
Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease

 , , , , ,
, , , , , 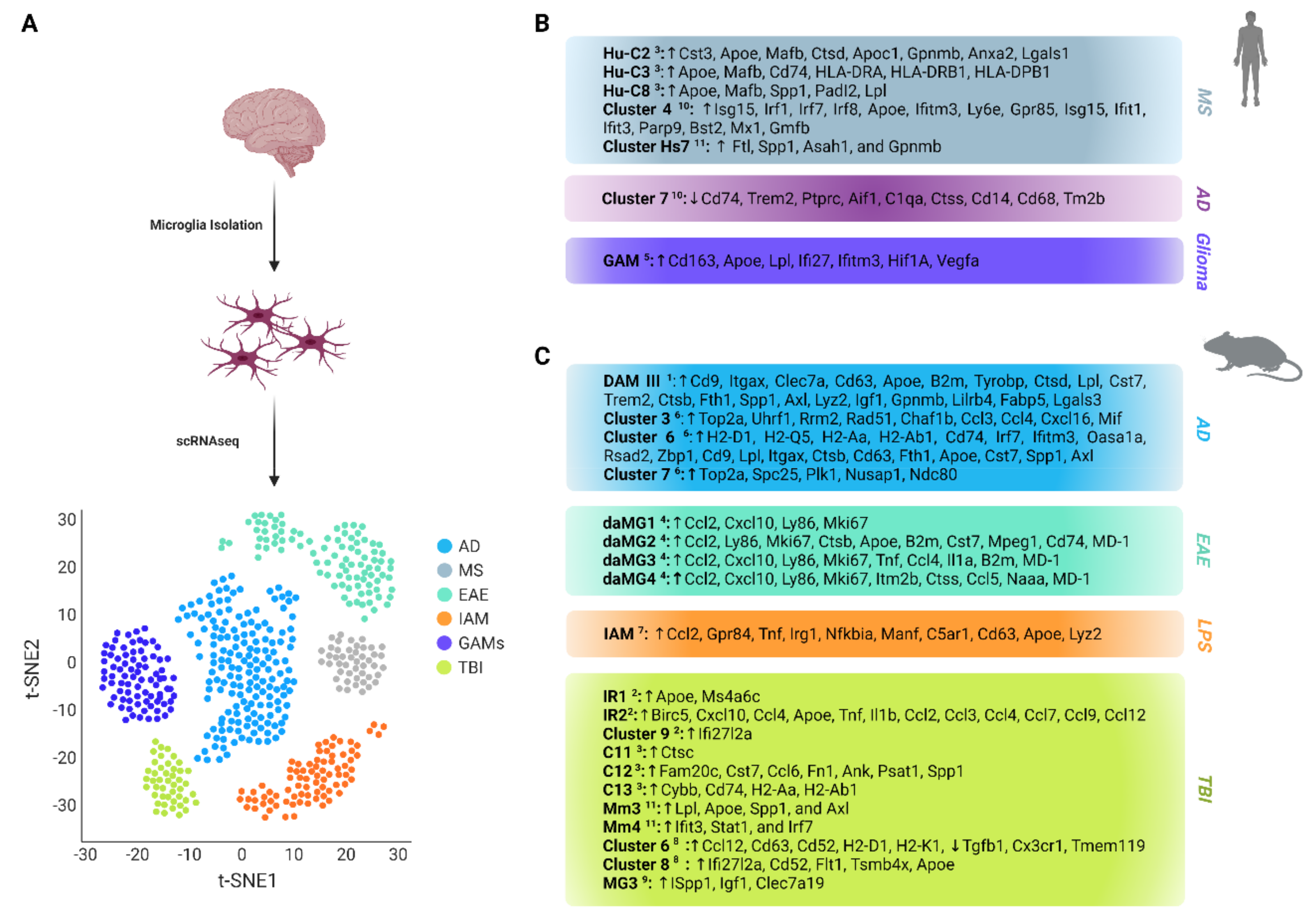{kind=link}
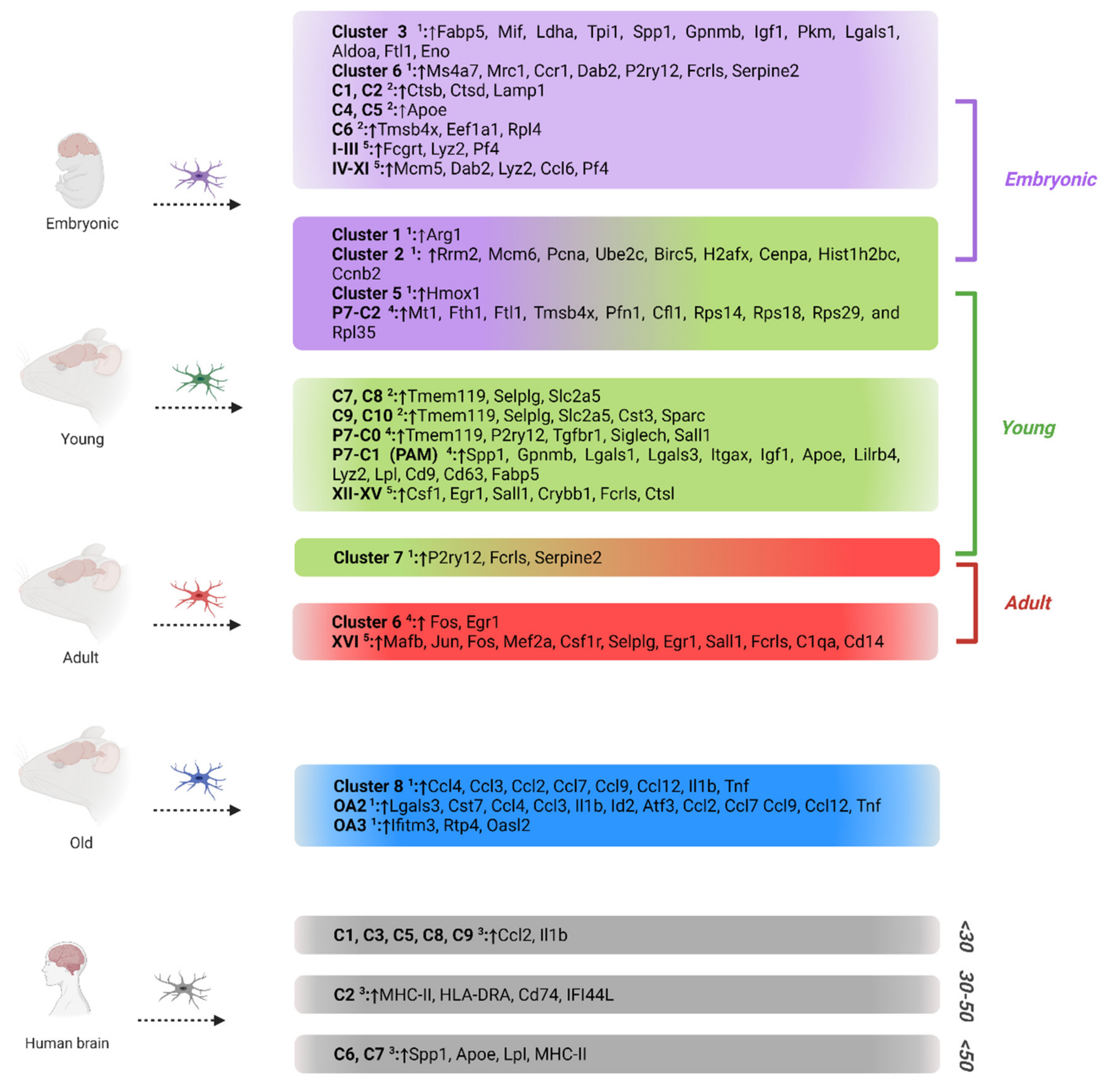{kind=link}
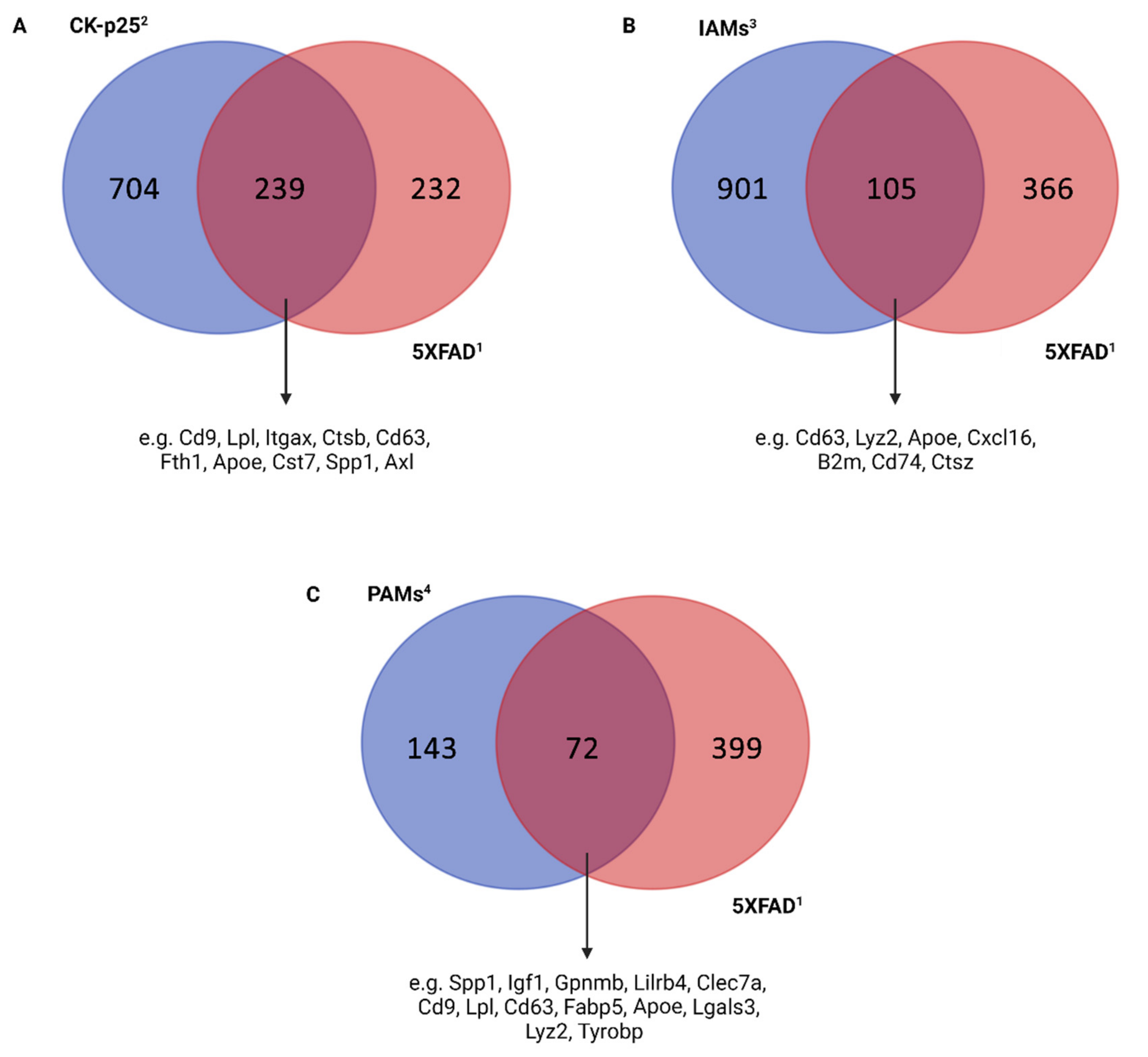{kind=link}
Abstract
:1. Introduction
2. Microglia Phenotypes in Homeostasis
3. Microglia Phenotypes in Diseases
3.1. Microglia in Multiple Sclerosis
3.2. Microglia in Alzheimer’s Disease
3.3. Microglia in Inflammation
3.4. Microglia in Traumatic Brain Injury
3.5. Microglia in Glioma
4. Microglia Phenotypes in Development and Aging
5. Microglia Phenotypes in Brain Regions
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reemst, K.; Noctor, S.C.; Lucassen, P.J.; Hol, E.M. The Indispensable Roles of Microglia and Astrocytes during Brain Development. Front. Hum. Neurosci. 2016, 10, 566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawson, L.J.; Perry, V.H.; Gordon, S. Turnover of Resident Microglia in the Normal Adult Mouse Brain. Neuroscience 1992, 48, 405–415. [Google Scholar] [CrossRef]
- Mittelbronn, M.; Dietz, K.; Schluesener, H.J.; Meyermann, R. Local Distribution of Microglia in the Normal Adult Human Central Nervous System Differs by up to One Order of Magnitude. Acta Neuropathol. 2001, 101, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Bessis, A.; Béchade, C.; Bernard, D.; Roumier, A. Microglial Control of Neuronal Death and Synaptic Properties. Glia 2007, 55, 233–238. [Google Scholar] [CrossRef]
- Marín-Teva, J.L.; Dusart, I.; Colin, C.; Gervais, A.; Van Rooijen, N.; Mallat, M. Microglia Promote the Death of Developing Purkinje Cells. Neuron 2004, 41, 535–547. [Google Scholar] [CrossRef]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia Promote Learning-Dependent Synapse Formation through Brain-Derived Neurotrophic Factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Bengtsson, M.; Ståhlberg, A.; Rorsman, P.; Kubista, M. Gene Expression Profiling in Single Cells from the Pancreatic Islets of Langerhans Reveals Lognormal Distribution of MRNA Levels. Genome Res. 2005, 15, 1388–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svensson, V.; Natarajan, K.N.; Ly, L.H.; Miragaia, R.J.; Labalette, C.; Macaulay, I.C.; Cvejic, A.; Teichmann, S.A. Power