
Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methodological Approaches
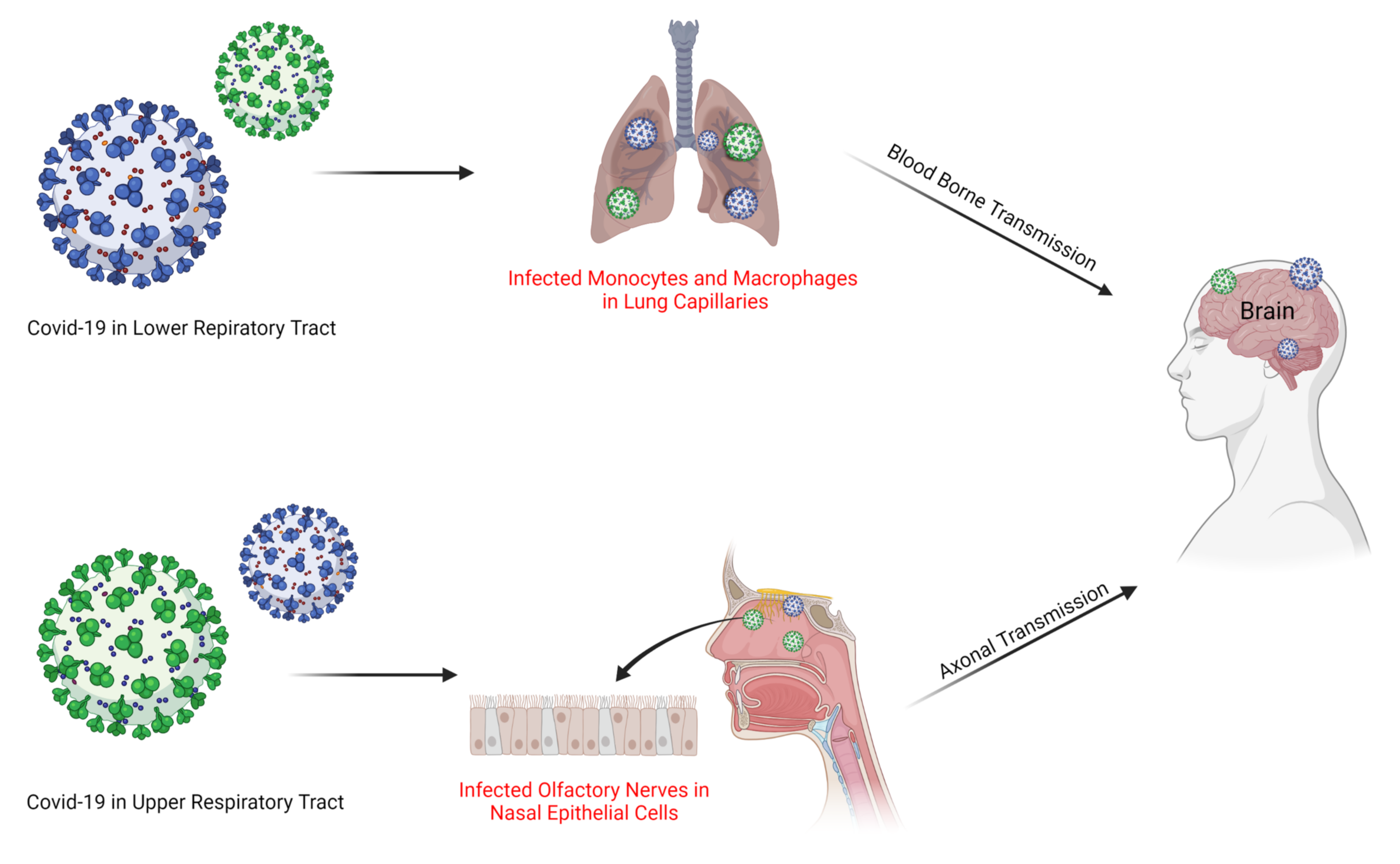
3. Possible Entry Mechanism of SARS-CoV-2 into the Brains
4. Role of Oxidative Stress in COVID-19 and Neurodegenerative Diseases
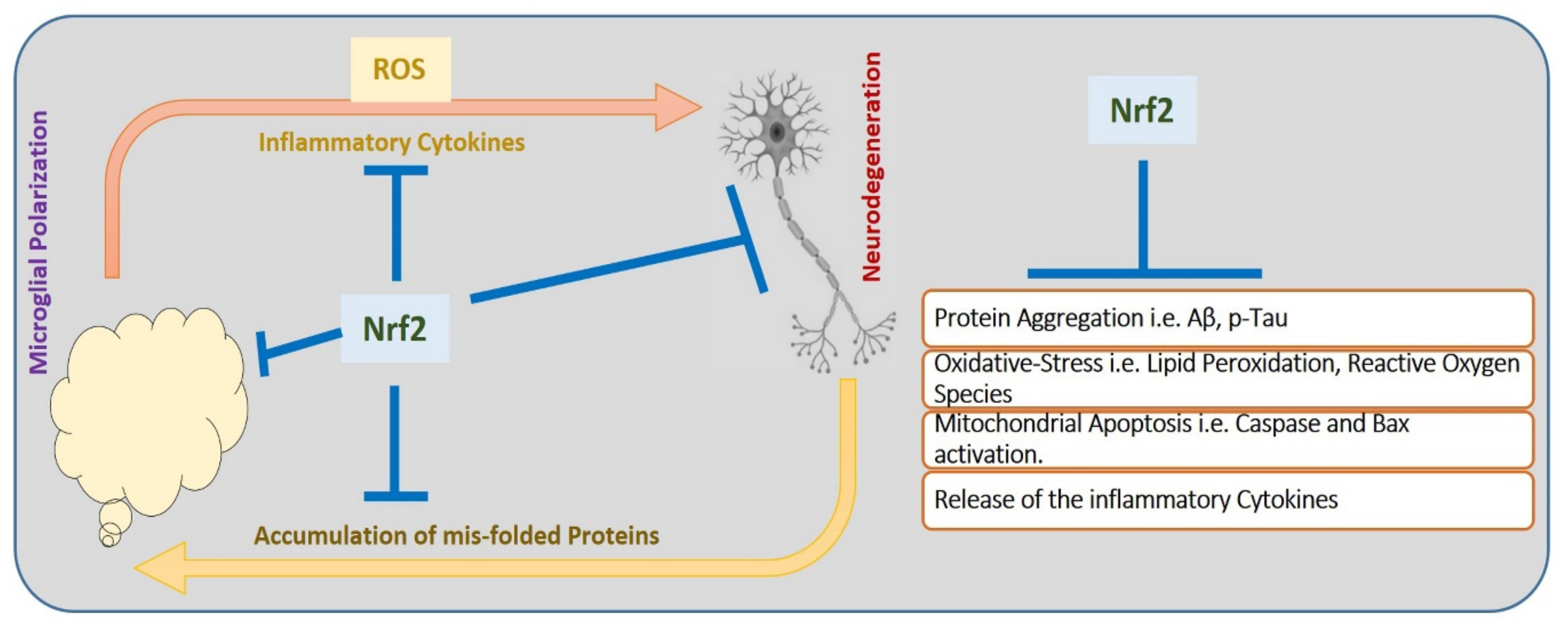
5. Oxidative Stress, Nrf-2, and Proteases Expression in COVID-19 and Neurodegenerative Conditions
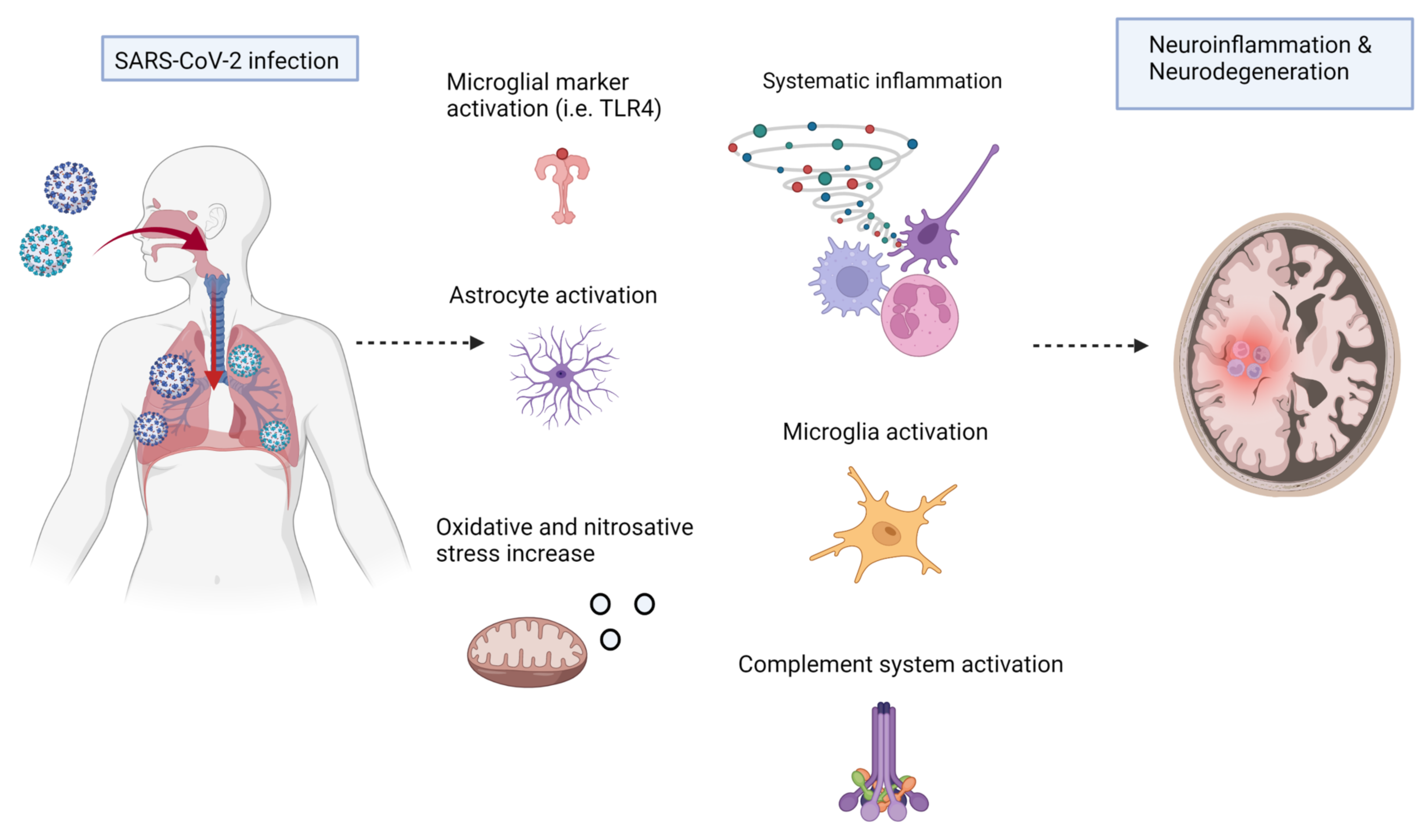
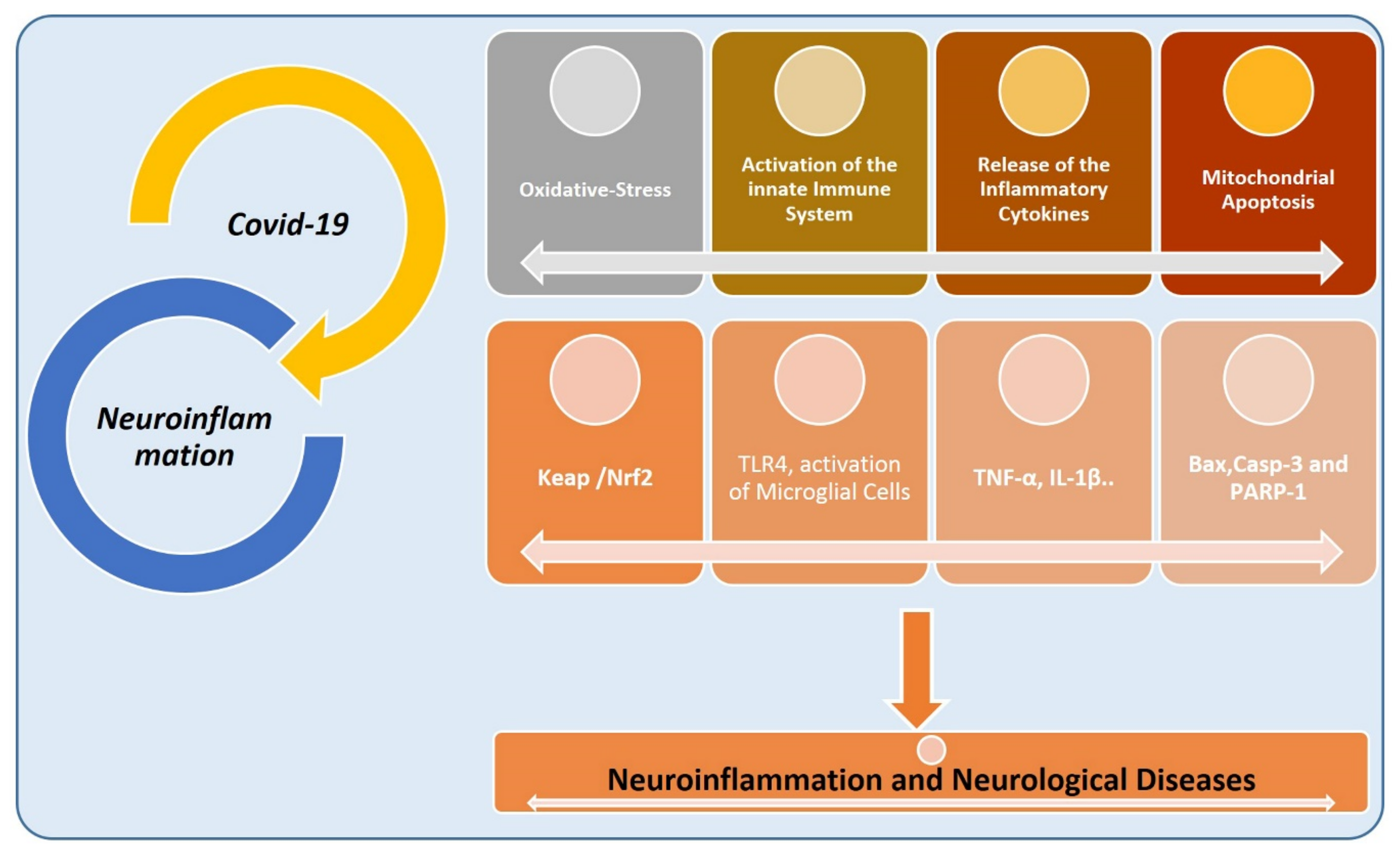
6. COVID-19, Inflammation, and Neurodegenerative Diseases
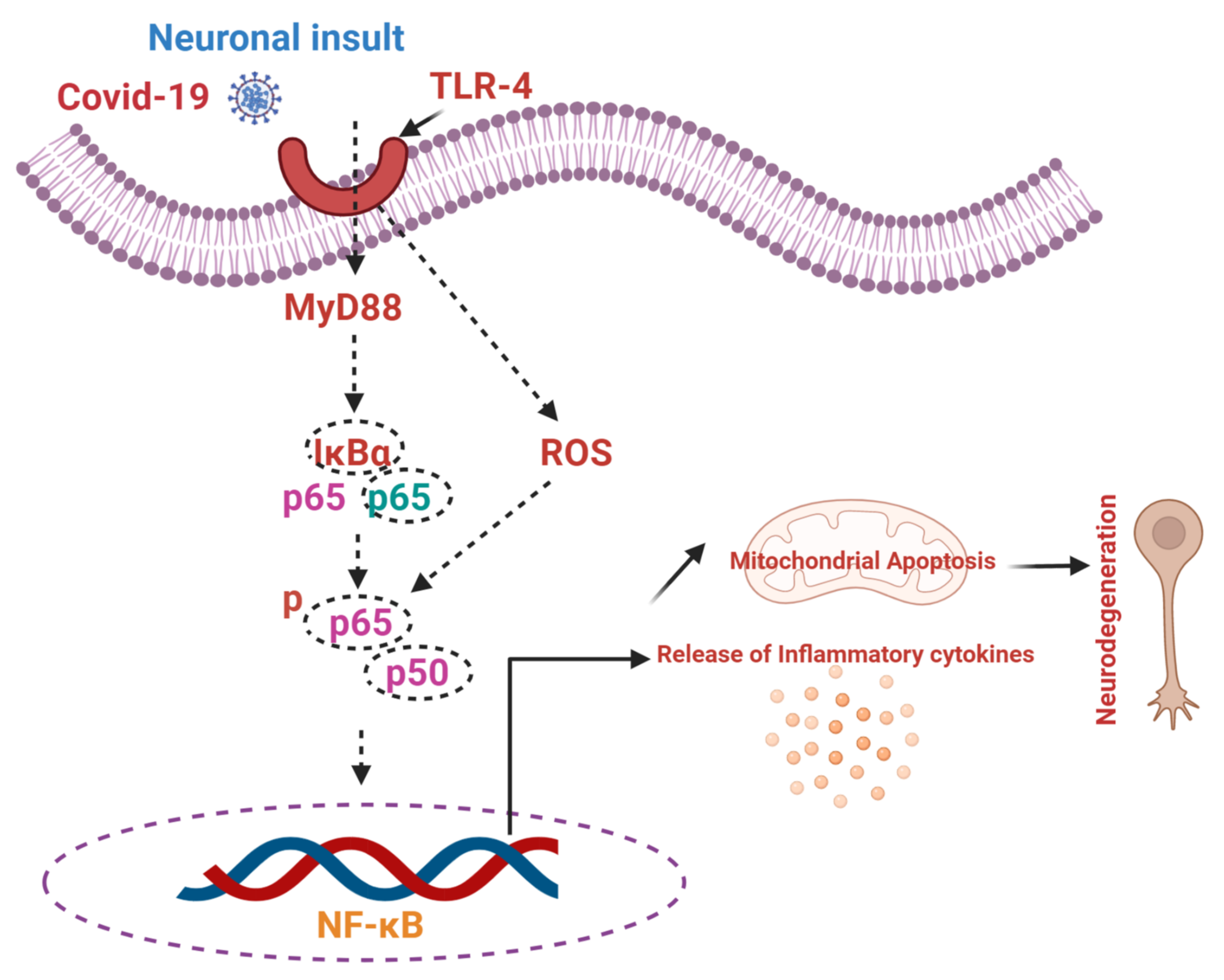
7. COVID-19, NF-κB, and Neurodegenerative Conditions
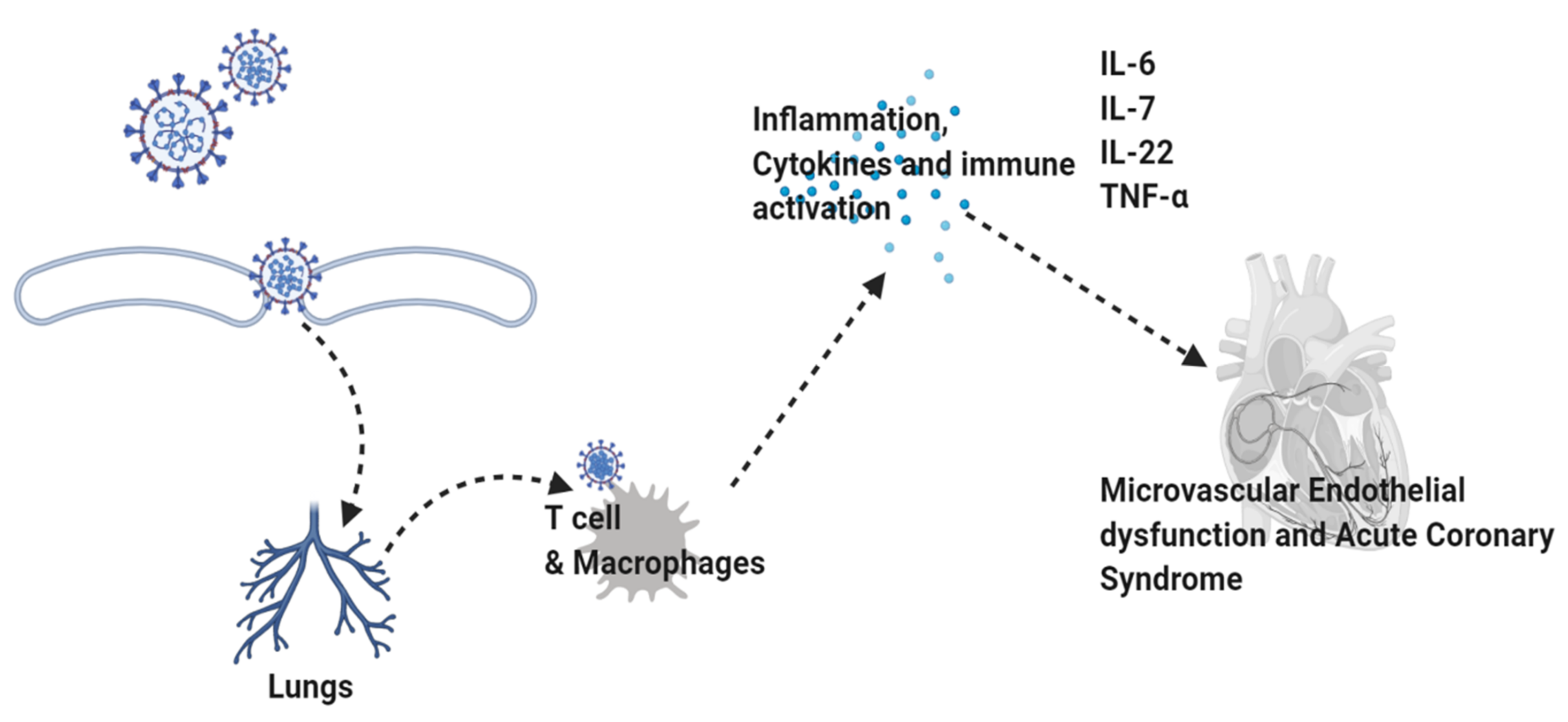
8. COVID-19, TNFα, and Neurodegenerative Conditions
9. COVID-19, mTOR, and Neurodegenerative Conditions
10. COVID-19, and Accumulation of Misfolded Proteins
11. Antioxidants and Their Effects against Neuroinflammation
12. Sulforaphane against Neurodegeneration and COVID-19
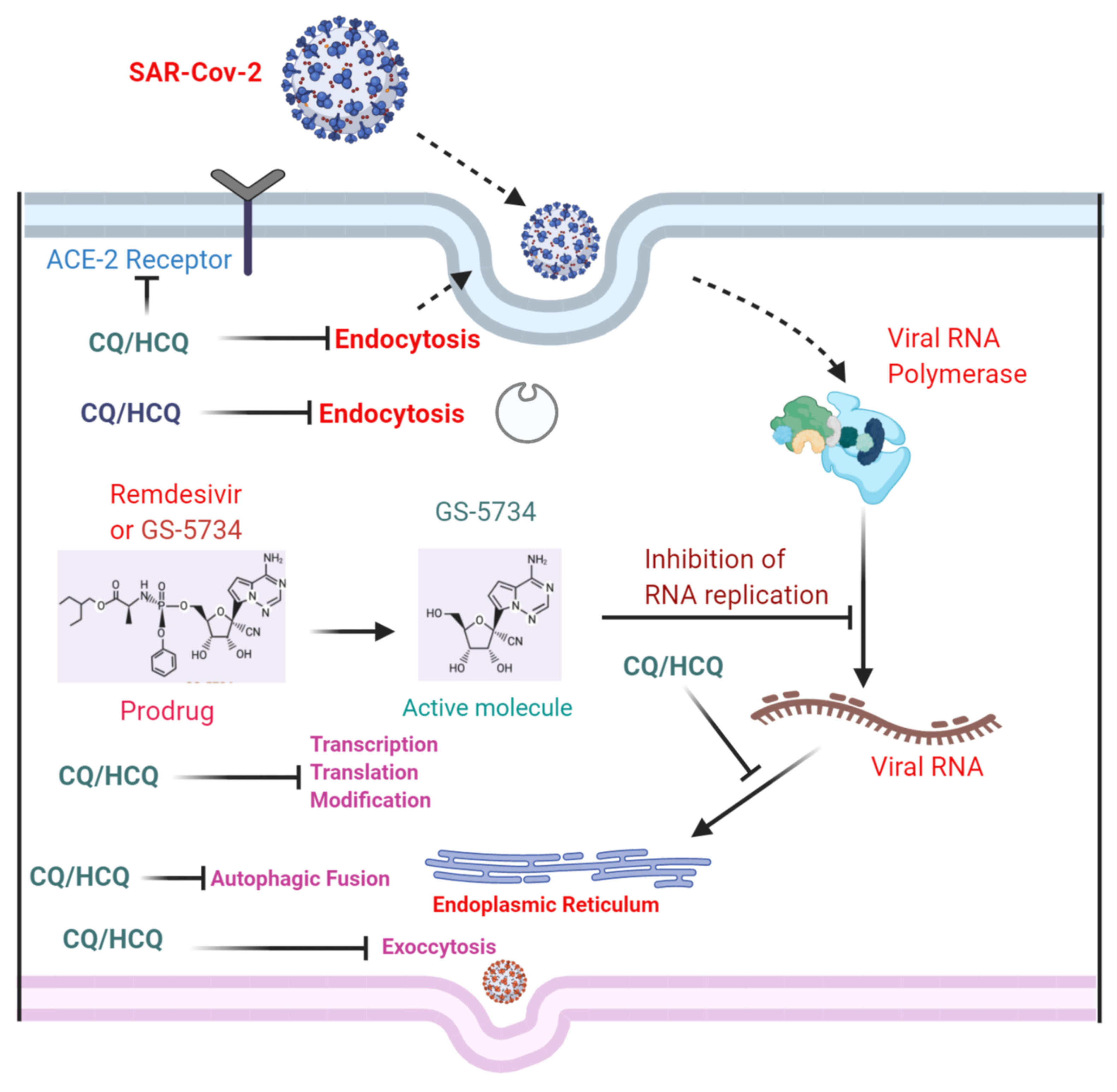
13. Chloroquine and Hydroxychloroquine
14. Melatonin against COVID-19 and Neurodegeneration
15. Conclusions, Gaps, and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bhat, E.A.; Khan, J.; Sajjad, N.; Ali, A.; Aldakeel, F.M.; Mateen, A.; Alqahtani, M.S.; Syed, R. SARS-CoV-2: Insight in genome structure, pathogenesis and viral receptor binding analysis—An updated review. Int. Immunopharmacol. 2021, 95, 107493. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Nabi, G.; Zhang, T.; Wu, Y.; Li, D. Potential Neurochemical and Neuroendocrine Effects of Social Distancing Amidst the COVID-19 Pandemic. Front. Endocrinol. 2020, 11, 582288. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [Green Version]
- Shumway, B.; Ibrahim, D.; Moss, W. Monitoring Returning Travelers During the Early Weeks of the COVID-19 Pandemic: One US County’s Experience. Am. J. Public Health 2020, 110, 962–963. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Heymann, D.L. Data sharing and outbreaks: Best practice exemplified. Lancet 2020, 395, 469–470. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Mubarak, A.; Alturaiki, W.; Hemida, M.G. Middle East Respiratory Syndrome Coronavirus (MERS-CoV): Infection, Immunological Response, and Vaccine Development. J. Immunol. Res. 2019, 2019, 6491738. [Google Scholar] [CrossRef]
- Nassar, M.S.; Bakhrebah, M.A.; Meo, S.A.; Alsuabeyl, M.S.; Zaher, W.A. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) infection: Epidemiology, pathogenesis and clinical characteristics. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 4956–4961. [Google Scholar] [CrossRef]
- Azhar, E.I.; Lanini, S.; Ippolito, G.; Zumla, A. The Middle East Respiratory Syndrome Coronavirus—A Continuing Risk to Global Health Security. Adv. Exp. Med. Biol. 2017, 972, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabi, Y.M.; Balkhy, H.H.; Hayden, F.G.; Bouchama, A.; Luke, T.; Baillie, J.K.; Al-Omari, A.; Hajeer, A.H.; Senga, M.; Denison, M.R.; et al. Middle East Respiratory Syndrome. N. Engl. J. Med. 2017, 376, 584–594. [Google Scholar] [CrossRef] [PubMed]
- Seddiq, N.; Al-Qahtani, M.; Al-Tawfiq, J.A.; Bukamal, N. First Confirmed Case of Middle East Respiratory Syndrome Coronavirus Infection in the Kingdom of Bahrain: In a Saudi Gentleman after Cardiac Bypass Surgery. Case Rep. Infect. Dis. 2017, 2017, 1262838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Peng, F.; Wang, R.; Guan, K.; Jiang, T.; Xu, G.; Sun, J.; Chang, C. The deadly coronaviruses: The 2003 SARS pandemic and the 2020 novel coronavirus epidemic in China. J. Autoimmun. 2020, 109, 102434. [Google Scholar] [CrossRef]
- Brian, D.A.; Baric, R.S. Coronavirus genome structure and replication. Curr. Top Microbiol. Immunol. 2005, 287, 1–30. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M. Coronavirus: Organization, replication and expression of genome. Annu. Rev. Microbiol. 1990, 44, 303–333. [Google Scholar] [CrossRef]
- Morales, L.; Mateos-Gomez, P.A.; Capiscol, C.; del Palacio, L.; Enjuanes, L.; Sola, I. Transmissible gastroenteritis coronavirus genome packaging signal is located at the 5′ end of the genome and promotes viral RNA incorporation into virions in a replication-independent process. J. Virol. 2013, 87, 11579–11590. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.L.; Guo, D.; Rottier, P.J. Coronavirus: Epidemiology, genome replication and the interactions with their hosts. Virol. Sin. 2016, 31, 1–2. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Yan, M.; Xu, H.; Liang, W.; Kan, B.; Zheng, B.; Chen, H.; Zheng, H.; Xu, Y.; Zhang, E.; et al. SARS-CoV infection in a restaurant from palm civet. Emerg. Infect. Dis. 2005, 11, 1860–1865. [Google Scholar] [CrossRef]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Simmons, G.; Zmora, P.; Gierer, S.; Heurich, A.; Pohlmann, S. Proteolytic activation of the SARS-coronavirus spike protein: Cutting enzymes at the cutting edge of antiviral research. Antiviral. Res. 2013, 100, 605–614. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Sharma, A.; Tiwari, S.; Deb, M.K.; Marty, J.L. Severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2): A global pandemic and treatment strategies. Int. J. Antimicrob. Agents 2020, 56, 106054. [Google Scholar] [CrossRef]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Gu, J.; Korteweg, C. Pathology and pathogenesis of severe acute respiratory syndrome. Am. J. Pathol. 2007, 170, 1136–1147. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Gu, J.; Gong, E.; Zhang, B.; Zheng, J.; Gao, Z.; Zhong, Y.; Zou, W.; Zhan, J.; Wang, S.; Xie, Z.; et al. Multiple organ infection and the pathogenesis of SARS. J. Exp. Med. 2005, 202, 415–424. [Google Scholar] [CrossRef]
