
A Genomic BSAseq Approach for the Characterization of QTLs Underlying Resistance to Fusarium oxysporum in Eggplant

 ,
,  ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Eggplant/Fusarium Oxysporum f. sp. Melongenae (Fom) Resistance Assessment
2.3. Statistical Analyses and QTL Detection
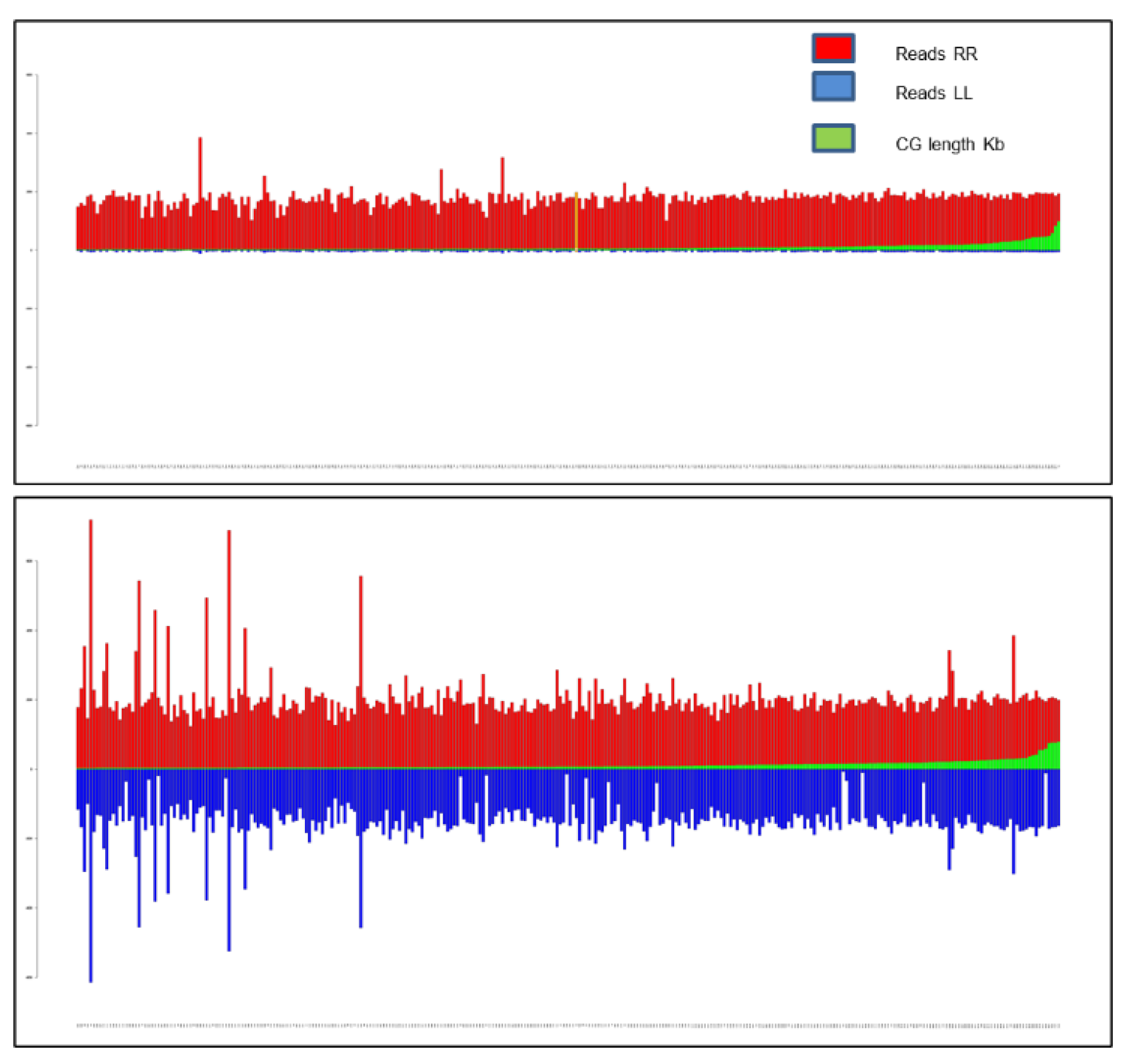
2.4. Composition of the Bulks of 5X Illumina Sequences
- BULK RR includes 28 fully resistant RILs with a calculated disease ratio of 100% and harboring the resistance locus Rfo-Sa1;
- BULK SS includes 18 fully susceptible RILs, with a disease resistance ratio of 0 and in which the locus Rfo-Sa1 is lacking;
- BULK PR includes 17 partially resistant RILs, in which the locus Rfo-Sa1 is lacking but with a resistance ratio ranging from 30 to 100%.