Analysis of Single-Cell RNA-Sequencing Experiments. Nat. Methods 2017, 14, 381–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalek, A.K.; Satija, R.; Adiconis, X.; Gertner, R.S.; Gaublomme, J.T.; Raychowdhury, R.; Schwartz, S.; Yosef, N.; Malboeuf, C.; Lu, D.; et al. Single-Cell Transcriptomics Reveals Bimodality in Expression and Splicing in Immune Cells. Nature 2013, 498, 236–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thrupp, N.; Frigerio, C.S.; Wolfs, L.; Mancuso, R.; Skene, N.G.; Fattorelli, N.; Poovathingal, S.; Fourne, Y.; Matthews, P.M.; Theys, T.; et al. Single-Nucleus RNA-Seq Is Not Suitable for Detection of Microglial Activation Genes in Humans. Cell Rep. 2020, 32, 108189. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290. [Google Scholar] [CrossRef]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Sankowski, R.; Staszewski, O.; Böttcher, C.; Amann, L.; Scheiwe, C.; Nessler, S.; Kunz, P.; van Loo, G.; Coenen, V.A.; et al. Spatial and Temporal Heterogeneity of Mouse and Human Microglia at Single-Cell Resolution. Nature 2019, 566, 388–392. [Google Scholar] [CrossRef]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Sagar, G.L.; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; et al. Neuroimmunology: Single-Cell Profiling Identifies Myeloid Cell Subsets with Distinct Fates during Neuroinflammation. Science 2019, 363, 6425. [Google Scholar] [CrossRef] [PubMed]
- Sankowski, R.; Böttcher, C.; Masuda, T.; Geirsdottir, L.; Sagar; Sindram, E.; Seredenina, T.; Muhs, A.; Scheiwe, C.; Shah, M.J.; et al. Mapping Microglia States in the Human Brain through the Integration of High-Dimensional Techniques. Nat. Neurosci. 2019, 22, 2098–2110. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [Green Version]
- Sousa, C.; Golebiewska, A.; Poovathingal, S.K.; Kaoma, T.; Pires-Afonso, Y.; Martina, S.; Coowar, D.; Azuaje, F.; Skupin, A.; Balling, R.; et al. Single-Cell Transcriptomics Reveals Distinct Inflammation-Induced Microglia Signatures. EMBO Rep. 2018, 19, e46171. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Chunchai, T.; Zhao, F.; O’Neil, S.M.; Gordillo, A.J.; Campbell, W.A.; McKim, D.B.; Liu, X.; Dziabis, J.E.; et al. Traumatic Brain Injury Causes Chronic Cortical Inflammation and Neuronal Dysfunction Mediated by Microglia. J. Neurosci. 