- Li, F. Receptor recognition and cross-species infections of SARS coronavirus. Antivir. Res. 2013, 100, 246–254. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Jing, X.; Xu, M.; Song, D.; Yue, T.; Wang, Y.; Zhang, P.; Zhong, Y.; Zhang, M.; Lam, T.T.; Faria, N.R.; et al. Association between inflammatory cytokines and anti-SARS-CoV-2 antibodies in hospitalized patients with COVID-19. Immun. Ageing 2022, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Ng Kee Kwong, K.C.; Mehta, P.R.; Shukla, G.; Mehta, A.R. COVID-19, SARS and MERS: A neurological perspective. J. Clin. Neurosci. 2020, 77, 13–16. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T.; Sun, W.; Wang, D.; Fu, B.; Zhou, Y.; Zheng, X.; Yang, Y.; Li, X.; et al. Effective treatment of severe COVID-19 patients with tocilizumab. Proc. Natl. Acad. Sci. USA 2020, 117, 10970–10975. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Curran, K.; Chung, S.S.; Stefanski, J.; Borquez-Ojeda, O.; Olszewska, M.; et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci. Transl. Med. 2014, 6, 224ra25. [Google Scholar] [CrossRef] [Green Version]
- Feldmann, M.; Maini, R.N.; Woody, J.N.; Holgate, S.T.; Winter, G.; Rowland, M.; Richards, D.; Hussell, T. Trials of anti-tumour necrosis factor therapy for COVID-19 are urgently needed. Lancet 2020, 395, 1407–1409. [Google Scholar] [CrossRef]
- Razmi, M.; Hashemi, F.; Gheytanchi, E.; Dehghan Manshadi, M.; Ghods, R.; Madjd, Z. Immunomodulatory-based therapy as a potential promising treatment strategy against severe COVID-19 patients: A systematic review. Int. Immunopharmacol. 2020, 88, 106942. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef]
- Jo, M.G.; Ikram, M.; Jo, M.H.; Yoo, L.; Chung, K.C.; Nah, S.-Y.; Hwang, H.; Rhim, H.; Kim, M.O. Gintonin Mitigates MPTP-Induced Loss of Nigrostriatal Dopaminergic Neurons and Accumulation of α-Synuclein via the Nrf2/HO-1 Pathway. Mol. Neurobiol. 2019, 56, 39–55. [Google Scholar] [CrossRef]
- Ikram, M.; Jo, M.G.; Park, T.J.; Kim, M.W.; Khan, I.; Jo, M.H.; Kim, M.O. Oral Administration of Gintonin Protects the Brains of Mice against Aβ-Induced Alzheimer Disease Pathology: Antioxidant and Anti-Inflammatory Effects. Oxidative Med. Cell. Longev. 2021, 2021, 6635552. [Google Scholar] [CrossRef]
- Muhammad, T.; Ikram, M.; Ullah, R.; Rehman, S.U.; Kim, M.O. Hesperetin, a citrus flavonoid, attenuates LPS-induced neuroinflammation, apoptosis and memory impairments by modulating TLR4/NF-κB signaling. Nutrients 2019, 11, 648. [Google Scholar] [CrossRef] [Green Version]
- More, S.V.; Kumar, H.; Kim, I.S.; Song, S.Y.; Choi, D.K. Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediat. Inflamm. 2013, 2013, 952375. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J. COVID-19 and Alzheimer’s disease: How one crisis worsens the other. Transl. Neurodegener 2021, 10, 15. [Google Scholar] [CrossRef]
- Miners, S.; Kehoe, P.G.; Love, S. Cognitive impact of COVID-19: Looking beyond the short term. Alzheimers Res. Ther. 2020, 12, 170. [Google Scholar] [CrossRef]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [Green Version]
- Arabi, Y.M.; Harthi, A.; Hussein, J.; Bouchama, A.; Johani, S.; Hajeer, A.H.; Saeed, B.T.; Wahbi, A.; Saedy, A.; AlDabbagh, T.; et al. Severe neurologic syndrome associated with Middle East respiratory syndrome corona virus (MERS-CoV). Infection 2015, 43, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Amin, F.U.; Hoshiar, A.K.; Do, T.D.; Noh, Y.; Shah, S.A.; Khan, M.S.; Yoon, J.; Kim, M.O.J.N. Osmotin-loaded magnetic nanoparticles with electromagnetic guidance for the treatment of Alzheimer’s disease. Nanoscale 2017, 9, 10619–10632. [Google Scholar] [CrossRef] [Green Version]
- Natoli, S.; Oliveira, V.; Calabresi, P.; Maia, L.F.; Pisani, A. Does SARS-CoV-2 invade the brain? Translational lessons from animal models. Eur. J. Neurol. 2020, 27, 1764–1773. [Google Scholar] [CrossRef]
- Serrano-Castro, P.J.; Estivill-Torrus, G.; Cabezudo-Garcia, P.; Reyes-Bueno, J.A.; Ciano Petersen, N.; Aguilar-Castillo, M.J.; Suarez-Perez, J.; Jimenez-Hernandez, M.D.; Moya-Molina, M.A.; Oliver-Martos, B.; et al. Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: A delayed pandemic? Neurologia 2020, 35, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.C.; Bai, W.Z.; Hashikawa, T. The neuroinvasive potential of SARS-CoV2 may play a role in the respiratory failure of COVID-19 patients. J. Med. Virol. 2020, 92, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Guan, J. Novel coronavirus and the central nervous system. Eur. J. Neurol. 2020, 27, e52. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xu, X.; Chen, Z.; Duan, J.; Hashimoto, K.; Yang, L.; Liu, C.; Yang, C. Nervous system involvement after infection with COVID-19 and other coronaviruses. Brain Behav. Immun. 2020, 87, 18–22. [Google Scholar] [CrossRef]
