
Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review



{kind=link}
{kind=link}
Abstract
:1. Introduction
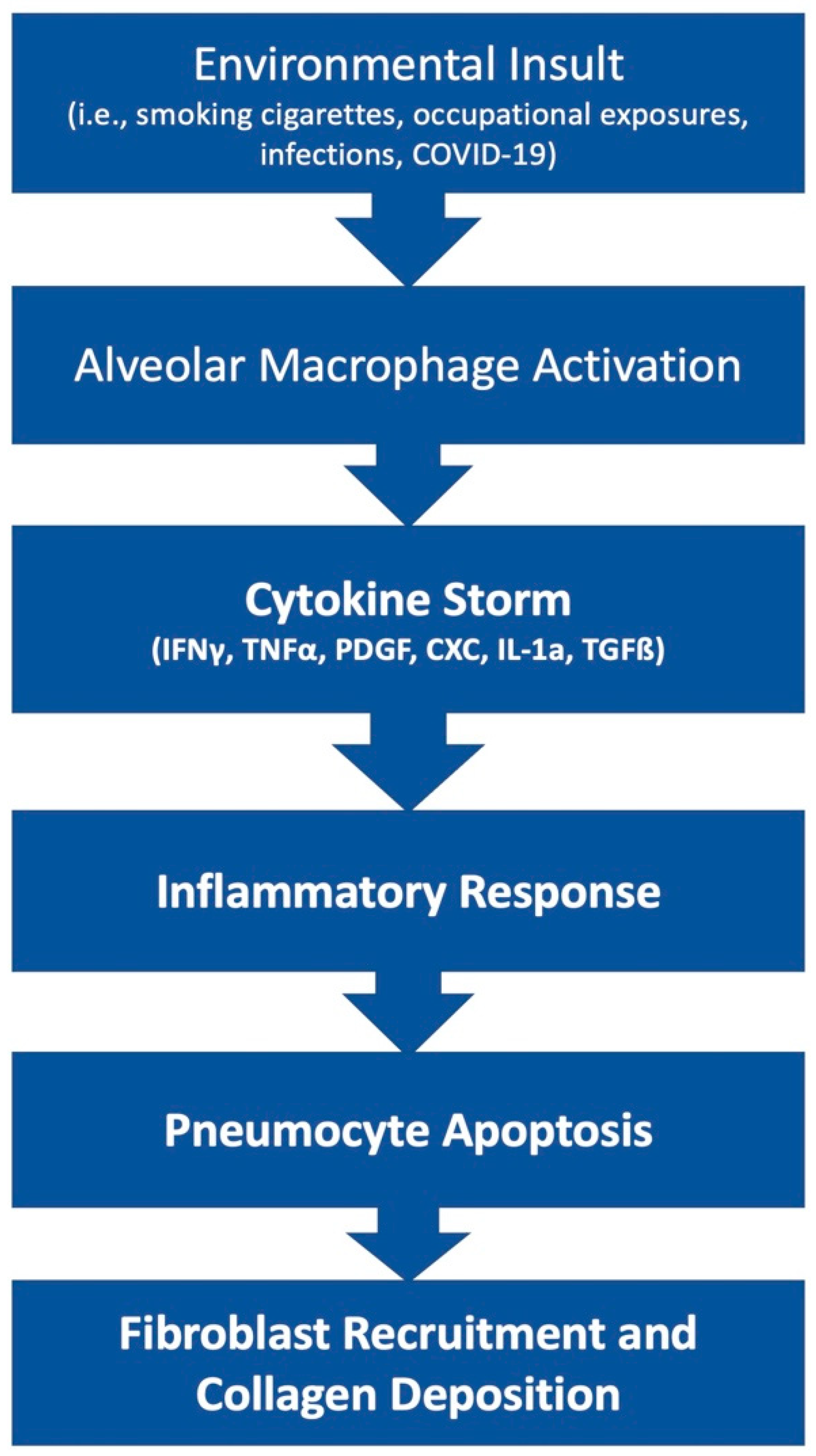
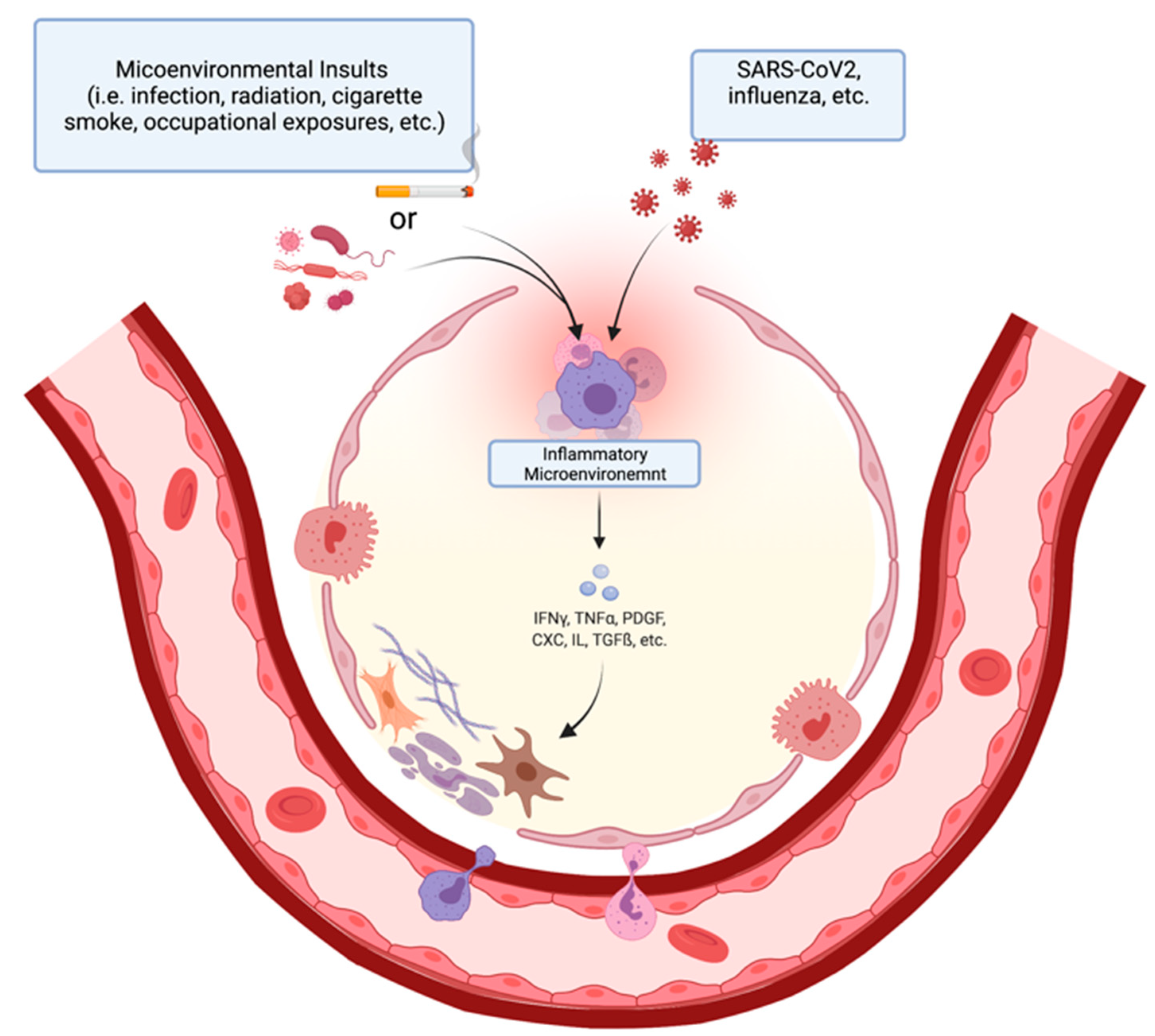
2. Pathophysiology of Pulmonary Fibrosis
3. COVID-19 and Lung Fibrosis
4. The Potential Prevention of Lung Fibrosis
5. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| PF | Pulmonary fibrosis |
| SARS | Severe acute respiratory syndrome |
| FVC | Forced vital capacity |
| TLC | Total lung capacity |
| DLco | Diffusion capacity for carbon monoxide |
| IPF | Idiopathic Pulmonary Fibrosis |
| ARDS | Acute respiratory distress syndrome |
| AECs | Alveolar epithelial cells |
| TGF | Transforming growth factor |
| ECM | Extracellular matrix |
| ERK | Extracellular-signal-regulated kinase |
| IFN | Interferon gamma |
| IL | Interleukin |
| TNF | Tumor necrosis factor |
| PDGF | Platelet-derived growth factor |
| ROS | Reactive oxygen species |
| TMRSS | Transmembrane Serine Protease |
| ACE | Angiotensin-converting enzyme |
| ICU | Intensive care unit |
| FDA | Food and Drug Administration |
References
- Gentile, F.; Aimo, A.; Forfori, F.; Catapano, G.; Clemente, A.; Cademartiri, F.; Emdin, M.; Giannoni, A. COVID-19 and risk of pulmonary fibrosis: The importance of planning ahead. Eur. J. Prev. Cardiol. 2020, 27, 1442–1446. [Google Scholar] [CrossRef] [PubMed]
- Hui, D.S.; Wong, K.T.; Ko, F.W.; Tam, L.S.; Chan, D.P.; Woo, J.; Sung, J.J. The 1-year impact of severe acute respiratory syndrome on pulmonary function, exercise capacity, and quality of life in a cohort of survivors. Chest 2005, 128, 2247–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngai, J.C.; Ko, F.W.; Ng, S.S.; To, K.W.; Tong, M.; Hui, D.S. The long-term impact of severe acute respiratory syndrome on pulmonary function, exercise capacity and health status. Respirology 2010, 15, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.C.; Yu, C.J.; Chang, S.C.; Galvin, J.R.; Liu, H.M.; Hsiao, C.H.; Kuo, P.H.; Chen, K.Y.; Franks, T.J.; Huang, K.M.; et al. Pulmonary sequelae in convalescent patients after severe acute respiratory syndrome: Evaluation with thin-section CT. Radiology 2005, 236, 1067–1075. [Google Scholar] [CrossRef] [PubMed]
