
Therapeutic Targeting Notch2 Protects Bone Micro-Vasculatures from Methotrexate Chemotherapy-Induced Adverse Effects in Rats




Abstract
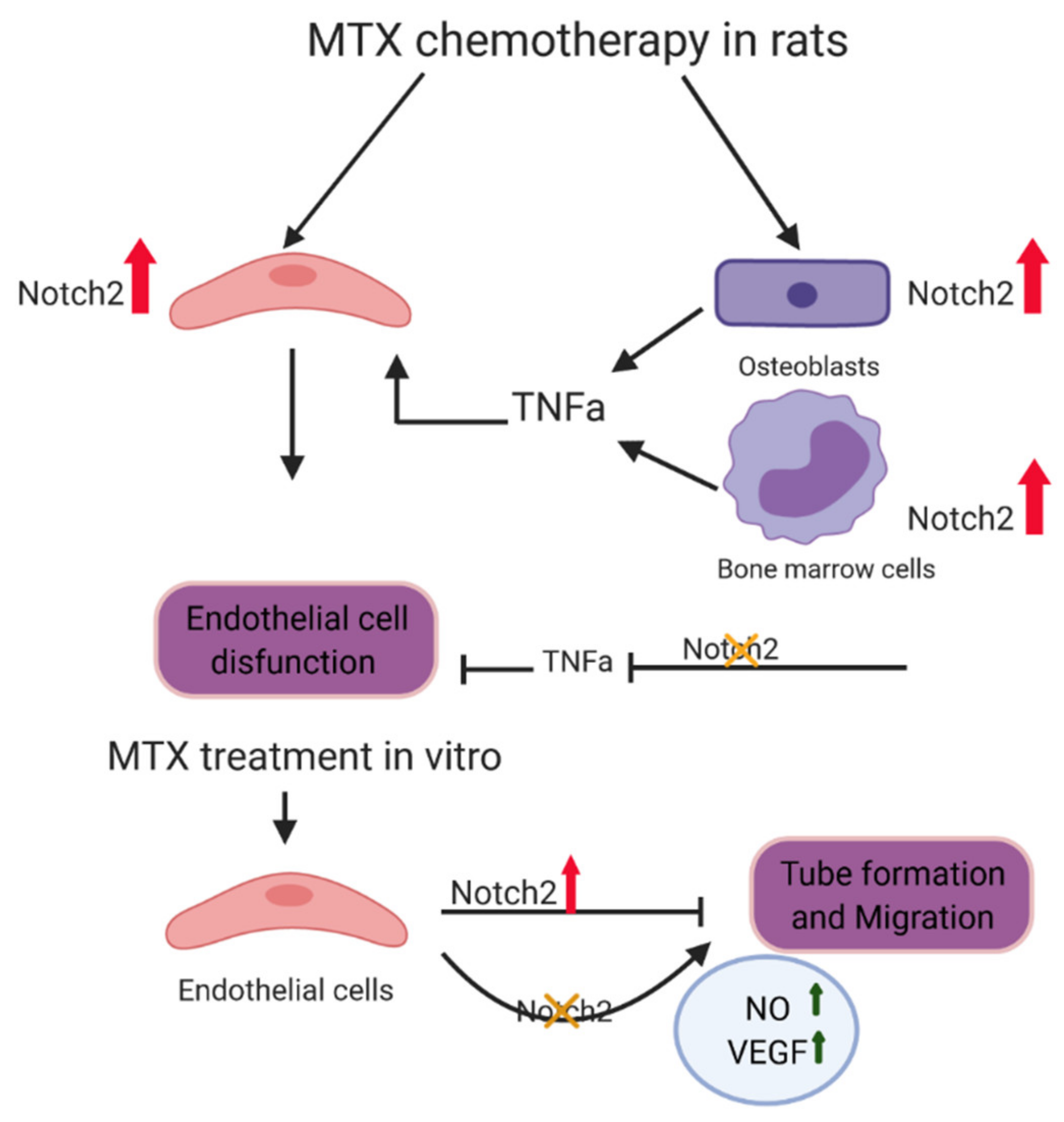
:1. Introduction
2. Materials and Methods
2.1. Rat MTX Chemotherapy Time-Course Study
2.2. MTX and Anti-Notch2 Antibody Treatment Animal Trial
2.3. Histological Analyses of Bone Marrow Sinusoids
2.4. Immunohistochemical Analyses
2.5. Micro-CT Quantification of Vasculature and Lacunar Porosities in Metaphyseal Cortical Bone
2.6. Cell Culture Study
2.7. Matrigel Tube Formation Assay
2.8. Trans-Well Migration Assay
2.9. Measurement of Nitric Oxide
2.10. Measurement of Pro-Inflammatory Cytokine TNFα in Rat Serum
2.11. RNA Isolation and Gene Expression Analyses
2.12. Statistical Analyses
3. Results
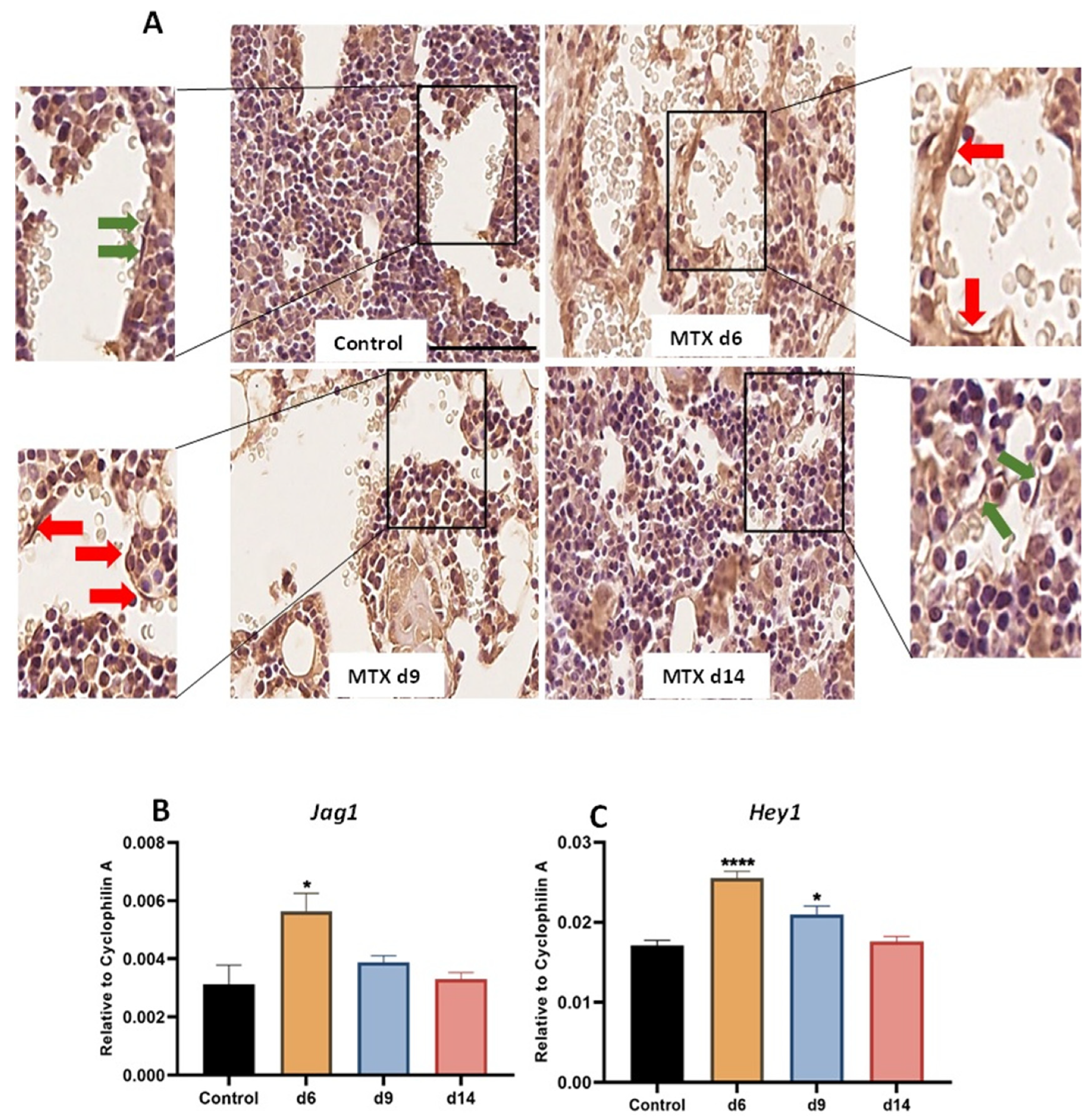
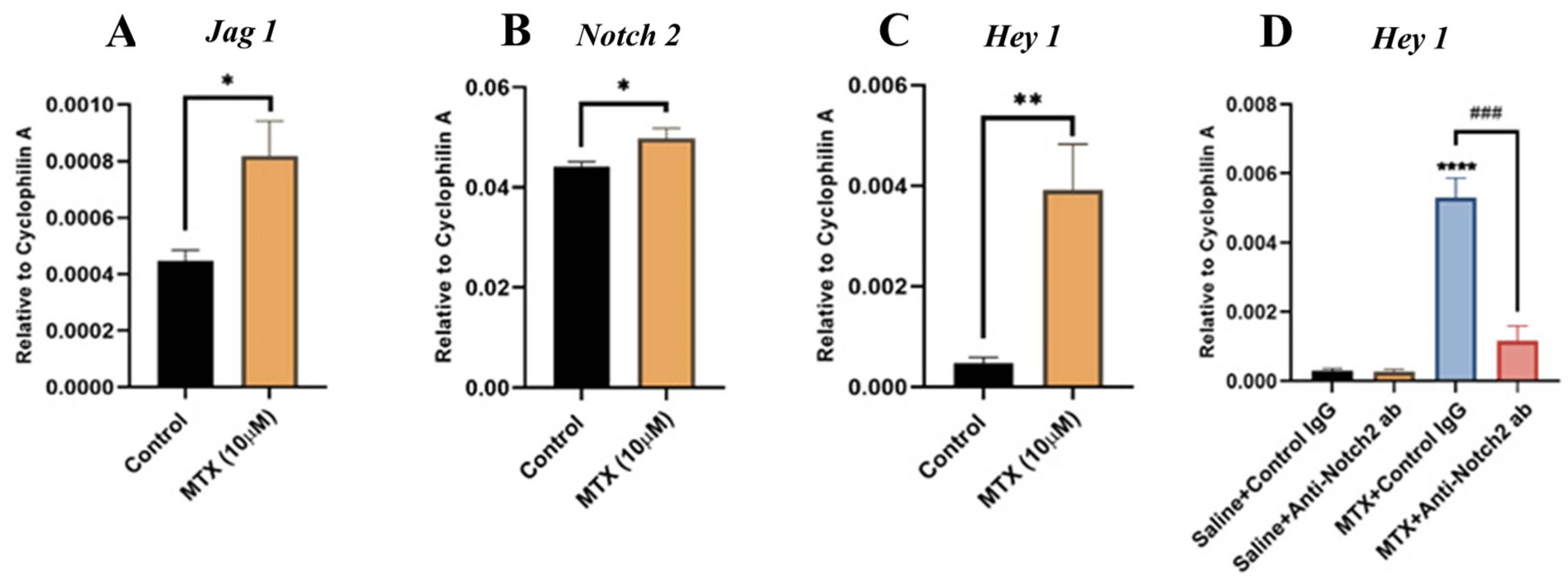
3.1. MTX Damaging Effect on Bone Marrow Vasculature Is Associated with Upregulation of NICD2, Notch Ligand Jag1 and Notch Target Gene Hey1 in Bone
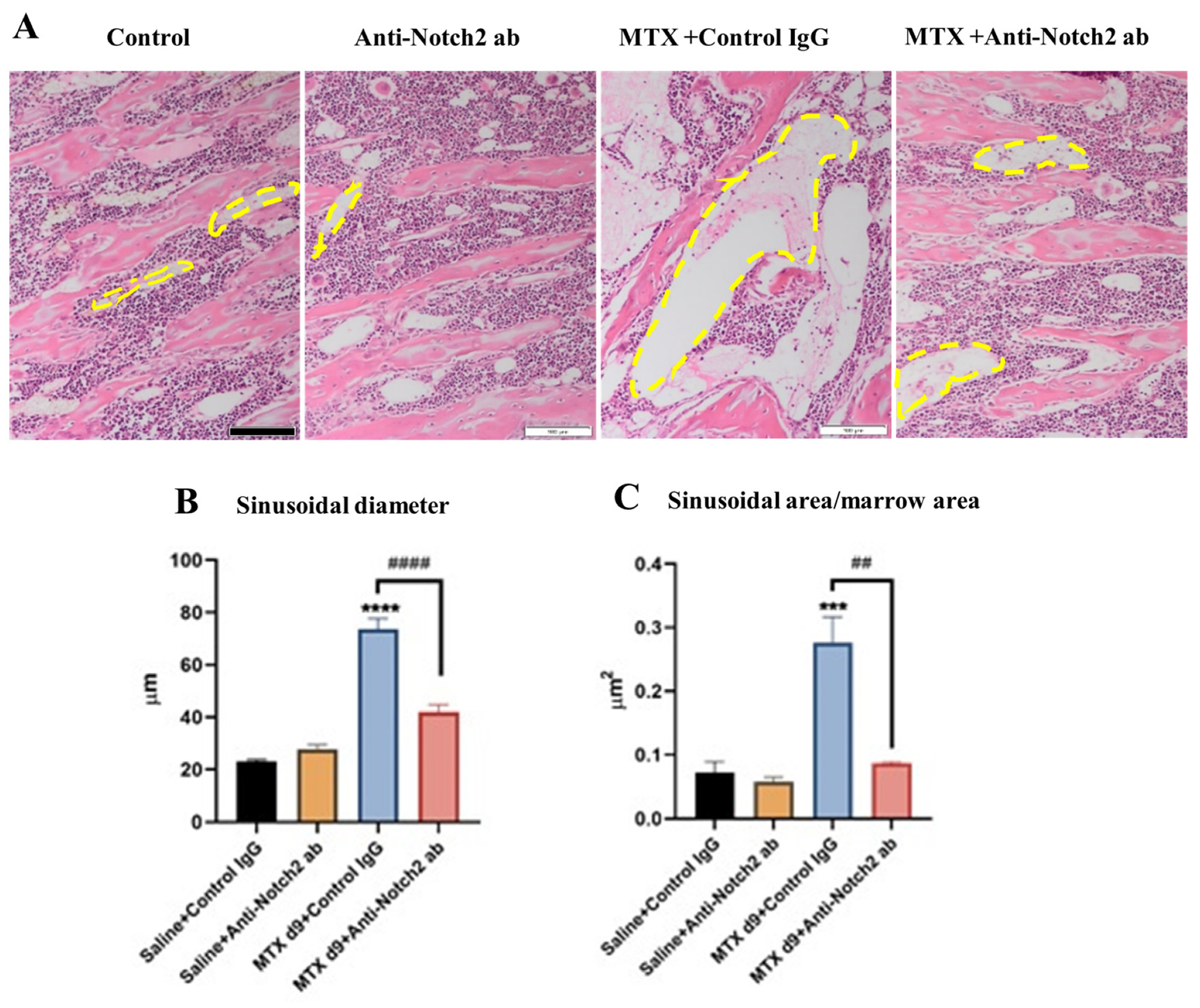
3.2. Anti-Notch2 Antibody Treatment Attenuated MTX-Induced Bone Marrow Sinusoidal Dilation
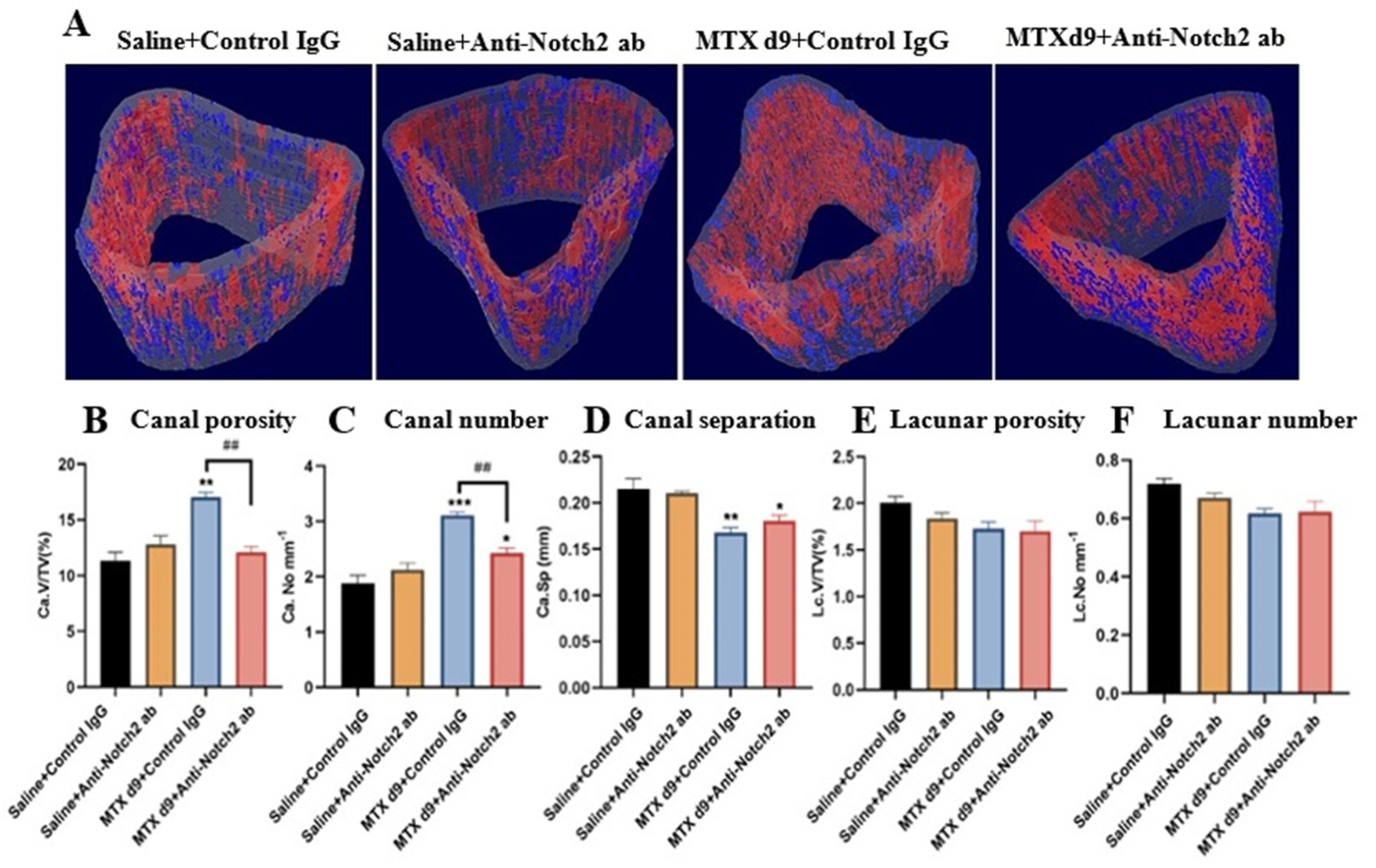
3.3. Micro-CT Assessments of Treatment Effects on Vasculature and Osteocyte Lacuna in Cortical Bone
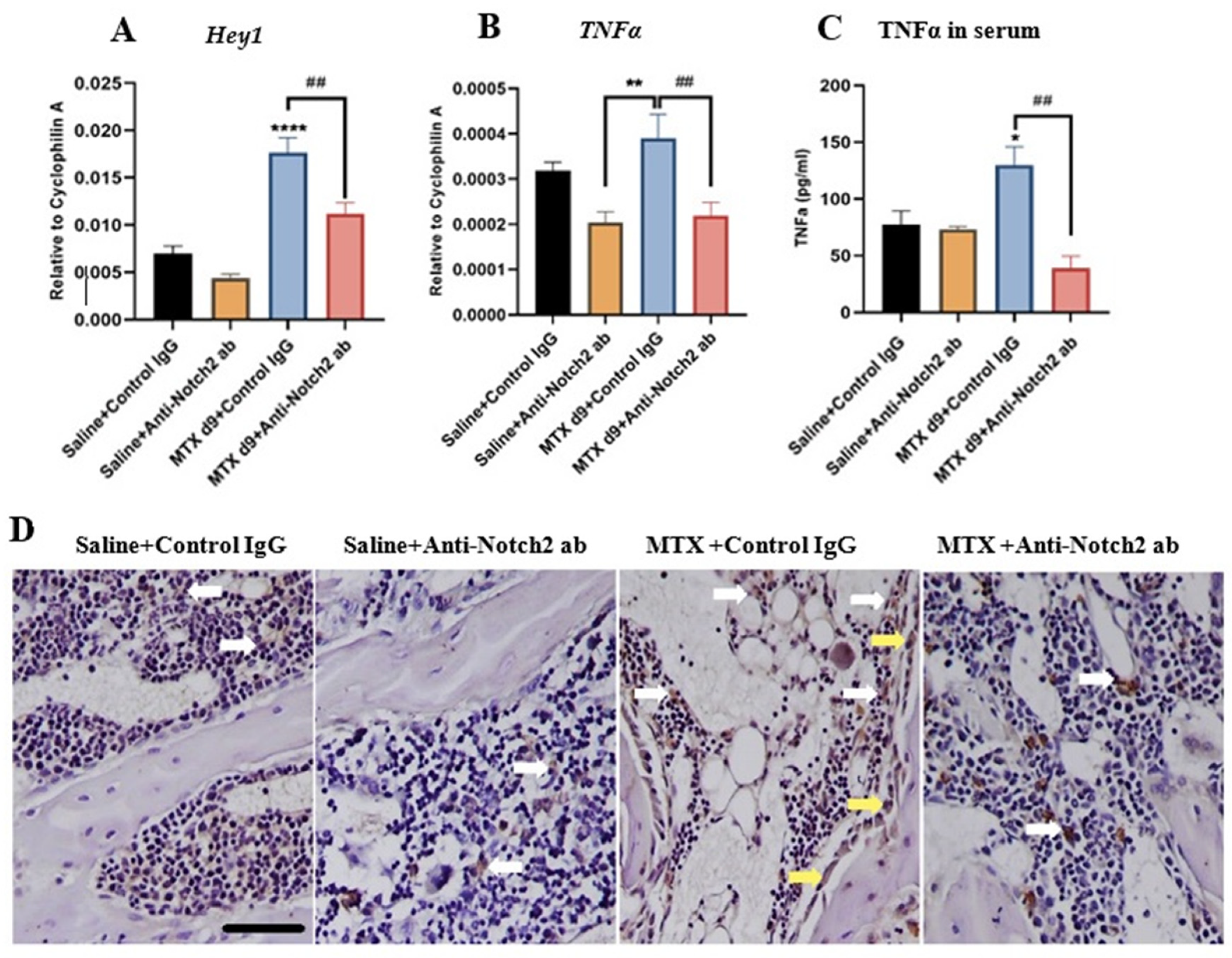
3.4. Notch2 Blockade Alters mRNA Expression of Notch Target Gene Hey1 and Attenuates MTX-Induced Increases in Inflammatory Cytokine TNFα Levels
3.5. MTX Treatment Alters Notch Signalling in Cultured Rat BMECs and Blocking Notch2 Attenuates MTX-Induced Notch Target Gene Hey1 Overexpression
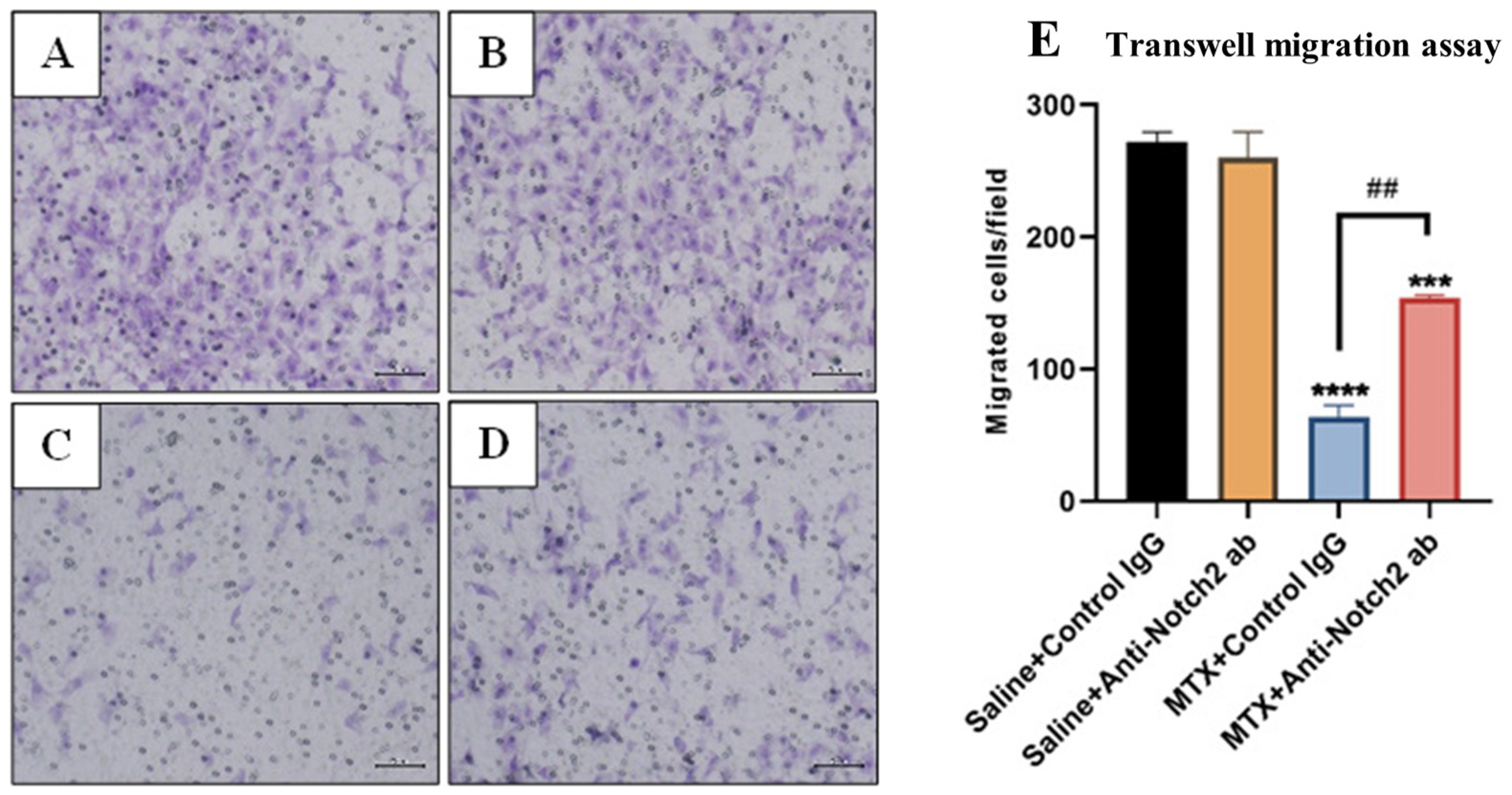
3.6. Treatment Effects on Migration Ability of Rat BMECs
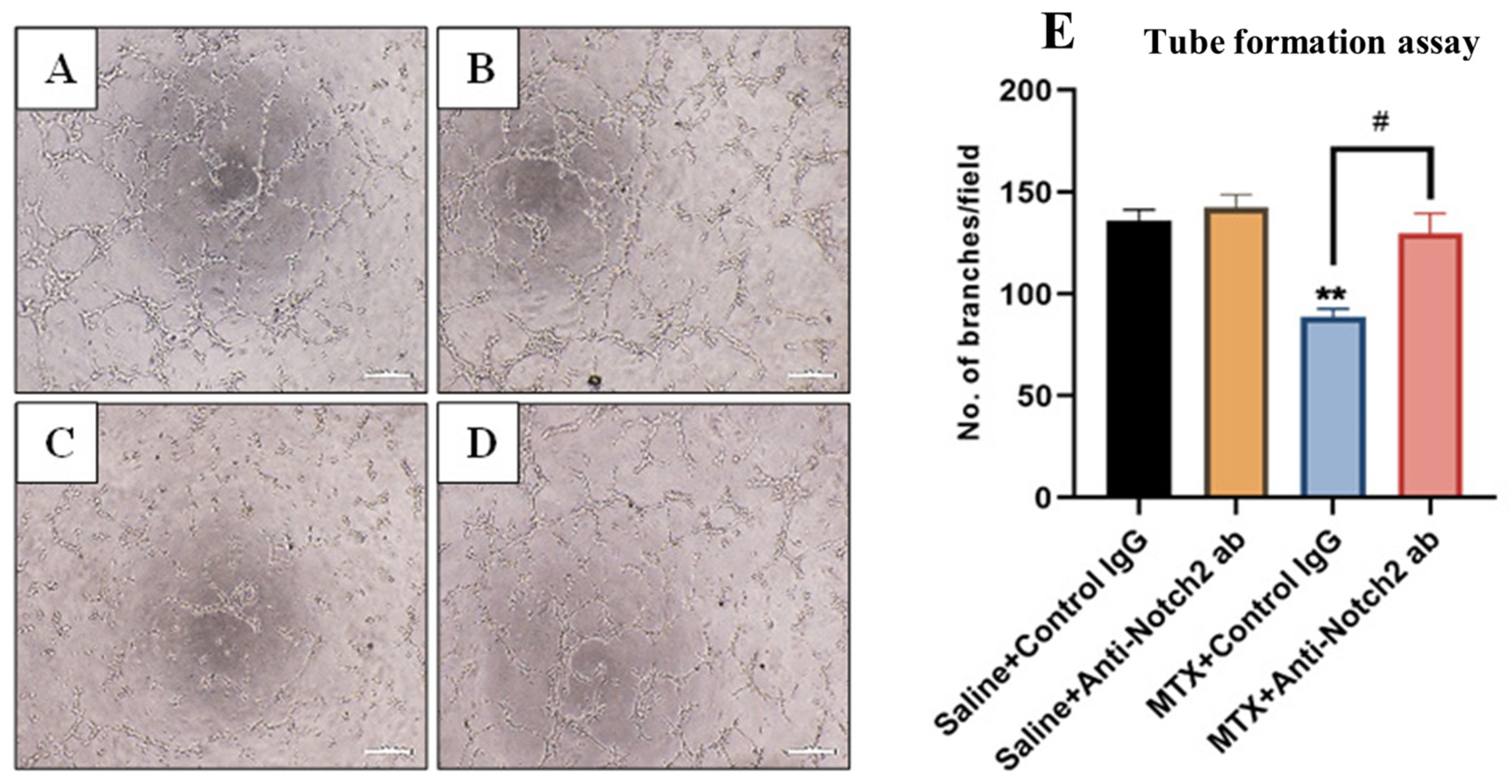
3.7. Treatment Effects on Endothelial Cell Tube Formation Ability
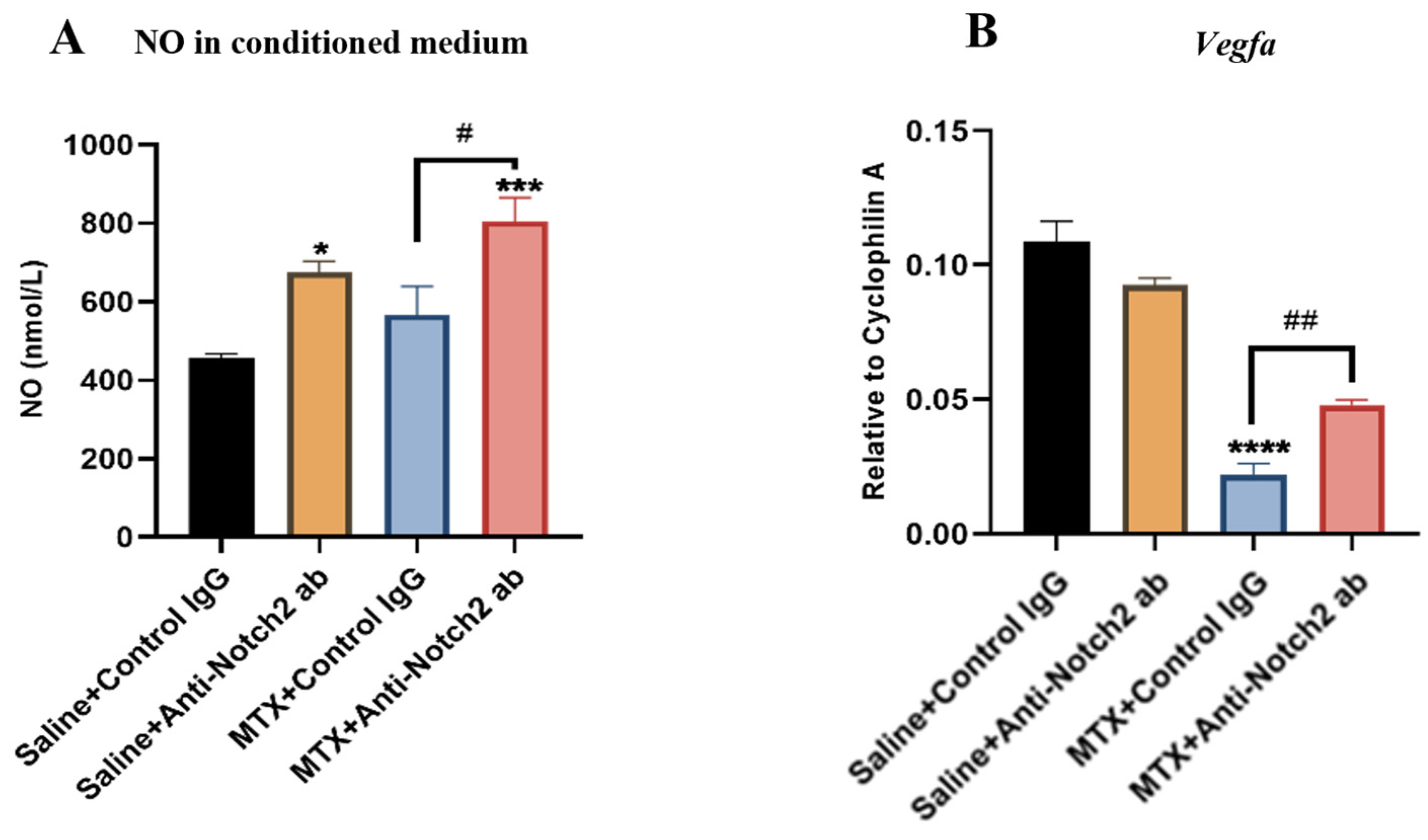
3.8. Notch2 Blockade Protecting Endothelial Cell Functionality against MTX Damage Is Accompanied by Induction of NO and VEGF
4. Discussion
4.1. Notch Signalling Alteration Is Associated with MTX-Induced Bone Micro-Vasculature Damage and Notch2 Blockade Ameliorates MTX-Induced Vasculature Damage in the Bone Marrow and Cortical Bone
4.2. Notch2 Blockade Alleviates MTX-Induced Increased Levels of Inflammatory Cytokine TNFα in Bone and Serum: A Possible Indirect Mechanism for Protecting Micro-Vasculature
4.3. MTX Treatment Alters Notch Signalling in Cultured Rat BMECs and Notch2 Blockade Mitigates MTX-Induced Damaging Effects on Endothelial Cell Functionality
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Georgiou, K.R.; King, T.J.; Scherer, M.A.; Zhou, H.; Foster, B.K.; Xian, C.J. Attenuated Wnt/beta-catenin signalling mediates methotrexate chemotherapy-induced bone loss and marrow adiposity in rats. Bone 2012, 50, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Crews, K.R.; Liu, T.; Rodriguez-Galindo, C.; Tan, M.; Meyer, W.H.; Panetta, J.C.; Link, M.P.; Daw, N.C. High-dose methotrexate pharmacokinetics and outcome of children and young adults with osteosarcoma. Cancer Interdiscip. Int. J. Am. Cancer Soc. 2004, 100, 1724–1733. [Google Scholar] [CrossRef] [PubMed]
- Papaconstantinou, H.T.; Xie, C.; Zhang, W.; Ansari, N.H.; Hellmich, M.R.; Townsend, C.M., Jr.; Ko, T.C. The role of caspases in methotrexate-induced gastrointestinal toxicity. Surgery 2001, 130, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Haddy, T.B.; Mosher, R.B.; Reaman, G.H. Osteoporosis in survivors of acute lymphoblastic leukemia. Oncologist 2001, 6, 278–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunstreich, M.; Kummer, S.; Laws, H.J.; Borkhardt, A.; Kuhlen, M. Osteonecrosis in children with acute lymphoblastic leukemia. Haematologica 2016, 101, 1295–1305. [Google Scholar] [CrossRef]