- Zhang, Y.; Geng, X.; Tan, Y.; Li, Q.; Xu, C.; Xu, J.; Hao, L.; Zeng, Z.; Luo, X.; Liu, F.; et al. New understanding of the damage of SARS-CoV-2 infection outside the respiratory system. Biomed. Pharmacother. 2020, 127, 110195. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, M.; Wang, J.; Gao, J. SARS-CoV-2: Underestimated damage to nervous system. Travel Med. Infect. Dis. 2020, 36, 101642. [Google Scholar] [CrossRef]
- Li, G.; Fan, Y.; Lai, Y.; Han, T.; Li, Z.; Zhou, P.; Pan, P.; Wang, W.; Hu, D.; Liu, X.; et al. Coronavirus infections and immune responses. J. Med. Virol. 2020, 92, 424–432. [Google Scholar] [CrossRef]
- Costa Sa, A.C.; Madsen, H.; Brown, J.R. Shared Molecular Signatures Across Neurodegenerative Diseases and Herpes Virus Infections Highlights Potential Mechanisms for Maladaptive Innate Immune Responses. Sci. Rep. 2019, 9, 8795. [Google Scholar] [CrossRef]
- Lester, S.N.; Li, K. Toll-like receptors in antiviral innate immunity. J. Mol. Biol. 2014, 426, 1246–1264. [Google Scholar] [CrossRef]
- Paik, S.; Kim, J.K.; Silwal, P.; Sasakawa, C.; Jo, E.K. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol. Immunol. 2021, 18, 1141–1160. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural dietary supplementation of anthocyanins via PI3K/Akt/Nrf2/HO-1 pathways mitigate oxidative stress, neurodegeneration, and memory impairment in a mouse model of Alzheimer’s disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- O’Connell, M.A.; Hayes, J.D. The Keap1/Nrf2 pathway in health and disease: From the bench to the clinic. Biochem. Soc. Trans. 2015, 43, 687–689. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Ma, Q. NRF2 cysteine residues are critical for oxidant/electrophile-sensing, Kelch-like ECH-associated protein-1-dependent ubiquitination-proteasomal degradation, and transcription activation. Mol. Pharmacol. 2009, 76, 1265–1278. [Google Scholar] [CrossRef] [Green Version]
- Das, B.N.; Kim, Y.W.; Keum, Y.S. Mechanisms of Nrf2/Keap1-dependent phase II cytoprotective and detoxifying gene expression and potential cellular targets of chemopreventive isothiocyanates. Oxid. Med. Cell. Longev. 2013, 2013, 839409. [Google Scholar] [CrossRef] [Green Version]
- Battino, M.; Giampieri, F.; Pistollato, F.; Sureda, A.; de Oliveira, M.R.; Pittala, V.; Fallarino, F.; Nabavi, S.F.; Atanasov, A.G.; Nabavi, S.M. Nrf2 as regulator of innate immunity: A molecular Swiss army knife! Biotechnol. Adv. 2018, 36, 358–370. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, A.; Ali, W.; Jo, M.H.; Park, J.; Ikram, M.; Kim, M.O. Fisetin rescues the mice brains against D-galactose-induced oxidative stress, neuroinflammation and memory impairment. Front. Pharmacol. 2021, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Shulla, A.; Heald-Sargent, T.; Subramanya, G.; Zhao, J.; Perlman, S.; Gallagher, T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011, 85, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuoka, S.; Ohnishi, T.; Kawano, S.; Tsuchihashi, S.; Ogawara, M.; Masuda, K.; Yamaoka, K.; Takahashi, M.; Sano, T. Purification, characterization, and localization of a novel trypsin-like protease found in the human airway. Am. J. Respir. Cell Mol. Biol. 1997, 16, 300–308. [Google Scholar] [CrossRef]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ikram, M.; Ullah, N.; Alam, S.I.; Park, H.Y.; Badshah, H.; Choe, K.; Ok Kim, M.J.C. Neurological enhancement effects of melatonin against brain injury-induced oxidative stress, neuroinflammation, and neurodegeneration via AMPK/CREB signaling. Cells 2019, 8, 760. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Lee, J.H.; Gaire, B.P.; Kim, S.Y. Sulforaphane Inhibits MGO-AGE-Mediated Neuroinflammation by Suppressing NF-kappaB, MAPK, and AGE-RAGE Signaling Pathways in Microglial Cells. Antioxidants 2020, 9, 792. [Google Scholar] [CrossRef]
- Casili, G.; Campolo, M.; Paterniti, I.; Lanza, M.; Filippone, A.; Cuzzocrea, S.; Esposito, E. Dimethyl Fumarate Attenuates Neuroinflammation and Neurobehavioral Deficits Induced by Experimental Traumatic Brain Injury. J. Neurotrauma 2018, 35, 1437–1451. [Google Scholar] [CrossRef]
- Lammi, C.; Arnoldi, A. Food-derived antioxidants and COVID-19. J. Food Biochem. 2020, 45, e13557. [Google Scholar] [CrossRef]
- Laurendon, T.; Radulesco, T.; Mugnier, J.; Gerault, M.; Chagnaud, C.; El Ahmadi, A.A.; Varoquaux, A. Bilateral transient olfactory bulb edema during COVID-19-related anosmia. Neurology 2020, 95, 224–225. [Google Scholar] [CrossRef]
- Balasubramanian, S.K.; Poh, K.W.; Ong, C.N.; Kreyling, W.G.; Ong, W.Y.; Yu, L.E. The effect of primary particle size on biodistribution of inhaled gold nano-agglomerates. Biomaterials 2013, 34, 5439–5452. [Google Scholar] [CrossRef]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Wang, B.; Yuan, T.; Chen, X.; Ao, Y.; Fitzpatrick, T.; Li, P.; Zhou, Y.; Lin, Y.F.; Duan, Q.; et al. Clinical characteristics of coronavirus disease 2019 (COVID-19) in China: A systematic review and meta-analysis. J. Infect. 2020, 80, 656–665. [Google Scholar] [CrossRef]