2021, 41, 1597–1616. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; He, X.; Kawaguchi, R.; Zhang, Y.; Wang, Q.; Monavarfeshani, A.; Yang, Z.; Chen, B.; Shi, Z.; Meng, H.; et al. Microglia-Organized Scar-Free Spinal Cord Repair in Neonatal Mice. Nature 2020, 587, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Olah, M.; Menon, V.; Habib, N.; Taga, M.F.; Ma, Y.; Yung, C.J.; Cimpean, M.; Khairallah, A.; Coronas-Samano, G.; Sankowski, R.; et al. Single Cell RNA Sequencing of Human Microglia Uncovers a Subset Associated with Alzheimer’s Disease. Nat. Commun. 2020, 11, 6129. [Google Scholar] [CrossRef] [PubMed]
- Miedema, A.; Gerrits, E.; Brouwer, N.; Jiang, Q.; Kracht, L.; Meijer, M.; Nutma, E.; Peferoen-Baert, R.; Pijnacker, A.T.E.; Wesseling, E.M.; et al. Brain Macrophages Acquire Distinct Transcriptomes in Multiple Sclerosis Lesions and Normal Appearing White Matter. Acta Neuropathol. Commun. 2022, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Ziegenhain, C.; Vieth, B.; Parekh, S.; Reinius, B.; Guillaumet-Adkins, A.; Smets, M.; Leonhardt, H.; Heyn, H.; Hellmann, I.; Enard, W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Mol. Cell 2017, 65, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Ning, B.; Shi, T. Single-Cell RNA-Seq Technologies and Related Computational Data Analysis. Front. Genet. 2019, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Van Der Maaten, L.; Hinton, G. Visualizing Data Using T-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Mcinnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2020. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.M.; Doykan, C.E.; et al. Identification of a Unique TGF-β Dependent Molecular and Functional Signature in Microglia. Nat. Neurosci. 2014, 17, 131. [Google Scholar] [CrossRef] [Green Version]
- Torkildsen, Ø.; Brunborg, L.A.; Myhr, K.M.; Bø, L. The Cuprizone Model for Demyelination. Acta Neurol. Scand. Suppl. 2008, 188, 72–76. [Google Scholar] [CrossRef]