- Tomashefski, J.F., Jr. Pulmonary pathology of acute respiratory distress syndrome. Clin. Chest Med. 2000, 21, 435–466. [Google Scholar] [CrossRef]
- Parimon, T.; Yao, C.; Stripp, B.R.; Noble, P.W.; Chen, P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int. J. Mol. Sci. 2020, 21, 2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzen, J.; Beers, M.F. Contributions of alveolar epithelial cell quality control to pulmonary fibrosis. J. Clin. Investig. 2020, 130, 5088–5099. [Google Scholar] [CrossRef]
- Yue, X.; Shan, B.; Lasky, J.A. TGF-β: Titan of Lung Fibrogenesis. Curr. Enzym. Inhib. 2010, 6, 67–77. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiao, H.; Wu, Y.; Sun, X. P120-catenin regulates pulmonary fibrosis and TGF-β induced lung fibroblast differentiation. Life Sci. 2019, 230, 35–44. [Google Scholar] [CrossRef]
- Chanda, D.; Otoupalova, E.; Smith, S.R.; Volckaert, T.; De Langhe, S.P.; Thannickal, V.J. Developmental pathways in the pathogenesis of lung fibrosis. Mol. Asp. Med. 2019, 65, 56–69. [Google Scholar] [CrossRef]
- Xiao, L.; Du, Y.; Shen, Y.; He, Y.; Zhao, H.; Li, Z. TGF-beta 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front. Biosci. 2012, 17, 2667–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, K.; Yatera, K.; Izumi, H.; Ishimoto, H.; Yamada, S.; Nakao, H.; Hanaka, T.; Ogoshi, T.; Noguchi, S.; Mukae, H. Profibrotic role of WNT10A via TGF-ß signaling in idiopathic pulmonary fibrosis. Respir. Res. 2016, 17, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, N.; Ishii, Y.; Morishima, Y.; Yageta, Y.; Haraguchi, N.; Itoh, K.; Yamamoto, M.; Hizawa, N. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir. Res. 2010, 11, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolahian, S.; Fernandez, I.E.; Eickelberg, O.; Hartl, D. Immune Mechanisms in Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 55, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Agostini, C.; Gurrieri, C. Chemokine/cytokine cocktail in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 357–363. [Google Scholar] [CrossRef]
- Upagupta, C.; Shimbori, C.; Alsilmi, R.; Kolb, M. Matrix abnormalities in pulmonary fibrosis. Eur. Respir. Rev. 2018, 27, 180033. [Google Scholar] [CrossRef] [Green Version]