- Bao, J.; Li, C.; Zhang, K.; Kang, H.; Chen, W.; Gu, B. Comparative analysis of laboratory indexes of severe and non-severe patients infected with COVID-19. Clin. Chim. Acta 2020, 509, 180–194. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, Y.; Zhang, F.; Wang, Q.; Li, T.; Liu, Z.; Wang, J.; Qin, Y.; Zhang, X.; Yan, X.; et al. The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): The Perspectives of clinical immunologists from China. Clin. Immunol. 2020, 214, 108393. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.S.; Muhammad, T.; Ikram, M.; Kim, M.O. Dietary supplementation of the antioxidant curcumin halts systemic LPS-induced neuroinflammation-associated neurodegeneration and memory/synaptic impairment via the JNK/NF-κB/Akt signaling pathway in adult rats. Oxidative Med. Cell. Longev. 2019, 2019, 7860650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amor, S.; Puentes, F.; Baker, D.; van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Arcuri, C.; Mecca, C.; Bianchi, R.; Giambanco, I.; Donato, R. The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front. Mol. Neurosci. 2017, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Minghetti, L. Role of inflammation in neurodegenerative diseases. Curr. Opin. Neurol. 2005, 18, 315–321. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kar, R.; Nand, P.; Swati, K.; Goswami, V.K. Nuclear factor-kappa beta as a therapeutic target for Alzheimer’s disease. J. Neurochem. 2019, 150, 113–137. [Google Scholar] [CrossRef] [Green Version]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Mattson, M.P.; Camandola, S. NF-kappaB in neuronal plasticity and neurodegenerative disorders. J. Clin. Investig. 2001, 107, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Kong, F.; Jiang, X.; Wang, R.; Zhai, S.; Zhang, Y.; Wang, D. Forsythoside B attenuates memory impairment and neuroinflammation via inhibition on NF-kappaB signaling in Alzheimer’s disease. J. Neuroinflamm. 2020, 17, 305. [Google Scholar] [CrossRef]
- Laforge, M.; Rodrigues, V.; Silvestre, R.; Gautier, C.; Weil, R.; Corti, O.; Estaquier, J. NF-kappaB pathway controls mitochondrial dynamics. Cell Death Differ. 2016, 23, 89–98. [Google Scholar] [CrossRef]
- Cheon, S.Y.; Kim, J.M.; Kam, E.H.; Ho, C.C.; Kim, E.J.; Chung, S.; Jeong, J.H.; Lee, D.D.; Lee, S.W.; Koo, B.N. Cell-penetrating interactomic inhibition of nuclear factor-kappa B in a mouse model of postoperative cognitive dysfunction. Sci. Rep. 2017, 7, 13482. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Zhou, W.; Liu, S.; Deng, Y.; Cai, F.; Tone, M.; Tone, Y.; Tong, Y.; Song, W. Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int. J. Neuropsychopharmacol. 2012, 15, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Liao, Q.J.; Ye, L.B.; Timani, K.A.; Zeng, Y.C.; She, Y.L.; Ye, L.; Wu, Z.H. Activation of NF-kappaB by the full-length nucleocapsid protein of the SARS coronavirus. Acta Biochim. Biophys. Sin. 2005, 37, 607–612. [Google Scholar] [CrossRef] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Regla-Nava, J.A.; Jimenez-Guardeno, J.M.; Fernandez-Delgado, R.; Fett, C.; Castano-Rodriguez, C.; Perlman, S.; Enjuanes, L. Inhibition of NF-kappaB-mediated inflammation in severe acute respiratory syndrome coronavirus-infected mice increases survival. J. Virol. 2014, 88, 913–924. [Google Scholar] [CrossRef] [Green Version]
- Day, C.W.; Baric, R.; Cai, S.X.; Frieman, M.; Kumaki, Y.; Morrey, J.D.; Smee, D.F.; Barnard, D.L. A new mouse-adapted strain of SARS-CoV as a lethal model for evaluating antiviral agents in vitro and in vivo. Virology 2009, 395, 210–222. [Google Scholar] [CrossRef] [Green Version]
- Dosch, S.F.; Mahajan, S.D.; Collins, A.R. SARS coronavirus spike protein-induced innate immune response occurs via activation of the NF-kappaB pathway in human monocyte macrophages in vitro. Virus Res. 2009, 142, 19–27. [Google Scholar] [CrossRef]
- De Wit, E.; van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [CrossRef]
- Santello, M.; Bezzi, P.; Volterra, A. TNFalpha controls glutamatergic gliotransmission in the hippocampal dentate gyrus. Neuron 2011, 69, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yao, C.H. The Physiological Role of Tumor Necrosis Factor in Human Immunity and Its Potential Implications in Spinal Manipulative Therapy: A Narrative Literature Review. J. Chiropr. Med. 2016, 15, 190–196. [Google Scholar] [CrossRef] [Green Version]
- Hanisch, U.K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Frei, K.; Siepl, C.; Groscurth, P.; Bodmer, S.; Schwerdel, C.; Fontana, A. Antigen presentation and tumor cytotoxicity by interferon-gamma-treated microglial cells. Eur. J. Immunol. 1987, 17, 1271–1278. [Google Scholar] [CrossRef] [PubMed]