- Brody, J.P.; Krause, J.R.; Penchansky, L. Bone marrow response to chemotherapy in acute lymphocytic leukaemia and acute non-lymphocytic leukaemia. Scand. J. Haematol. 1985, 35, 240–245. [Google Scholar] [CrossRef]
- Kopp, H.G.; Avecilla, S.T.; Hooper, A.T.; Shmelkov, S.V.; Ramos, C.A.; Zhang, F.; Rafii, S. Tie2 activation contributes to hemangiogenic regeneration after myelosuppression. Blood 2005, 106, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Narayan, K.; Juneja, S.; Garcia, C. Effects of 5-fluorouracil or total-body irradiation on murine bone marrow microvasculature. Exp. Hematol. 1994, 22, 142–148. [Google Scholar]
- Chim, S.M.; Tickner, J.; Chow, S.T.; Kuek, V.; Guo, B.; Zhang, G.; Rosen, V.; Erber, W.; Xu, J. Angiogenic factors in bone local environment. Cytokine Growth Factor Rev. 2013, 24, 297–310. [Google Scholar] [CrossRef]
- Pazzaglia, U.E.; Congiu, T.; Raspanti, M.; Ranchetti, F.; Quacci, D. Anatomy of the intracortical canal system: Scanning electron microscopy study in rabbit femur. Clin. Orthop. Relat. Res. 2009, 467, 2446–2456. [Google Scholar] [CrossRef] [Green Version]
- Grüneboom, A.; Hawwari, I.; Weidner, D.; Culemann, S.; Müller, S.; Henneberg, S.; Brenzel, A.; Merz, S.; Bornemann, L.; Zec, K.; et al. A network of trans-cortical capillaries as mainstay for blood circulation in long bones. Nat. Metab. 2019, 1, 236–250. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Larriera, A.I.; Palacio-Mancheno, P.E.; Gatti, V.; Fritton, J.C.; Bromage, T.G.; Cardoso, L.; Doty, S.B.; Fritton, S.P. The effects of estrogen deficiency on cortical bone microporosity and mineralization. Bone 2018, 110, 1–10. [Google Scholar] [CrossRef]
- Zebaze, R.M.; Ghasem-Zadeh, A.; Bohte, A.; Iuliano-Burns, S.; Mirams, M.; Price, R.I.; Mackie, E.J.; Seeman, E. Intracortical remodelling and porosity in the distal radius and post-mortem femurs of women: A cross-sectional study. Lancet 2010, 375, 1729–1736. [Google Scholar] [CrossRef]
- Kusumbe, A.P.; Ramasamy, S.K.; Adams, R.H. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 2014, 507, 323. [Google Scholar] [CrossRef] [PubMed]
- Tavassoli, M. Structure and function of sinusoidal endothelium of bone marrow. Prog. Clin. Biol. Res. 1981, 59, 249–256. [Google Scholar]
- Kopp, H.-G.; Avecilla, S.T.; Hooper, A.T.; Rafii, S. The Bone Marrow Vascular Niche: Home of HSC Differentiation and Mobilization. Physiology 2005, 20, 349–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassanshahi, M.; Su, Y.-W.; Fan, C.-M.; Khabbazi, S.; Hassanshahi, A.; Xian, C.J. Methotrexate chemotherapy–induced damages in bone marrow sinusoids: An in vivo and in vitro study. J. Cell. Biochem. 2019, 120, 3220–3231. [Google Scholar] [CrossRef]
- Hofmann, J.J.; Iruela-Arispe, M.L. Notch signaling in blood vessels: Who is talking to whom about what? Circ. Res. 2007, 100, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Iso, T.; Hamamori, Y.; Kedes, L. Notch signaling in vascular development. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 543–553. [Google Scholar] [CrossRef] [Green Version]
- Zanotti, S.; Canalis, E. Notch and its ligands. In Principles of Bone Biology; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1083–1112. [Google Scholar]
