive for HIV DNA + HIV-mRNA + p24 | |||||||||||
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Dilution (number of HeLa cells) | 103 | 100 | 76 | 90 | 100 | 100 | 100 | 91 | 90 | 100 | 100 |
| 104 | 81 | 83 | 93 | 100 | 91 | 100 | 94 | 90 | 92 | 100 | |
| 105 | 86 | 80 | 91 | 100 | 88 | 100 | 86 | 89 | 90 | 92 | |
| 106 | 100 | 73 | 96 | 100 | 100 | 94 | 90 | 90 | 91 | 91 | |
| 107 | 100 | 83 | 100 | 100 | 90 | 92 | 93 | 100 | 92 | 88 | |
| 108 | 100 | 82 | 92 | 98 | 100 | 93 | 89 | 100 | 90 | 89 | |
| 109 | 100 | 83 | 100 | 100 | 82 | 100 | 86 | 88 | 90 | 100 | |
| 1010 | 100 | 80 | 89 | 100 | 88 | 97 | 75 | 75 | 95 | 100 | |
| 1011 | 83 | 83 | 94 | 100 | 100 | 93 | 87 | 94 | 90 | 99 | |
| 1012 | 67 | 82 | 88 | 100 | 84 | 100 | 100 | 100 | 100 | 100 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Donoso, M.; D’Amico, D.; Valdebenito, S.; Hernandez, C.A.; Prideaux, B.; Eugenin, E.A. Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain. Cells 2022, 11, 2379. https://doi.org/10.3390/cells11152379
Donoso M, D’Amico D, Valdebenito S, Hernandez CA, Prideaux B, Eugenin EA. Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain. Cells. 2022; 11(15):2379. https://doi.org/10.3390/cells11152379
Chicago/Turabian StyleDonoso, Maribel, Daniela D’Amico, Silvana Valdebenito, Cristian A. Hernandez, Brendan Prideaux, and Eliseo A. Eugenin. 2022. "Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain" Cells 11, no. 15: 2379. https://doi.org/10.3390/cells11152379
APA StyleDonoso, M., D’Amico, D., Valdebenito, S., Hernandez, C. A., Prideaux, B., & Eugenin, E. A. (2022). Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain. Cells, 11(15), 2379. https://doi.org/10.3390/cells11152379