- Byun, K.; Young Kim, J.; Bayarsaikhan, E.; Kim, D.; Jeong, G.B.; Yun, K.N.; Kyeong Min, H.; Kim, S.U.; Yoo, J.S.; Lee, B. Quantitative proteomic analysis reveals that lipopolysaccharide induces mitogen-activated protein kinase-dependent activation in human microglial cells. Electrophoresis 2012, 33, 3756–3763. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Zhang, L.; Lian, G.; Wang, X.; Zhang, H.; Yao, X.; Yang, J.; Wu, C. Sildenafil attenuates LPS-induced pro-inflammatory responses through down-regulation of intracellular ROS-related MAPK/NF-kappaB signaling pathways in N9 microglia. Int. Immunopharmacol. 2011, 11, 468–474. [Google Scholar] [CrossRef]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef] [Green Version]
- Chau, B.N.; Chen, T.T.; Wan, Y.Y.; DeGregori, J.; Wang, J.Y. Tumor necrosis factor alpha-induced apoptosis requires p73 and c-ABL activation downstream of RB degradation. Mol. Cell. Biol. 2004, 24, 4438–4447. [Google Scholar] [CrossRef] [Green Version]
- Decourt, B.; Lahiri, D.K.; Sabbagh, M.N. Targeting Tumor Necrosis Factor Alpha for Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 412–425. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Zheng, Y.; Chen, X. Drugs for Autoimmune Inflammatory Diseases: From Small Molecule Compounds to Anti-TNF Biologics. Front. Pharmacol. 2017, 8, 460. [Google Scholar] [CrossRef]
- Wallis, R.S.; Broder, M.S.; Wong, J.Y.; Hanson, M.E.; Beenhouwer, D.O. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin. Infect. Dis. 2004, 38, 1261–1265. [Google Scholar] [CrossRef] [Green Version]
- Roda, G.; Jharap, B.; Neeraj, N.; Colombel, J.F. Loss of Response to Anti-TNFs: Definition, Epidemiology, and Management. Clin. Transl. Gastroenterol. 2016, 7, e135. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Zhang, Q.; Tan, L.; Xu, Y.; Xie, X.; Zhao, Y. The Regulatory Effects of mTOR Complexes in the Differentiation and Function of CD4(+) T Cell Subsets. J. Immunol. Res. 2020, 2020, 3406032. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Reis, F.; Mishra, P.K. mTOR Signaling in Cardiometabolic Disease, Cancer, and Aging 2018. Oxid. Med. Cell. Longev. 2019, 2019, 9692528. [Google Scholar] [CrossRef]
- Ranadheera, C.; Coombs, K.M.; Kobasa, D. Comprehending a Killer: The Akt/mTOR Signaling Pathways Are Temporally High-Jacked by the Highly Pathogenic 1918 Influenza Virus. EBioMedicine 2018, 32, 142–163. [Google Scholar] [CrossRef]
- Taylor, H.E.; Calantone, N.; Lichon, D.; Hudson, H.; Clerc, I.; Campbell, E.M.; D’Aquila, R.T. mTOR Overcomes Multiple Metabolic Restrictions to Enable HIV-1 Reverse Transcription and Intracellular Transport. Cell Rep. 2020, 31, 107810. [Google Scholar] [CrossRef]
- Kindrachuk, J.; Ork, B.; Hart, B.J.; Mazur, S.; Holbrook, M.R.; Frieman, M.B.; Traynor, D.; Johnson, R.F.; Dyall, J.; Kuhn, J.H.; et al. Antiviral potential of ERK/MAPK and PI3K/AKT/mTOR signaling modulation for Middle East respiratory syndrome coronavirus infection as identified by temporal kinome analysis. Antimicrob. Agents Chemother. 2015, 59, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Lehrer, S. Inhaled biguanides and mTOR inhibition for influenza and coronavirus (Review). World Acad. Sci. J. 2020, 2, 1. [Google Scholar] [CrossRef]
- Appelberg, S.; Gupta, S.; Svensson Akusjarvi, S.; Ambikan, A.T.; Mikaeloff, F.; Saccon, E.; Vegvari, A.; Benfeitas, R.; Sperk, M.; Stahlberg, M.; et al. Dysregulation in Akt/mTOR/HIF-1 signaling identified by proteo-transcriptomics of SARS-CoV-2 infected cells. Emerg. Microbes Infect. 2020, 9, 1748–1760. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef]
- Khan, N.; Chen, X.; Geiger, J.D. Possible Therapeutic Use of Natural Compounds against COVID-19. J. Cell. Signal. 2021, 2, 63–79. [Google Scholar]
- Martin, A.R.; Pollack, R.A.; Capoferri, A.; Ambinder, R.F.; Durand, C.M.; Siliciano, R.F. Rapamycin-mediated mTOR inhibition uncouples HIV-1 latency reversal from cytokine-associated toxicity. J. Clin. Investig. 2017, 127, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Pereira, G.; Leao, A.; Erustes, A.G.; Morais, I.B.M.; Vrechi, T.A.M.; Zamarioli, L.D.S.; Pereira, C.A.S.; Marchioro, L.O.; Sperandio, L.P.; Lins, I.V.F.; et al. Pharmacological Modulators of Autophagy as a Potential Strategy for the Treatment of COVID-19. Int. J. Mol. Sci. 2021, 22, 4067. [Google Scholar] [CrossRef]
- Munoz-Esparza, N.C.; Latorre-Moratalla, M.L.; Comas-Baste, O.; Toro-Funes, N.; Veciana-Nogues, M.T.; Vidal-Carou, M.C. Polyamines in Food. Front. Nutr. 2019, 6, 108. [Google Scholar] [CrossRef]
- Jobgen, W.S.; Fried, S.K.; Fu, W.J.; Meininger, C.J.; Wu, G. Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J. Nutr. Biochem. 2006, 17, 571–588. [Google Scholar] [CrossRef]
- Xu, T.T.; Li, H.; Dai, Z.; Lau, G.K.; Li, B.Y.; Zhu, W.L.; Liu, X.Q.; Liu, H.F.; Cai, W.W.; Huang, S.Q.; et al. Spermidine and spermine delay brain aging by inducing autophagy in SAMP8 mice. Aging 2020, 12, 6401–6414. [Google Scholar] [CrossRef]
- Sigrist, S.J.; Carmona-Gutierrez, D.; Gupta, V.K.; Bhukel, A.; Mertel, S.; Eisenberg, T.; Madeo, F. Spermidine-triggered autophagy ameliorates memory during aging. Autophagy 2014, 10, 178–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.H.; Park, H.Y. Anti-inflammatory effects of spermidine in lipopolysaccharide-stimulated BV2 microglial cells. J. Biomed. Sci. 2012, 19, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skatchkov, S.N.; Woodbury-Farina, M.A.; Eaton, M. The role of glia in stress: Polyamines and brain disorders. Psychiatr. Clin. N. Am. 2014, 37, 653–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chen, S.; Zhang, Y.; Lin, X.; Song, Y.; Xue, Z.; Qian, H.; Wang, S.; Wan, G.; Zheng, X.; et al. Induction of autophagy by spermidine is neuroprotective via inhibition of caspase 3-mediated Beclin 1 cleavage. Cell Death Dis. 2017, 8, e2738. [Google Scholar] [CrossRef]