- Quillard, T.; Devalliere, J.; Chatelais, M.; Coulon, F.; Séveno, C.; Romagnoli, M.; Barillé Nion, S.; Charreau, B. Notch2 Signaling Sensitizes Endothelial Cells to Apoptosis by Negatively Regulating the Key Protective Molecule Survivin. PLoS ONE 2009, 4, e8244. [Google Scholar] [CrossRef] [Green Version]
- Benedito, R.; Rocha, S.F.; Woeste, M.; Zamykal, M.; Radtke, F.; Casanovas, O.; Duarte, A.; Pytowski, B.; Adams, R.H. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF–VEGFR2 signalling. Nature 2012, 484, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Tian, D.Y.; Jin, X.R.; Zeng, X.; Wang, Y. Notch Signaling in Endothelial Cells: Is It the Therapeutic Target for Vascular Neointimal Hyperplasia? Int. J. Mol. Sci. 2017, 18, 1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, A.C.Y.; Patenaude, A.; Lu, K.; Fuller, M.; Ly, M.; Kyle, A.; Golbidi, S.; Wang, Y.; Walley, K.; Minchinton, A.; et al. Notch-Dependent Regulation of the Ischemic Vasodilatory Response—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 510–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, X.; Xie, Y.; Xian, C.J.; Chen, L. Role of FGFs/FGFRs in skeletal development and bone regeneration. J. Cell Physiol. 2012, 227, 3731–3743. [Google Scholar] [CrossRef] [PubMed]
- Dufraine, J.; Funahashi, Y.; Kitajewski, J. Notch signaling regulates tumor angiogenesis by diverse mechanisms. Oncogene 2008, 27, 5132–5137. [Google Scholar] [CrossRef] [Green Version]
- Quillard, T.; Devallière, J.; Coupel, S.; Charreau, B. Inflammation dysregulates Notch signaling in endothelial cells: Implication of Notch2 and Notch4 to endothelial dysfunction. Biochem. Pharmacol. 2010, 80, 2032–2041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulday, G.N.; Coupel, S.P.; Coulon, F.; Soulillou, J.-P.; Charreau, B.A. Antigraft antibody-mediated expression of metalloproteinases on endothelial cells: Differential expression of TIMP-1 and ADAM-10 depends on antibody specificity and isotype. Circ. Res. 2001, 88, 430–437. [Google Scholar] [CrossRef] [Green Version]
- Six, E.; Ndiaye, D.; Laâbi, Y.; Brou, C.; Gupta-Rossi, N.; Israël, A.; Logeat, F. The Notch ligand Delta1 is sequentially cleaved by an ADAM protease and γ-secretase. Proc. Natl. Acad. Sci. USA 2003, 100, 7638–7643. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, L.l.C.; Edelmann, R.J.; Krüger, S.; Diéguez-Hurtado, R.; Shah, A.; Stav-Noraas, T.E.; Renzi, A.; Szymanska, M.; Wang, J.; Ehling, M.; et al. Inhibition of Endothelial NOTCH1 Signaling Attenuates Inflammation by Reducing Cytokine-Mediated Histone Acetylation at Inflammatory Enhancers. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 854–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, D.; Aikawa, E.; Swirski, F.K.; Novobrantseva, T.I.; Kotelianski, V.; Gorgun, C.Z.; Chudnovskiy, A.; Yamazaki, H.; Croce, K.; Weissleder, R.; et al. Notch ligand delta-like 4 blockade attenuates atherosclerosis and metabolic disorders. Proc. Natl. Acad. Sci. USA 2012, 109, E1868–E1877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.S.; Kim, S.H.; Kim, K.; Jin, C.H.; Choi, K.Y.; Jang, J.; Choi, Y.; Gwon, A.R.; Baik, S.H.; Yun, U.J.; et al. Inhibition of notch signalling ameliorates experimental inflammatory arthritis. Ann. Rheum. Dis. 2015, 74, 267–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xian, C.J.; Cool, J.C.; Scherer, M.A.; Macsai, C.E.; Fan, C.; Covino, M.; Foster, B.K. Cellular mechanisms for methotrexate chemotherapy-induced bone growth defects. Bone 2007, 41, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, G.E.; Tross, R.B.; Doganis, A.C.; Kirkwood, J.M.; Baron, R. Effects of chemotherapeutic agents on bone. I. Short-term methotrexate and doxorubicin (adriamycin) treatment in a rat model. J. Bone Jt. Surg. Am. 1984, 66, 602–607. [Google Scholar] [CrossRef]