- Wei, P.; Xie, Y.; Abel, P.W.; Huang, Y.; Ma, Q.; Li, L.; Hao, J.; Wolff, D.W.; Wei, T.; Tu, Y. Transforming growth factor (TGF)-β1-induced miR-133a inhibits myofibroblast differentiation and pulmonary fibrosis. Cell Death Dis. 2019, 10, 670. [Google Scholar] [CrossRef]
- Li, H.; Zhao, C.; Tian, Y.; Lu, J.; Zhang, G.; Liang, S.; Chen, D.; Liu, X.; Kuang, W.; Zhu, M. Src family kinases and pulmonary fibrosis: A review. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110183. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Suh, Y.J.; Hong, H.; Ohana, M.; Bompard, F.; Revel, M.P.; Valle, C.; Gervaise, A.; Poissy, J.; Susen, S.; Hékimian, G.; et al. Pulmonary Embolism and Deep Vein Thrombosis in COVID-19: A Systematic Review and Meta-Analysis. Radiology 2021, 298, E70–E80. [Google Scholar] [CrossRef]
- Tale, S.; Ghosh, S.; Meitei, S.P.; Kolli, M.; Garbhapu, A.K.; Pudi, S. Post-COVID-19 pneumonia pulmonary fibrosis. QJM 2020, 113, 837–838. [Google Scholar] [CrossRef] [PubMed]
- Seyed Hosseini, E.; Riahi Kashani, N.; Nikzad, H.; Azadbakht, J.; Hassani Bafrani, H.; Haddad Kashani, H. The novel coronavirus Disease-2019 (COVID-19): Mechanism of action, detection and recent therapeutic strategies. Virology 2020, 551, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Harrison, A.G.; Lin, T.; Wang, P. Mechanisms of SARS-CoV-2 Transmission and Pathogenesis. Trends Immunol. 2020, 41, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Ghazavi, A.; Ganji, A.; Keshavarzian, N.; Rabiemajd, S.; Mosayebi, G. Cytokine profile and disease severity in patients with COVID-19. Cytokine 2021, 137, 155323. [Google Scholar] [CrossRef]
- Camargo, L.D.N.; Righetti, R.F.; Aristóteles, L.R.D.C.R.B.; Dos Santos, T.M.; De Souza, F.C.R.; Fukuzaki, S.; Cruz, M.M.; Alonso-Vale, M.I.C.; Saraiva-Romanholo, B.M.; Prado, C.M.; et al. Effects of Anti-IL-17 on Inflammation, Remodeling, and Oxidative Stress in an Experimental Model of Asthma Exacerbated by LPS. Front. Immunol. 2018, 8, 1835. [Google Scholar] [CrossRef]
- Bialek, S.; Boundy, E.; Bowen, V.; Chow, N.; Cohn, A.; Dowling, N.; Ellington, S. Severe Outcomes Among Patients with Coronavirus Disease 2019 (COVID-19)—United States, February 12–16 March 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 343–346. [Google Scholar]