- Li, G.; Ding, H.; Yu, X.; Meng, Y.; Li, J.; Guo, Q.; Zhou, H.; Shen, N. Spermidine Suppresses Inflammatory DC Function by Activating the FOXO3 Pathway and Counteracts Autoimmunity. iScience 2020, 23, 100807. [Google Scholar] [CrossRef] [Green Version]
- Salehi, B.; Mishra, A.P.; Nigam, M.; Sener, B.; Kilic, M.; Sharifi-Rad, M.; Fokou, P.V.T.; Martins, N.; Sharifi-Rad, J. Resveratrol: A Double-Edged Sword in Health Benefits. Biomedicines 2018, 6, 91. [Google Scholar] [CrossRef] [Green Version]
- Romero-Perez, A.I.; Ibern-Gomez, M.; Lamuela-Raventos, R.M.; de La Torre-Boronat, M.C. Piceid, the major resveratrol derivative in grape juices. J. Agric. Food Chem. 1999, 47, 1533–1536. [Google Scholar] [CrossRef]
- Jiang, H.; Shang, X.; Wu, H.; Gautam, S.C.; Al-Holou, S.; Li, C.; Kuo, J.; Zhang, L.; Chopp, M. Resveratrol downregulates PI3K/Akt/mTOR signaling pathways in human U251 glioma cells. J. Exp. Ther. Oncol. 2009, 8, 25–33. [Google Scholar]
- Tian, Y.; Song, W.; Li, D.; Cai, L.; Zhao, Y. Resveratrol as a Natural Regulator of Autophagy for Prevention and Treatment of Cancer. Onco. Targets Ther. 2019, 12, 8601–8609. [Google Scholar] [CrossRef] [Green Version]
- Ter Ellen, B.M.; Kumar, N.D.; Bouma, E.M.; Troost, B.; van de Pol, D.P.; van der Ende-Metselaar, H.H.; Apperloo, L.; van Gosliga, D.; van den Berge, M.; Nawijn, M.C. Resveratrol and pterostilbene potently inhibit SARS-CoV-2 infection in vitro. bioRxiv 2020. [Google Scholar] [CrossRef]
- Marinella, M.A. Indomethacin and resveratrol as potential treatment adjuncts for SARS-CoV-2/COVID-19. Int. J. Clin. Pract. 2020, 74, e13535. [Google Scholar] [CrossRef]
- Russell, B.; Moss, C.; Rigg, A.; Van Hemelrijck, M. COVID-19 and treatment with NSAIDs and corticosteroids: Should we be limiting their use in the clinical setting? Ecancermedicalscience 2020, 14, 1023. [Google Scholar] [CrossRef] [Green Version]
- Kelleni, M.T. Early use of non-steroidal anti-inflammatory drugs in COVID-19 might reverse pathogenesis, prevent complications and improve clinical outcomes. Biomed. Pharmacother. 2021, 133, 110982. [Google Scholar] [CrossRef]
- Lipton, S.A.; Gu, Z.; Nakamura, T. Inflammatory mediators leading to protein misfolding and uncompetitive/fast off-rate drug therapy for neurodegenerative disorders. Int. Rev. Neurobiol. 2007, 82, 1–27. [Google Scholar] [CrossRef]
- Dziechciaz, M.; Filip, R. Biological psychological and social determinants of old age: Bio-psycho-social aspects of human aging. Ann. Agric. Environ. Med. 2014, 21, 835–838. [Google Scholar] [CrossRef]
- Tan, B.L.; Norhaizan, M.E.; Liew, W.P.; Sulaiman Rahman, H. Antioxidant and Oxidative Stress: A Mutual Interplay in Age-Related Diseases. Front. Pharmacol. 2018, 9, 1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Sanchez, A.; Miranda-Diaz, A.G.; Cardona-Munoz, E.G. The Role of Oxidative Stress in Physiopathology and Pharmacological Treatment with Pro- and Antioxidant Properties in Chronic Diseases. Oxid. Med. Cell. Longev. 2020, 2020, 2082145. [Google Scholar] [CrossRef]
- Yun, H.R.; Jo, Y.H.; Kim, J.; Shin, Y.; Kim, S.S.; Choi, T.G. Roles of Autophagy in Oxidative Stress. Int. J. Mol. Sci. 2020, 21, 3289. [Google Scholar] [CrossRef]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Kotiadis, V.N.; Duchen, M.R.; Osellame, L.D. Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 2014, 1840, 1254–1265. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Ru, X.; Wen, T. NRF2, a Transcription Factor for Stress Response and Beyond. Int. J. Mol. Sci. 2020, 21, 4777. [Google Scholar] [CrossRef]
- Brewer, L.; Rogers, S. Fumaric acid esters in the management of severe psoriasis. Clin. Exp. Dermatol. 2007, 32, 246–249. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Haghikia, A. Dimethyl fumarate in multiple sclerosis: Latest developments, evidence and place in therapy. Ther. Adv. Chronic Dis. 2016, 7, 198–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O.J.M.N. Hesperetin confers neuroprotection by regulating Nrf2/TLR4/NF-κB signaling in an Aβ mouse model. Mol. Neurobiol. 2019, 56, 6293–6309. [Google Scholar] [CrossRef]
- Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine modulates cadmium-induced oxidative stress, neuroinflammation, and cognitive impairments by regulating Nrf-2/HO-1 in vivo and in vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef] [Green Version]
- Houghton, C.A.; Fassett, R.G.; Coombes, J.S. Sulforaphane and Other Nutrigenomic Nrf2 Activators: Can the Clinician’s Expectation Be Matched by the Reality? Oxid. Med. Cell. Longev. 2016, 2016, 7857186. [Google Scholar] [CrossRef] [Green Version]
- Santin-Marquez, R.; Alarcon-Aguilar, A.; Lopez-Diazguerrero, N.E.; Chondrogianni, N.; Konigsberg, M. Sulforaphane—Role in aging and neurodegeneration. Geroscience 2019, 41, 655–670. [Google Scholar] [CrossRef]