2.5. BSA-Seq Alignment of Bulked Sequences to Reference ‘67/3’
2.6. De Novo Assembly of ‘305E40’ Line
2.7. BSA-Seq Alignment of Bulked Sequences to the Reference Genome Asm_305
2.8. Expression Analysis of Candidate Genes
3. Results
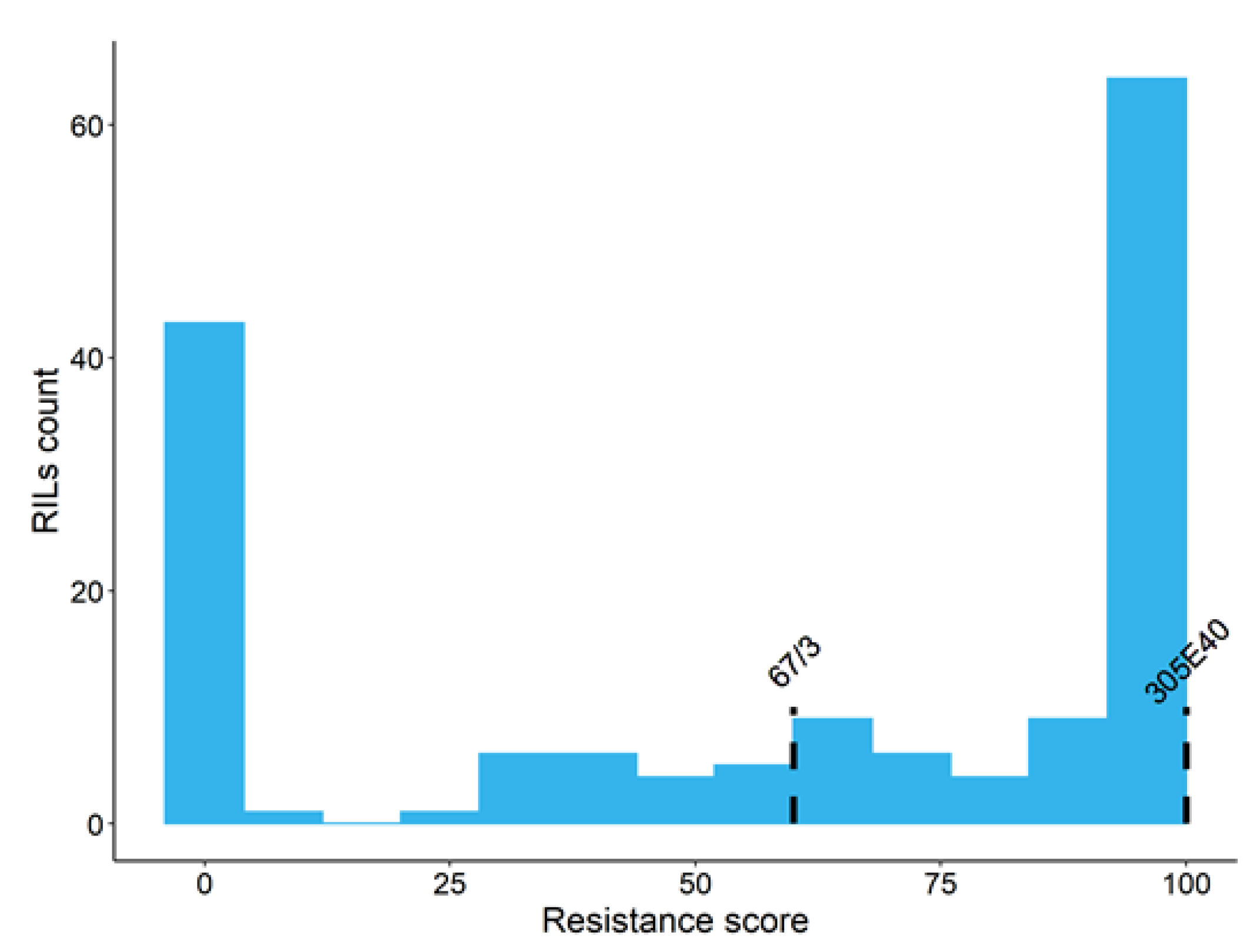
3.1. Phenotypical Score and Statistical Analysis
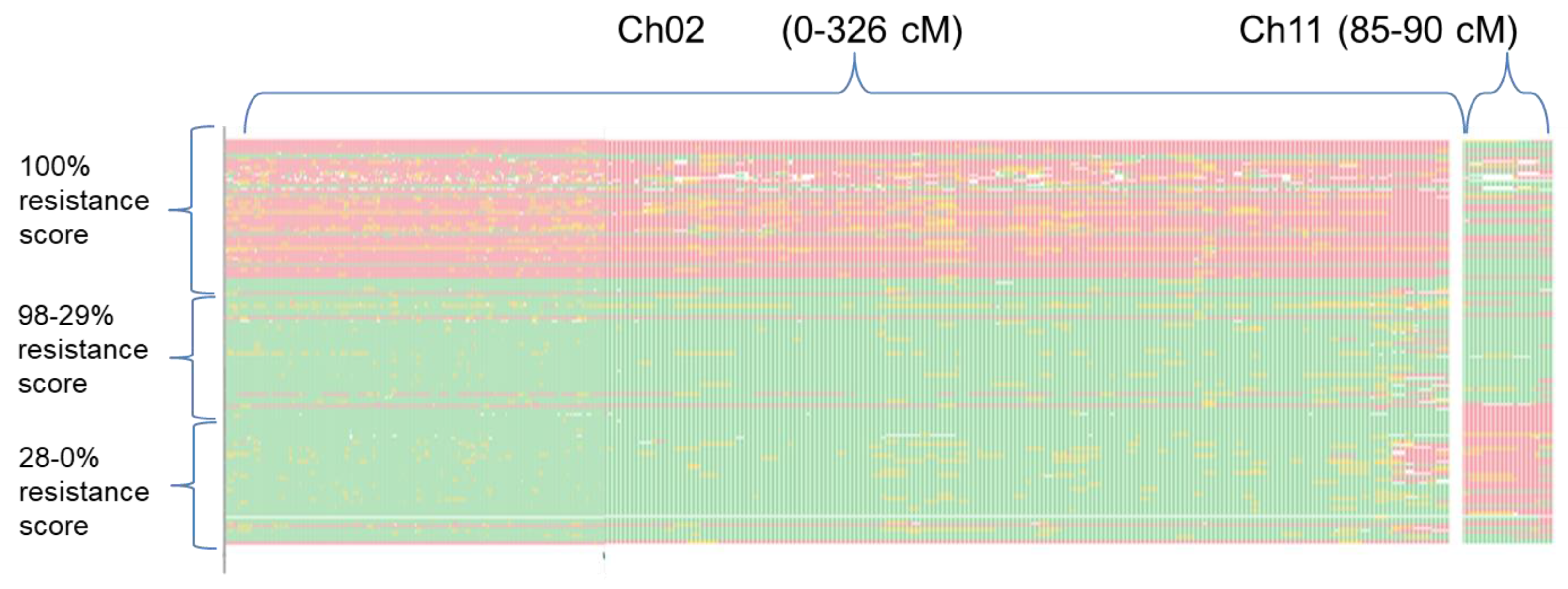
3.2. QTL Mapping
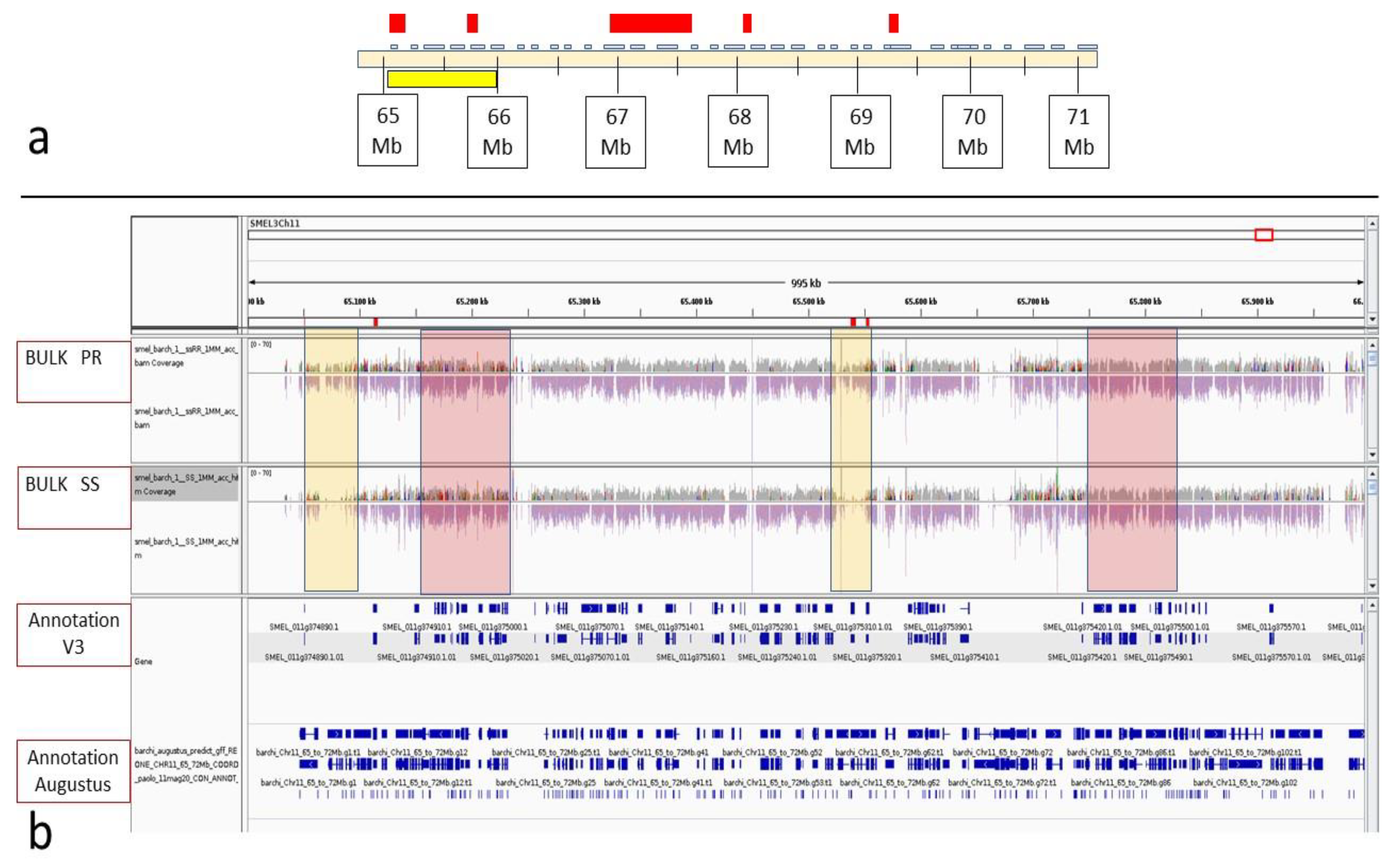
3.3. BSA-Seq Analysis and Candidate Gene Identification in the FomCH11 Region
3.4. De Novo Assembly of ‘305E40’, BSAseq and Candidate Gene Identification in the QTL FomCH02 Region

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Candidate Genes | Query | Scaffold on S. aethiopicum transcriptome | Identities | Predicted Function Based on Domains Analysis Performed via NCBI Platform | Scaffold on S. aethiopicum Pangenome |
|---|---|---|---|---|---|