- Gulati, S.; Thannickal, V.J. The Aging Lung and Idiopathic Pulmonary Fibrosis. Am. J. Med. Sci. 2019, 357, 384–389. [Google Scholar] [CrossRef]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Das, K.M.; Lee, E.Y.; Singh, R.; Enani, M.A.; Al Dossari, K.; Van Gorkom, K.; Larsson, S.G.; Langer, R.D. Follow-up chest radiographic findings in patients with MERS-CoV after recovery. Indian J. Radiol. Imaging 2017, 27, 342–349. [Google Scholar] [CrossRef]
- Cabrera-Benitez, N.E.; Laffey, J.G.; Parotto, M.; Spieth, P.M.; Villar, J.; Zhang, H.; Slutsky, A.S. Mechanical ventilation-associated lung fibrosis in acute respiratory distress syndrome: A significant contributor to poor outcome. Anesthesiology 2014, 121, 189–198. [Google Scholar] [CrossRef] [Green Version]
- George, P.M.; Wells, A.U.; Jenkins, R.G. Pulmonary fibrosis and COVID-19: The potential role for antifibrotic therapy. Lancet Respir. Med. 2020, 8, 807. [Google Scholar] [CrossRef]
- Guan, W.; Ni, Z.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- Seifirad, S. Pirfenidone: A novel hypothetical treatment for COVID-19. Med. Hypotheses 2020, 144, 110005. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves Esbriet (Pirfenidone) for Idiopathic Pulmonary Fibrosis. Available online: https://www.drugs.com/newdrugs/fda-approves-esbriet-pirfenidone-idiopathic-pulmonary-fibrosis-4099.html (accessed on 31 October 2021).
- FDA Approves First Treatment for Group of Progressive Interstitial Lung Diseases|FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-group-progressive-interstitial-lung-diseases (accessed on 31 October 2021).
- Matsumura, T.; Fujimoto, T.; Futakuchi, A.; Takihara, Y.; Watanabe-Kitamura, F.; Takahashi, E.; Inoue-Mochita, M.; Tanihara, H.; Inoue, T. TGF-β-induced activation of conjunctival fibroblasts is modulated by FGF-2 and substratum stiffness. PLoS ONE 2020, 15, e0242626. [Google Scholar] [CrossRef]
- Shihab, F.S.; Bennett, W.M.; Yi, H.; Andoh, T.F. Pirfenidone treatment decreases transforming growth factor-beta1 and matrix proteins and ameliorates fibrosis in chronic cyclosporine nephrotoxicity. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2002, 2, 111–119. [Google Scholar] [CrossRef]