- Barker, H.E.; Cox, T.R.; Erler, J.T. The rationale for targeting the LOX family in cancer. Nat. Rev. Cancer 2012, 12, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Cain-Hom, C.; Choy, L.; Hagenbeek, T.J.; de Leon, G.P.; Chen, Y.; Finkle, D.; Venook, R.; Wu, X.; Ridgway, J.; et al. Therapeutic antibody targeting of individual Notch receptors. Nature 2010, 464, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Canalis, E.; Sanjay, A.; Yu, J.; Zanotti, S. An Antibody to Notch2 Reverses the Osteopenic Phenotype of Hajdu-Cheney Mutant Male Mice. Endocrinology 2017, 158, 730–742. [Google Scholar] [CrossRef]
- Lafkas, D.; Shelton, A.; Chiu, C.; de Leon Boenig, G.; Chen, Y.; Stawicki, S.S.; Siltanen, C.; Reichelt, M.; Zhou, M.; Wu, X.; et al. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 2015, 528, 127–131. [Google Scholar] [CrossRef]
- Georgiou, K.R.; Scherer, M.A.; Fan, C.-M.; Cool, J.C.; King, T.J.; Foster, B.K.; Xian, C.J. Methotrexate chemotherapy reduces osteogenesis but increases adipogenic potential in the bone marrow. J. Cell. Physiol. 2012, 227, 909–918. [Google Scholar] [CrossRef]
- Hassanshahi, M.; Su, Y.-W.; Khabbazi, S.; Fan, C.-M.; Chen, K.-M.; Wang, J.-F.; Qian, A.; Howe, P.R.; Yan, D.-W.; Zhou, H.-D.; et al. Flavonoid genistein protects bone marrow sinusoidal blood vessels from damage by methotrexate therapy in rats. J. Cell. Physiol. 2019, 234, 11276–11286. [Google Scholar] [CrossRef]
- Zhou, F.H.; Foster, B.K.; Sander, G.; Xian, C.J. Expression of proinflammatory cytokines and growth factors at the injured growth plate cartilage in young rats. Bone 2004, 35, 1307–1315. [Google Scholar] [CrossRef]
- Palacio-Mancheno, P.E.; Larriera, A.I.; Doty, S.B.; Cardoso, L.; Fritton, S.P. 3D assessment of cortical bone porosity and tissue mineral density using high-resolution µCT: Effects of resolution and threshold method. J. Bone Miner. Res. 2014, 29, 142–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Law, H.K.; Lau, Y.L.; Chan, G.C. Differential damage and recovery of human mesenchymal stem cells after expo-sure to chemotherapeutic agents. Br. J. Haematol. 2004, 127, 326–334. [Google Scholar] [CrossRef]
- Peymanfar, Y.; Su, Y.-W.; Hassanshahi, M.; Xian, C.J. Methotrexate treatment suppresses osteoblastic differentiation by inducing Notch2 signaling and blockade of Notch2 rescues osteogenesis by preserving Wnt/β-catenin signaling. J. Orthop. Res. 2021. Online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Falk, R.; Falk, A.; Dyson, M.R.; Melidoni, A.N.; Parthiban, K.; Young, J.L.; Roake, W.; McCafferty, J. Generation of anti-Notch antibodies and their application in blocking Notch signalling in neural stem cells. Methods 2012, 58, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-W.; Chen, Y.-F.; Wong, J.-M.; Weng, C.-W.; Chen, H.-Y.; Yu, S.-L.; Chen, H.-W.; Yuan, A.; Chen, J.J.W. Cancer cells increase endothelial cell tube formation and survival by activating the PI3K/Akt signalling pathway. J. Exp. Clin. Cancer Res. 2017, 36, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhai, Y.