| RES1 | SMEL_002g157480.1 (Miyatake et al. [20], orthologous of SOLYC02G032200.2 | SAUC48279_TC01 Length = 3745 | 3392/3418 (99%) Strand = +/+ | Encoding a putative TMV resistance protein N-like LOC102604931, transcript variant X2 | scaffold3814_cov65 (978531-977633) lenght:898 strand (+/−) |
| RES2 | C7021905__121_4977 (348 letters) | SAUC67459_TC01 Length = 578 | 348/348 (100%) Strand = +/− | chaperonin | scafold150403_cov62 (904702-904125) lenght:577 strand (+/−) |
| RES3 | C7104747__218_9061 (1767 letters) | SAUC05724_TC02 Length = 1311 | 915/915 (100%) Strand = +/+ | cysteine-rich RLK (RECEPTOR-like protein kinase) 8 | scafold150406_cov62 (122904-121605) lenght:1299 strand (+/−) |
| RES4 | C7123897__247_10432 (1176 letters) | SAUC85719_TC01 Length = 1281 | 194/194(100%) + 807/807(100%) Strand: +/+ | Putative late blight resistance protein homolog R1A-3 | scaffold149207_cov61 (473206-471925) lenght:1281 strand (+/−) |
| RES5 | scaffold131120__543_23320 (630 letters) | SAUC54187_TC01 Length = 581 | 312/312 (100%) Strand: +/+ | scaffold4400_cov64 (319724-320299) lenght:575 strand (+/+) | |