- Li, G.; Ren, J.; Hu, Q.; Deng, Y.; Chen, G.; Guo, K.; Li, R.; Li, Y.; Wu, L.; Wang, G.; et al. Oral pirfenidone protects against fibrosis by inhibiting fibroblast proliferation and TGF-β signaling in a murine colitis model. Biochem. Pharmacol. 2016, 117, 57–67. [Google Scholar] [CrossRef]
- Zhang, F.; Wei, Y.; He, L.; Zhang, H.; Hu, Q.; Yue, H.; He, J.; Dai, H. A trial of pirfenidone in hospitalized adult patients with severe coronavirus disease 2019. Chin. Med. J. 2022, 135, 368–370. [Google Scholar] [CrossRef]
- Umemura, Y.; Mitsuyama, Y.; Minami, K.; Nishida, T.; Watanabe, A.; Okada, N.; Yamakawa, K.; Nochioka, K.; Fujimi, S. Efficacy and safety of nintedanib for pulmonary fibrosis in severe pneumonia induced by COVID-19: An interventional study. Int. J. Infect. Dis. 2021, 108, 454–460. [Google Scholar] [CrossRef]
- Echeverría-Esnal, D.; Martin-Ontiyuelo, C.; Navarrete-Rouco, M.E.; De-Antonio Cusco, M.; Ferrández, O.; Horcajada, J.P.; Grau, S. Azithromycin in the treatment of COVID-19: A review. Expert Rev. Anti-Infect. Ther. 2021, 19, 147–163. [Google Scholar] [CrossRef]
- Recovery Collaborative Group. Azithromycin in patients admitted to hospital with COVID-19 (Recovery): A randomised, controlled, open-label, platform trial. Lancet 2021, 397, 605–612. [Google Scholar] [CrossRef]
- Oldenburg, C.E.; Pinsky, B.A.; Brogdon, J.; Chen, C.; Ruder, K.; Zhong, L.; Nyatigo, F.; Cook, C.A.; Hinterwirth, A.; Lebas, E.; et al. Effect of Oral Azithromycin vs Placebo on COVID-19 Symptoms in Outpatients With SARS-CoV-2 Infection: A Randomized Clinical Trial. JAMA 2021, 326, 490–498. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of Covid-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef]
- Nujić, K.; Banjanac, M.; Munić, V.; Polančec, D.; Eraković Haber, V. Impairment of lysosomal functions by azithromycin and chloroquine contributes to anti-inflammatory phenotype. Cell. Immunol. 2012, 279, 78–86. [Google Scholar] [CrossRef]
- Sivashanmugam, K.; Kandasamy, M.; Subbiah, R.; Ravikumar, V. Repurposing of histone deacetylase inhibitors: A promising strategy to combat pulmonary fibrosis promoted by TGF-β signalling in COVID-19 survivors. Life Sci. 2021, 266, 118883. [Google Scholar]