- Olmstead, D.N.; Mesnard-Hoaglin, N.A.; Batka, R.J.; Haulcomb, M.M.; Miller, W.M.; Jones, K.J. Facial Nerve Axotomy in Mice: A Model to Study Motoneuron Response to Injury. J. Vis. Exp. 2015, 23, e52382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental Autoimmune Encephalomyelitis (EAE) as a Model for Multiple Sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med. 2011, 3, 77sr1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the Aged Brain: Primed to Be Activated and Resistant to Regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef]
- Langlois, J.A.; Rutland-Brown, W.; Wald, M.M. The Epidemiology and Impact of Traumatic Brain Injury: A Brief Overview. J. Head Trauma Rehabil. 2006, 21, 375–378. [Google Scholar] [CrossRef] [Green Version]
- Hyder, A.A.; Wunderlich, C.A.; Puvanachandra, P.; Gururaj, G.; Kobusingye, O.C. The Impact of Traumatic Brain Injuries: A Global Perspective. NeuroRehabilitation 2007, 22, 341–353. [Google Scholar] [CrossRef] [Green Version]
- Donat, C.K.; Scott, G.; Gentleman, S.M.; Sastre, M. Microglial Activation in Traumatic Brain Injury. Front. Aging Neurosci. 2017, 9, 208. [Google Scholar] [CrossRef] [Green Version]
- Ximerakis, M.; Lipnick, S.L.; Innes, B.T.; Simmons, S.K.; Adiconis, X.; Dionne, D.; Mayweather, B.A.; Nguyen, L.; Niziolek, Z.; Ozek, C.; et al. Single-Cell Transcriptomic Profiling of the Aging Mouse Brain. Nat. Neurosci. 2019, 22, 1696–1708. [Google Scholar] [CrossRef]
- Li, Q.; Cheng, Z.; Zhou, L.; Darmanis, S.; Neff, N.F.; Okamoto, J.; Gulati, G.; Bennett, M.L.; Sun, L.O.; Clarke, L.E.; et al. Developmental Heterogeneity of Microglia and Brain Myeloid Cells Revealed by Deep Single-Cell RNA Sequencing. Neuron 2019, 101, 207–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matcovitch-Natan, O.; Winter, D.R.; Giladi, A.; Aguilar, S.V.; Spinrad, A.; Sarrazin, S.; Ben-Yehuda, H.; David, E.; González, F.Z.; Perrin, P.; et al. Microglia Development Follows a Stepwise Program to Regulate Brain Homeostasis. Science 2016, 353, aad8670. [Google Scholar] [CrossRef]