-K.; Guo, X.-Y.; Ge, B.-F.; Zhen, P.; Ma, X.-N.; Zhou, J.; Ma, H.-P.; Xian, C.J.; Chen, K.-M. Icariin stimulates the osteogenic differentiation of rat bone marrow stromal cells via activating the PI3K–AKT–eNOS–NO–cGMP–PKG. Bone 2014, 66, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Xian, C.J.; Cool, J.C.; Scherer, M.A.; Fan, C.; Foster, B.K. Folinic acid attenuates methotrexate chemotherapy-induced damages on bone growth mechanisms and pools of bone marrow stromal cells. J. Cell Physiol. 2008, 214, 777–785. [Google Scholar] [CrossRef]
- Fan, C.-M.; Foster, B.K.; Hui, S.K.; Xian, C.J. Prevention of Bone Growth Defects, Increased Bone Resorption and Marrow Adiposity with Folinic Acid in Rats Receiving Long-Term Methotrexate. PLoS ONE 2012, 7, e46915. [Google Scholar] [CrossRef] [Green Version]
- Peymanfar, Y.; Su, Y.W.; Xian, C.J. Notch2 Blockade Mitigates Methotrexate Chemotherapy-Induced Bone Loss and Marrow Adiposity. Cells 2022, 11, 1521. [Google Scholar] [CrossRef]
- Cooper, D.M.; Turinsky, A.L.; Sensen, C.W.; Hallgrímsson, B. Quantitative 3D analysis of the canal network in cortical bone by micro-computed tomography. Anat. Rec. B New Anat. 2003, 274, 169–179. [Google Scholar] [CrossRef]
- Lafage-Proust, M.H.; Roche, B.; Langer, M.; Cleret, D.; Vanden Bossche, A.; Olivier, T.; Vico, L. Assessment of bone vascularization and its role in bone remodeling. Bonekey Rep. 2015, 4, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharpfenecker, M.; Kruse, J.J.; Sprong, D.; Russell, N.S.; Ten Dijke, P.; Stewart, F.A. Ionizing radiation shifts the PAI-1/ID-1 balance and activates notch signaling in endothelial cells. Int. J. Radiat. Oncol. Biol. Phys. 2009, 73, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Schumacher, N.; Maier, M.; Sendtner, M.; Gessler, M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004, 18, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa, L.; Cathelin, S.; D’Altri, T.; Trimarchi, T.; Statnikov, A.; Guiu, J.; Rodilla, V.; Inglés-Esteve, J.; Nomdedeu, J.; Bellosillo, B.; et al. The Notch/Hes1 pathway sustains NF-κB activation through CYLD repression in T cell leukemia. Cancer Cell 2010, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, I.; Fang, T.; Pear, W.S. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu. Rev. Immunol. 2005, 23, 945–974. [Google Scholar] [CrossRef]
- Yin, J.; Huang, F.; Yi, Y.; Yin, L.; Peng, D. EGCG attenuates atherosclerosis through the Jagged-1/Notch pathway. Int. J. Mol. Med. 2016, 37, 398–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacLauchlan, S.; Yu, J.; Parrish, M.; Asoulin, T.A.; Schleicher, M.; Krady, M.M.; Zeng, J.; Huang, P.L.; Sessa, W.C.; Kyriakides, T.R. Endothelial nitric oxide synthase controls the expression of the angiogenesis inhibitor thrombospondin 2. Proc. Natl. Acad. Sci. USA 2011, 108, E1137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, T.W.; Isenberg, J.S.; Roberts, D.D. Molecular regulation of tumor angiogenesis and perfusion via redox signaling. Chem. Rev. 2009, 109, 3099–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Chen, T.T.; Barber, C.L.; Jordan, M.C.; Murdock, J.; Desai, S.; Ferrara, N.; Nagy, A.; Roos, K.P.; Iruela-Arispe, M.L. Autocrine VEGF signaling is required for vascular homeostasis. Cell 2007, 130, 691–703. [Google Scholar] [CrossRef] [Green Version]