- Subedi, L.; Cho, K.; Park, Y.U.; Choi, H.J.; Kim, S.Y. Sulforaphane-Enriched Broccoli Sprouts Pretreated by Pulsed Electric Fields Reduces Neuroinflammation and Ameliorates Scopolamine-Induced Amnesia in Mouse Brain through Its Antioxidant Ability via Nrf2-HO-1 Activation. Oxid. Med. Cell Longev. 2019, 2019, 3549274. [Google Scholar] [CrossRef] [Green Version]
- Eren, E.; Tufekci, K.U.; Isci, K.B.; Tastan, B.; Genc, K.; Genc, S. Sulforaphane Inhibits Lipopolysaccharide-Induced Inflammation, Cytotoxicity, Oxidative Stress, and miR-155 Expression and Switches to Mox Phenotype through Activating Extracellular Signal-Regulated Kinase 1/2-Nuclear Factor Erythroid 2-Related Factor 2/Antioxidant Response Element Pathway in Murine Microglial Cells. Front. Immunol. 2018, 9, 36. [Google Scholar] [CrossRef] [Green Version]
- Subedi, L.; Lee, J.H.; Yumnam, S.; Ji, E.; Kim, S.Y. Anti-Inflammatory Effect of Sulforaphane on LPS-Activated Microglia Potentially through JNK/AP-1/NF-kappaB Inhibition and Nrf2/HO-1 Activation. Cells 2019, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Ehianeta, T.S.; Akinyeye, R.O.; Orege, J.I.; Ejeromedoghene, O.; Adebule, A.P.; Okonkwo, B.O. Old drugs for a new indication: A review of chloroquine and analogue in COVID-19 treatment. Porto. Biomed. J. 2021, 6, e132. [Google Scholar] [CrossRef] [PubMed]
- Gasmi, A.; Peana, M.; Noor, S.; Lysiuk, R.; Menzel, A.; Gasmi Benahmed, A.; Bjorklund, G. Chloroquine and hydroxychloroquine in the treatment of COVID-19: The never-ending story. Appl. Microbiol. Biotechnol. 2021, 105, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Tripathy, S.; Dassarma, B.; Roy, S.; Chabalala, H.; Matsabisa, M.G. A review on possible modes of action of chloroquine/hydroxychloroquine: Repurposing against SAR-CoV-2 (COVID-19) pandemic. Int. J. Antimicrob. Agents 2020, 56, 106028. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef]
- Savarino, A.; Di Trani, L.; Donatelli, I.; Cauda, R.; Cassone, A. New insights into the antiviral effects of chloroquine. Lancet Infect. Dis. 2006, 6, 67–69. [Google Scholar] [CrossRef]
- Yan, Y.; Zou, Z.; Sun, Y.; Li, X.; Xu, K.F.; Wei, Y.; Jin, N.; Jiang, C. Anti-malaria drug chloroquine is highly effective in treating avian influenza A H5N1 virus infection in an animal model. Cell Res. 2013, 23, 300–302. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [Green Version]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Han, S.; Liu, R.; Meng, L.; He, H.; Zhang, Y.; Wang, C.; Lv, Y.; Wang, J.; Li, X.; et al. Chloroquine and hydroxychloroquine as ACE2 blockers to inhibit viropexis of 2019-nCoV Spike pseudotyped virus. Phytomedicine 2020, 79, 153333. [Google Scholar] [CrossRef]
- Zou, L.; Dai, L.; Zhang, X.; Zhang, Z.; Zhang, Z. Hydroxychloroquine and chloroquine: A potential and controversial treatment for COVID-19. Arch. Pharm. Res. 2020, 43, 765–772. [Google Scholar] [CrossRef]
- Hong, Z.; Jiang, Z.; Liangxi, W.; Guofu, D.; Ping, L.; Yongling, L.; Wendong, P.; Minghai, W. Chloroquine protects mice from challenge with CpG ODN and LPS by decreasing proinflammatory cytokine release. Int. Immunopharmacol. 2004, 4, 223–234. [Google Scholar] [CrossRef]
- Long, Y.; Liu, X.; Wang, N.; Zhou, H.; Zheng, J. Chloroquine attenuates LPS-mediated macrophage activation through miR-669n-regulated SENP6 protein translation. Am. J. Transl. Res. 2015, 7, 2335–2345. [Google Scholar]
- Hirata, Y.; Yamamoto, H.; Atta, M.S.; Mahmoud, S.; Oh-hashi, K.; Kiuchi, K. Chloroquine inhibits glutamate-induced death of a neuronal cell line by reducing reactive oxygen species through sigma-1 receptor. J. Neurochem. 2011, 119, 839–847. [Google Scholar] [CrossRef]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Reiter, R.J.; Ma, Q.; Sharma, R.J.M.R. Treatment of Ebola and other infectious diseases: Melatonin “goes viral”. Melatonin Res. 2020, 3, 43–57. [Google Scholar] [CrossRef]
- Hosseinzadeh, A.; Kamrava, S.K.; Joghataei, M.T.; Darabi, R.; Shakeri-Zadeh, A.; Shahriari, M.; Reiter, R.J.; Ghaznavi, H.; Mehrzadi, S. Apoptosis signaling pathways in osteoarthritis and possible protective role of melatonin. J. Pineal. Res. 2016, 61, 411–425. [Google Scholar] [CrossRef]
- Bahrami, N.; Goudarzi, M.; Hosseinzadeh, A.; Sabbagh, S.; Reiter, R.J.; Mehrzadi, S. Evaluating the protective effects of melatonin on di(2-ethylhexyl) phthalate-induced testicular injury in adult mice. Biomed. Pharmacother. 2018, 108, 515–523. [Google Scholar] [CrossRef]
- Dehdashtian, E.; Pourhanifeh, M.H.; Hemati, K.; Mehrzadi, S.; Hosseinzadeh, A. Therapeutic application of nutraceuticals in diabetic nephropathy: Current evidence and future implications. Diabetes Metab. Res. Rev. 2020, 36, e3336. [Google Scholar] [CrossRef]
- Daryani, A.; Montazeri, M.; Pagheh, A.S.; Sharif, M.; Sarvi, S.; Hosseinzadeh, A.; Reiter, R.J.; Hadighi, R.; Joghataei, M.T.; Ghaznavi, H.; et al. The potential use of melatonin to treat protozoan parasitic infections: A review. Biomed. Pharmacother. 2018, 97, 948–957. [Google Scholar] [CrossRef]