| RES6 | scaffold151247__813_35520 (1935 letters) | SAUC60998_TC03 Length = 2636 | 986/986(100%) + 719/721(99%) Strand = +/+ | scaffold149470_cov62 (278029-278886) lenght:857 strand (+/+) | |
| RES7 | scaffold161031_1035_45870 (1362 letters) | SAUC62185_TC01 Length = 517 | 517/517 (100%) Strand = +/+ | Protein transparent testa 12-like | scaffold149494_cov61 (246517-247031) lenght:514 strand (+/+) |
| RES8 | scaffold4270_1160_51181 (3702 letters) | SAUC68094_TC01 Length = 4264 | 2697/2697(100%) + 974/974 (100%) Strand = +/− | Putative late blight resistance protein homolog R1B-14-like [Solanum lycopersicum] | scaffold872_cov63 (353791-349798) lenght:3993 strand (+/−) |
| RES9 | scaffold160330_1010_44805 (327 letters) | SAUC18225_TC01 Length = 741 | 327/327(100%) Strand = +/− | scaffold149207_cov61 (183171-183700) lenght:529 strand (+/+) | |
| RES10 | scaffold83272__1378_60779 (375 letters) | SAUC44781_TC01 Length = 543 | 364/364(100%) Strand = +/− | scaffold150551_cov61 (32877-32417) lenght:460 strand (+/−) |
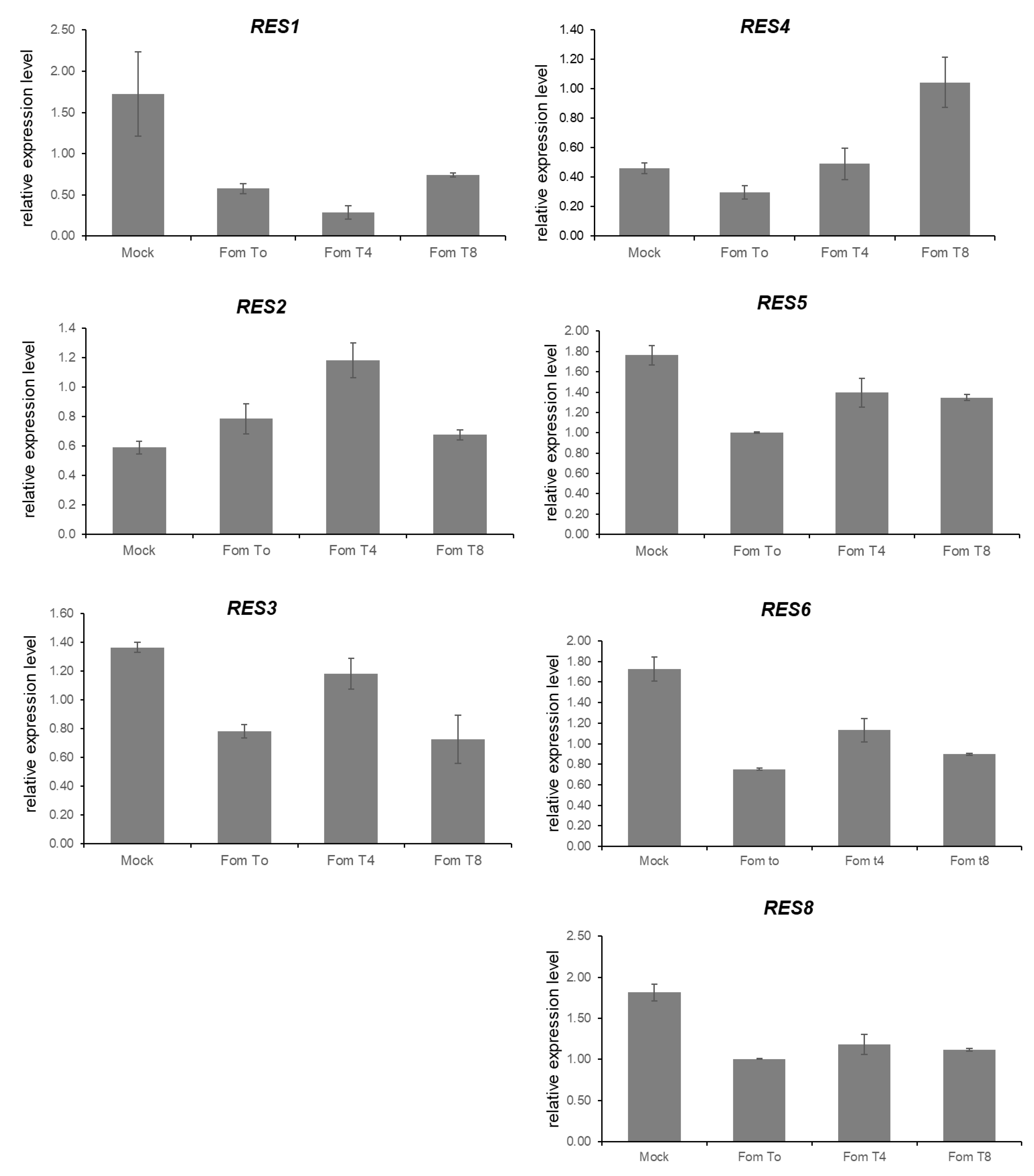