- Zachary, I.; Gliki, G. Signaling transduction mechanisms mediating biological actions of the vascular endothelial growth factor family. Cardiovasc. Res. 2001, 49, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Cheng, G.; Zhou, Y.; Xu, G. Thymosin beta4 induces angiogenesis through Notch signaling in endothelial cells. Mol. Cell. Biochem. 2013, 381, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karsan, A. The role of notch in modeling and maintaining the vasculature. Can. J. Physiol. Pharmacol. 2005, 83, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Goulet, G.C.; Hamilton, N.; Cooper, D.; Coombe, D.; Tran, D.; Martinuzzi, R.; Zernicke, R.F. Influence of vascular porosity on fluid flow and nutrient transport in loaded cortical bone. J. Biomech. 2008, 41, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, S.K.; Kusumbe, A.P.; Schiller, M.; Zeuschner, D.; Bixel, M.G.; Milia, C.; Gamrekelashvili, J.; Limbourg, A.; Medvinsky, A.; Santoro, M.M.; et al. Blood flow controls bone vascular function and osteogenesis. Nat. Commun. 2016, 7, 13601. [Google Scholar] [CrossRef] [Green Version]
- Lindner, H.; Holler, E.; Ertl, B.; Multhoff, G.; Schreglmann, M.; Klauke, I.; Schultz-Hector, S.; Eissner, G.N. Peripheral Blood Mononuclear Cells Induce Programmed Cell Death in Human Endothelial Cells and May Prevent Repair: Role of Cytokines. Blood 1997, 89, 1931–1938. [Google Scholar] [CrossRef]
- Eissner, G.; Kohlhuber, F.; Grell, M.; Ueffing, M.; Scheurich, P.; Hieke, A.; Multhoff, G.; Bornkamm, G.W.; Holler, E. Critical involvement of transmembrane tumor necrosis factor-alpha in endothelial programmed cell death mediated by ionizing radiation and bacterial endotoxin. Blood 1995, 86, 4184–4193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.J.; Georgiou, K.R.; Cool, J.C.; Scherer, M.A.; Ang, E.S.; Foster, B.K.; Xu, J.; Xian, C.J. Methotrexate chemotherapy promotes osteoclast formation in the long bone of rats via increased pro-inflammatory cytokines and enhanced NF-κB activation. Am. J. Pathol. 2012, 181, 121–129. [Google Scholar] [CrossRef]
- Sievert, W.; Trott, K.-R.; Azimzadeh, O.; Tapio, S.; Zitzelsberger, H.; Multhoff, G. Late proliferating and inflammatory effects on murine microvascular heart and lung endothelial cells after irradiation. Radiother. Oncol. 2015, 117, 376–381. [Google Scholar] [CrossRef]
- Schultz-Hector, S.; Trott, K.-R. Radiation-induced cardiovascular diseases: Is the epidemiologic evidence compatible with the radiobiologic data? Int. J. Radiat. Oncol. Biol. Phys. 2007, 67, 10–18. [Google Scholar] [CrossRef]
- Mantovani, A.; Bussolino, F.; Introna, M. Cytokine regulation of endothelial cell function: From molecular level to the bedside. Immunol. Today 1997, 18, 231–240. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Zhang, H.; Hill, M.A.; Zhang, C.; Park, Y. Interaction of IL-6 and TNF-α contributes to endothelial dysfunction in type 2 diabetic mouse hearts. PLoS ONE 2017, 12, e0187189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picchi, A.; Gao, X.; Belmadani, S.; Potter, B.J.; Focardi, M.; Chilian, W.M.; Zhang, C. Tumor Necrosis Factor-α Induces Endothelial Dysfunction in the Prediabetic Metabolic Syndrome. Circ. Res. 2006, 99, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrache, I.; Verin, A.D.; Crow, M.T.; Birukova, A.; Liu, F.; Garcia, J.G.N. Differential effect of MLC kinase in TNF-α-induced endothelial cell apoptosis and barrier dysfunction. Am. J. Physiol. -Lung Cell. Mol. Physiol. 2001, 280, L1168–L1178. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, E.; Zocchi, M.R.; Magni, E.; Panzeri, M.C.; Curnis, F.; Rugarli, C.; Ferrero, M.E.; Corti, A. Roles of tumor necrosis factor p55 and p75 receptors in TNF-α-induced vascular permeability. Am. J. Physiol.-Cell Physiol. 2001, 281, C1173–C1179. [Google Scholar] [CrossRef] [PubMed]