- Calandra, T.; Roger, T. Macrophage Migration Inhibitory Factor: A Regulator of Innate Immunity. Nat. Rev. Immunol. 2003, 3, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Kannan-Thulasiraman, P.; Seachrist, D.D.; Mahabeleshwar, G.H.; Jain, M.K.; Noy, N. Fatty Acid-Binding Protein 5 and PPARβ/δ Are Critical Mediators of Epidermal Growth Factor Receptor-Induced Carcinoma Cell Growth. J. Biol. Chem. 2011, 286, 36161. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.Z.; Mita, R.; Beaulieu, M.; Gao, Z.; Godbout, R. Fatty Acid Binding Proteins in Brain Development and Disease. Int. J. Dev. Biol. 2010, 54, 1229–1239. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Wieghofer, P.; Jordão, M.J.C.; Prutek, F.; Hagemeyer, N.; Frenzel, K.; Amann, L.; Staszewski, O.; Kierdorf, K.; Krueger, M.; et al. Origin, Fate and Dynamics of Macrophages at Central Nervous System Interfaces. Nat. Immunol. 2016, 17, 797–805. [Google Scholar] [CrossRef]
- Baruch, K.; Deczkowska, A.; David, E.; Castellano, J.M.; Miller, O.; Kertser, A.; Berkutzki, T.; Barnett-Itzhaki, Z.; Bezalel, D.; Wyss-Coray, T.; et al. Aging-Induced Type I Interferon Response at the Choroid Plexus Negatively Affects Brain Function. Science 2014, 346, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Kenneth Baillie, J.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial Brain Region−dependent Diversity and Selective Regional Sensitivities to Aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef] [Green Version]
- Hagemeyer, N.; Hanft, K.M.; Akriditou, M.A.; Unger, N.; Park, E.S.; Stanley, E.R.; Staszewski, O.; Dimou, L.; Prinz, M. Microglia Contribute to Normal Myelinogenesis and to Oligodendrocyte Progenitor Maintenance during Adulthood. Acta Neuropathol. 2017, 134, 441–458. [Google Scholar] [CrossRef] [Green Version]
- Leyns, C.E.G.; Holtzman, D.M. Glial Contributions to Neurodegeneration in Tauopathies. Mol. Neurodegener. 2017, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and Microglia Mediate Early Synapse Loss in Alzheimer Mouse Models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [Green Version]
- Ritzel, R.M.; Patel, A.R.; Pan, S.; Crapser, J.; Hammond, M.; Jellison, E.; McCullough, L.D. Age- and Location-Related Changes in Microglial Function. Neurobiol. Aging 2015, 36, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Denisenko, E.; Guo, B.B.; Jones, M.; Hou, R.; De Kock, L.; Lassmann, T.; Poppe, D.; Poppe, D.; Clément, O.; Simmons, R.K.; et al. Systematic Assessment of Tissue Dissociation and Storage Biases in Single-Cell and Single-Nucleus RNA-Seq Workflows. Genome Biol. 2020, 21, 130. [Google Scholar] [CrossRef]