- Glenisson, W.; Castronovo, V.; Waltregny, D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim. Biophys. Acta 2007, 1773, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Andugulapati, S.B.; Gourishetti, K.; Tirunavalli, S.K.; Shaikh, T.B.; Sistla, R. Biochanin-A ameliorates pulmonary fibrosis by suppressing the TGF-β mediated EMT, myofibroblasts differentiation and collagen deposition in in vitro and in vivo systems. Phytomedicine Int. J. Phytother. Phytopharm. 2020, 78, 153298. [Google Scholar] [CrossRef]
- Mrityunjaya, M.; Pavithra, V.; Neelam, R.; Janhavi, P.; Halami, P.M.; Ravindra, P.V. Immune-Boosting, Antioxidant and Anti-inflammatory Food Supplements Targeting Pathogenesis of COVID-19. Front. Immunol. 2020, 11, 570122. [Google Scholar] [CrossRef]
- Teymoori-Rad, M.; Shokri, F.; Salimi, V.; Marashi, S.M. The interplay between vitamin D and viral infections. Rev. Med. Virol. 2019, 29, e2032. [Google Scholar] [CrossRef]
- Zdrenghea, M.T.; Makrinioti, H.; Bagacean, C.; Bush, A.; Johnston, S.L.; Stanciu, L.A. Vitamin D modulation of innate immune responses to respiratory viral infections. Rev. Med. Virol. 2017, 27, e1909. [Google Scholar] [CrossRef]
- Martineau, A.R.; Jolliffe, D.A.; Hooper, R.L.; Greenberg, L.; Aloia, J.F.; Bergman, P.; Dubnov-Raz, G.; Esposito, S.; Ganmaa, D.; Ginde, A.A.; et al. Vitamin D supplementation to prevent acute respiratory tract infections: Systematic review and meta-analysis of individual participant data. BMJ 2017, 356, i6583. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, S.; Ksajikian, A.; Overbey, J.; Li, P.; Li, Y. Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review. Cells 2022, 11, 2489. https://doi.org/10.3390/cells11162489
Tran S, Ksajikian A, Overbey J, Li P, Li Y. Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review. Cells. 2022; 11(16):2489. https://doi.org/10.3390/cells11162489
Chicago/Turabian StyleTran, Son, Andre Ksajikian, Juliana Overbey, Patrick Li, and Yong Li. 2022. "Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review" Cells 11, no. 16: 2489. https://doi.org/10.3390/cells11162489
APA StyleTran, S., Ksajikian, A., Overbey, J., Li, P., & Li, Y. (2022). Pathophysiology of Pulmonary Fibrosis in the Context of COVID-19 and Implications for Treatment: A Narrative Review. Cells, 11(16), 2489. https://doi.org/10.3390/cells11162489