- Leibovich, S.J.; Polverini, P.J.; Shepard, H.M.; Wiseman, D.M.; Shively, V.; Nuseir, N. Macrophage-induced angiogenesis is mediated by tumour necrosis factor-α. Nature 1987, 329, 630–632. [Google Scholar] [CrossRef]
- Yabe, Y.; Matsumoto, T.; Tsurumoto, T.; Shindo, H. Immunohistological localization of Notch receptors and their ligands Delta and Jagged in synovial tissues of rheumatoid arthritis. J. Orthop. Sci. 2005, 10, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Ando, K.; Kanazawa, S.; Tetsuka, T.; Ohta, S.; Jiang, X.; Tada, T.; Kobayashi, M.; Matsui, N.; Okamoto, T. Induction of Notch signaling by tumor necrosis factor in rheumatoid synovial fibroblasts. Oncogene 2003, 22, 7796–7803. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, T.; Takeshita, K.; Kikuchi, R.; Yamamoto, K.; Cheng, X.W.; Liao, J.K.; Murohara, T. gamma-Secretase inhibitor reduces diet-induced atherosclerosis in apolipoprotein E-deficient mice. Biochem. Biophys. Res. Commun. 2009, 383, 216–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, A.M.; Wang, S.-J.; Taylor, A.C.; Aitkenhead, M.; Hughes, C.C. The basic helix-loop-helix transcription factor HESR1 regulates endothelial cell tube formation. J. Biol. Chem. 2001, 276, 6169–6176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobov, I.; Renard, R.; Papadopoulos, N.; Gale, N.; Thurston, G.; Yancopoulos, G.; Wiegand, S. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc. Natl. Acad. Sci. USA 2007, 104, 3219–3224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siekmann, A.F.; Lawson, N.D. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature 2007, 445, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Tang, S.-B.; Hu, J.; Gao, Y.; Zhang, G.; Lin, S.-F.; Chen, J.-H.; Li, B.-J. Protective effects of transcription factor HESR1 on retinal vasculature. Microvasc. Res. 2006, 72, 146–152. [Google Scholar] [CrossRef]
- Chang, A.C.; Fu, Y.; Garside, V.C.; Niessen, K.; Chang, L.; Fuller, M.; Setiadi, A.; Smrz, J.; Kyle, A.; Minchinton, A.; et al. Notch initiates the endothelial-to-mesenchymal transition in the atrioventricular canal through autocrine activation of soluble guanylyl cyclase. Dev. Cell 2011, 21, 288–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Muto, A.; Kudo, F.A.; Model, L.; Eghbalieh, S.; Chowdhary, P.; Dardik, A. Age-related Notch-4 quiescence is associated with altered wall remodeling during vein graft adaptation. J. Surg. Res. 2011, 171, e149–e160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tousoulis, D.; Kampoli, A.M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Pearce, C.G.; Najjar, S.F.; Kapadia, M.R.; Murar, J.; Eng, J.; Lyle, B.; Aalami, O.O.; Jiang, Q.; Hrabie, J.A.; Saavedra, J.E.; et al. Beneficial effect of a short-acting NO donor for the prevention of neointimal hyperplasia. Free. Radic. Biol. Med. 2008, 44, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murohara, T.; Witzenbichler, B.; Spyridopoulos, I.; Asahara, T.; Ding, B.; Sullivan, A.; Losordo, D.W.; Isner, J.M. Role of Endothelial Nitric Oxide Synthase in Endothelial Cell Migration. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 1156–1161. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Huang, L.; Fang, Y.; Guo, R.; Yin, Y.; Zhao, X. Transplantation of endothelial progenitor cells overexpressing endothelial nitric oxide synthase enhances inhibition of neointimal hyperplasia and restores endothelium-dependent vasodilatation. Microvasc. Res. 2011, 81, 143–150. [Google Scholar] [CrossRef]