- Juybari, K.B.; Hosseinzadeh, A.; Ghaznavi, H.; Kamali, M.; Sedaghat, A.; Mehrzadi, S.; Naseripour, M. Melatonin As a Modulator of Degenerative and Regenerative Signaling Pathways in Injured Retinal Ganglion Cells. Curr. Pharm. Des. 2019, 25, 3057–3073. [Google Scholar] [CrossRef]
- Srinivasan, V.; Spence, D.W.; Pandi-Perumal, S.R.; Trakht, I.; Cardinali, D.P. Therapeutic actions of melatonin in cancer: Possible mechanisms. Integr. Cancer Ther. 2008, 7, 189–203. [Google Scholar] [CrossRef] [PubMed]
- Carbajo-Pescador, S.; Garcia-Palomo, A.; Martin-Renedo, J.; Piva, M.; Gonzalez-Gallego, J.; Mauriz, J.L. Melatonin modulation of intracellular signaling pathways in hepatocarcinoma HepG2 cell line: Role of the MT1 receptor. J. Pineal. Res. 2011, 51, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.H.; Lin, R.C.; Yang, J.S.; Yang, W.E.; Reiter, R.J.; Yang, S.F. Molecular and Cellular Mechanisms of Melatonin in Osteosarcoma. Cells 2019, 8, 1618. [Google Scholar] [CrossRef] [Green Version]
- Luchetti, F.; Canonico, B.; Betti, M.; Arcangeletti, M.; Pilolli, F.; Piroddi, M.; Canesi, L.; Papa, S.; Galli, F. Melatonin signaling and cell protection function. FASEB J. 2010, 24, 3603–3624. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Cao, X.J.; Wei, W. Melatonin decreases TLR3-mediated inflammatory factor expression via inhibition of NF-kappa B activation in respiratory syncytial virus-infected RAW264.7 macrophages. J. Pineal Res. 2008, 45, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.H.; Cao, X.J.; Liu, W.; Shi, X.Y.; Wei, W. Inhibitory effect of melatonin on lung oxidative stress induced by respiratory syncytial virus infection in mice. J. Pineal Res. 2010, 48, 109–116. [Google Scholar] [CrossRef]
- Hazra, S.; Chaudhuri, A.G.; Tiwary, B.K.; Chakrabarti, N. Matrix metallopeptidase 9 as a host protein target of chloroquine and melatonin for immunoregulation in COVID-19: A network-based meta-analysis. Life Sci. 2020, 257, 118096. [Google Scholar] [CrossRef]
- Habtemariam, S.; Daglia, M.; Sureda, A.; Selamoglu, Z.; Gulhan, M.F.; Nabavi, S.M. Melatonin and Respiratory Diseases: A Review. Curr. Top. Med. Chem. 2017, 17, 467–488. [Google Scholar] [CrossRef]
- Martin Gimenez, V.M.; Inserra, F.; Tajer, C.D.; Mariani, J.; Ferder, L.; Reiter, R.J.; Manucha, W. Lungs as target of COVID-19 infection: Protective common molecular mechanisms of vitamin D and melatonin as a new potential synergistic treatment. Life Sci. 2020, 254, 117808. [Google Scholar] [CrossRef]
- Shim, D.W.; Shin, H.J.; Han, J.W.; Ji, Y.E.; Jang, C.H.; Koppula, S.; Kang, T.B.; Lee, K.H. A novel synthetic derivative of melatonin, 5-hydroxy-2′-isobutyl-streptochlorin (HIS), inhibits inflammatory responses via regulation of TRIF-dependent signaling and inflammasome activation. Toxicol. Appl. Pharmacol. 2015, 284, 227–235. [Google Scholar] [CrossRef]
- Reiter, R.J.; Sharma, R.; Ma, Q.; Dominquez-Rodriguez, A.; Marik, P.E.; Abreu-Gonzalez, P. Melatonin Inhibits COVID-19-induced Cytokine Storm by Reversing Aerobic Glycolysis in Immune Cells: A Mechanistic Analysis. Med. Drug Discov. 2020, 6, 100044. [Google Scholar] [CrossRef]
- Bouhafs, R.K.; Jarstrand, C. Effects of antioxidants on surfactant peroxidation by stimulated human polymorphonuclear leukocytes. Free Radic. Res. 2002, 36, 727–734. [Google Scholar] [CrossRef]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. 2019, 14, 278–294. [Google Scholar] [CrossRef]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef]
- Shah, S.A.; Khan, M.; Jo, M.H.; Jo, M.G.; Amin, F.U.; Kim, M.O. Melatonin Stimulates the SIRT1/Nrf2 Signaling Pathway Counteracting Lipopolysaccharide (LPS)-Induced Oxidative Stress to Rescue Postnatal Rat Brain. CNS Neurosci. Ther. 2017, 23, 33–44. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflam. 2018, 15, 119. [Google Scholar] [CrossRef]
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef]
- Sarkar, I.; Sen, G.; Bhattacharya, M.; Bhattacharyya, S.; Sen, A. In silico inquest reveals the efficacy of Cannabis in the treatment of post-COVID-19 related neurodegeneration. J. Biomol. Struct. Dyn. 2021, 1–10. [Google Scholar] [CrossRef]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 252, 117652. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choe, K.; Park, H.Y.; Ikram, M.; Lee, H.J.; Park, T.J.; Ullah, R.; Kim, M.O. Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants. Cells 2022, 11, 1298. https://doi.org/10.3390/cells11081298
Choe K, Park HY, Ikram M, Lee HJ, Park TJ, Ullah R, Kim MO. Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants. Cells. 2022; 11(8):1298. https://doi.org/10.3390/cells11081298
Chicago/Turabian StyleChoe, Kyonghwan, Hyun Young Park, Muhammad Ikram, Hyeon Jin Lee, Tae Ju Park, Rahat Ullah, and Myeong Ok Kim. 2022. "Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants" Cells 11, no. 8: 1298. https://doi.org/10.3390/cells11081298
APA StyleChoe, K., Park, H. Y., Ikram, M., Lee, H. J., Park, T. J., Ullah, R., & Kim, M. O. (2022). Systematic Review of the Common Pathophysiological Mechanisms in COVID-19 and Neurodegeneration: The Role of Bioactive Compounds and Natural Antioxidants. Cells, 11(8), 1298. https://doi.org/10.3390/cells11081298