- Marsh, S.E.; Walker, A.J.; Kamath, T.; Dissing-Olesen, L.; Hammond, T.R.; de Soysa, T.Y.; Young, A.M.H.; Murphy, S.; Abdulraouf, A.; Nadaf, N.; et al. Dissection of Artifactual and Confounding Glial Signatures by Single-Cell Sequencing of Mouse and Human Brain. Nat. Neurosci. 2022, 25, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Geirsdottir, L.; David, E.; Keren-Shaul, H.; Weiner, A.; Bohlen, S.C.; Neuber, J.; Balic, A.; Giladi, A.; Sheban, F.; Dutertre, C.A.; et al. Cross-Species Single-Cell Analysis Reveals Divergence of the Primate Microglia Program. Cell 2019, 179, 1609–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esaulova, E.; Cantoni, C.; Shchukina, I.; Zaitsev, K.; Bucelli, R.C.; Wu, G.F.; Artyomov, M.N.; Cross, A.H.; Edelson, B.T. Single-Cell RNA-Seq Analysis of Human CSF Microglia and Myeloid Cells in Neuroinflammation. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e732. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pettas, S.; Karagianni, K.; Kanata, E.; Chatziefstathiou, A.; Christoudia, N.; Xanthopoulos, K.; Sklaviadis, T.; Dafou, D. Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease. Cells 2022, 11, 2383. https://doi.org/10.3390/cells11152383
Pettas S, Karagianni K, Kanata E, Chatziefstathiou A, Christoudia N, Xanthopoulos K, Sklaviadis T, Dafou D. Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease. Cells. 2022; 11(15):2383. https://doi.org/10.3390/cells11152383
Chicago/Turabian StylePettas, Spyros, Korina Karagianni, Eirini Kanata, Athanasia Chatziefstathiou, Nikoletta Christoudia, Konstantinos Xanthopoulos, Theodoros Sklaviadis, and Dimitra Dafou. 2022. "Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease" Cells 11, no. 15: 2383. https://doi.org/10.3390/cells11152383
APA StylePettas, S., Karagianni, K., Kanata, E., Chatziefstathiou, A., Christoudia, N., Xanthopoulos, K., Sklaviadis, T., & Dafou, D. (2022). Profiling Microglia through Single-Cell RNA Sequencing over the Course of Development, Aging, and Disease. Cells, 11(15), 2383. https://doi.org/10.3390/cells11152383