- Patenaude, A.; Fuller, M.; Chang, L.; Wong, F.; Paliouras, G.; Shaw, R.; Kyle, A.H.; Umlandt, P.; Baker, J.H.; Diaz, E.; et al. Endothelial-specific Notch blockade inhibits vascular function and tumor growth through an eNOS-dependent mechanism. Cancer Res. 2014, 74, 2402–2411. [Google Scholar] [CrossRef] [Green Version]
- Duan, J.L.; Ruan, B.; Yan, X.C.; Liang, L.; Song, P.; Yang, Z.Y.; Liu, Y.; Dou, K.F.; Han, H.; Wang, L. Endothelial Notch activation reshapes the angiocrine of sinusoidal endothelia to aggravate liver fibrosis and blunt regeneration in mice. Hepatology 2018, 68, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ubezio, B.; Blanco, R.A.; Geudens, I.; Stanchi, F.; Mathivet, T.; Jones, M.L.; Ragab, A.; Bentley, K.; Gerhardt, H. Synchronization of endothelial Dll4-Notch dynamics switch blood vessels from branching to expansion. eLife 2016, 5, e12167. [Google Scholar] [CrossRef] [PubMed]
- Pitulescu, M.E.; Schmidt, I.; Giaimo, B.D.; Antoine, T.; Berkenfeld, F.; Ferrante, F.; Park, H.; Ehling, M.; Biljes, D.; Rocha, S.F.; et al. Dll4 and Notch signalling couples sprouting angiogenesis and artery formation. Nat. Cell Biol. 2017, 19, 915–927. [Google Scholar] [CrossRef] [Green Version]
- Pancewicz, J.; Nicot, C. Current views on the role of Notch signaling and the pathogenesis of human leukemia. BMC Cancer 2011, 11, 502. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-H.; Zhao, X.-M.; Yoon, I.; Lee, J.Y.; Kwon, N.H.; Wang, Y.-Y.; Lee, K.-M.; Lee, M.-J.; Kim, J.; Moon, H.-G. Integrative analysis of mutational and transcriptional profiles reveals driver mutations of metastatic breast cancers. Cell Discov. 2016, 2, 16025. [Google Scholar] [CrossRef] [PubMed] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|
| Cyclophilin A | GAGCTGTTTGCAGACAAAGTTC | CCCTGGCACATGAATCCTG |
| Hey1 | GGAGAGCGCAGACGAGAATG | CTCGATGATGCCTCTCCGTC |
| Notch2 | ATGCCGGGTTTCAAAGGTGT | ATGTCGATCTGGCACACTGG |
| Jagged-1 | CATCGGGGGCAATACCTTCA | GCAAAGTGTAGGACCTCGGC |
| VEGF-A | ATCTTCAAGCCGTCCTGTGTG | TGAGGTTTGATCCGCATGATC |
| TNFa | ATGGCCCAGACCCTCACACTCAGA | CTCCGCTTGGTGGTTTGCTACGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peymanfar, Y.; Su, Y.-W.; Hassanshahi, M.; Xian, C.J. Therapeutic Targeting Notch2 Protects Bone Micro-Vasculatures from Methotrexate Chemotherapy-Induced Adverse Effects in Rats. Cells 2022, 11, 2382. https://doi.org/10.3390/cells11152382
Peymanfar Y, Su Y-W, Hassanshahi M, Xian CJ. Therapeutic Targeting Notch2 Protects Bone Micro-Vasculatures from Methotrexate Chemotherapy-Induced Adverse Effects in Rats. Cells. 2022; 11(15):2382. https://doi.org/10.3390/cells11152382
Chicago/Turabian StylePeymanfar, Yaser, Yu-Wen Su, Mohammadhossein Hassanshahi, and Cory J. Xian. 2022. "Therapeutic Targeting Notch2 Protects Bone Micro-Vasculatures from Methotrexate Chemotherapy-Induced Adverse Effects in Rats" Cells 11, no. 15: 2382. https://doi.org/10.3390/cells11152382
APA StylePeymanfar, Y., Su, Y. -W., Hassanshahi, M., & Xian, C. J. (2022). Therapeutic Targeting Notch2 Protects Bone Micro-Vasculatures from Methotrexate Chemotherapy-Induced Adverse Effects in Rats. Cells, 11(15), 2382. https://doi.org/10.3390/cells11152382