3.5. Expression Analysis of Candidate Genes for FomCH02
4. Discussion
4.1. QTL Mapping
4.2. BSA-Seq Approach
4.3. Differentially Enriched Regions and Candidate Gene Identification within FomCH11 QTL
4.4. De Novo Assembly of ‘305E40’, BSAseq and Candidate Gene Identification within FomCH02 QTL
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Weese, T.L.; Bohs, L. Eggplant origins: Out of Africa, into the Orient. Taxon 2010, 59, 49–56. [Google Scholar] [CrossRef]
- Meyer, R.S.; Karol, K.G.; Little, D.P.; Nee, M.H.; Litt, A. Phylogeographic relationships among Asian eggplants and new perspectives on eggplant domestication. Mol. Phylogenet. Evol. 2012, 63, 685–701. [Google Scholar] [CrossRef] [PubMed]
- Knapp, S.; Vorontsova, M.S.; Prohens, J. Wild Relatives of the Eggplant (Solanum melongenan L.: Solanaceae): New Understanding of Species Names in a Complex Group. PLoS ONE 2013, 8, e57039. [Google Scholar] [CrossRef] [PubMed]
- Page, A.; Gibson, J.; Meyer, R.S.; Chapman, M.A. Eggplant Domestication: Pervasive Gene Flow, Feralization, and Transcriptomic Divergence. Mol. Biol. Evol. 2019, 36, 1359–1372. [Google Scholar] [CrossRef]
- Arnoux, S.; Fraïsse, C.; Sauvage, C. Genomic inference of complex domestication histories in three Solanaceae species. J. Evol. Biol. 2021, 34, 270–283. [Google Scholar] [CrossRef]
- Faostat Data. 2021. Available online: http://www.fao.org/faostat/en/#data/QC (accessed on 10 March 2022).
- Stravato, V.M.; Cappelli, C.; Polverari, A. Attacchi di Fusarium oxysporum f. sp. melongenae agente della tracheofusariosi della melanzana in Italia centrale. Inf. Fitopatol. 1993, 4310, 51–54. [Google Scholar]
- Sihachakr, D.; Daunay, M.C.; Serraf, L.; Chaput, M.H.; Mussio, I.; Haicour, R.; Rossignol, L.; Ducreux, G. Somatic hybridization of eggplant (Solanum melongena L), with its close and wilt relatives. In Biotechnology in Agriculture and Forestry, Somatic Hybridization in Crop Improvement; Bajaj, Y.P.S., Ed.; Springer: Berlin, Germany, 1994. [Google Scholar]
- Urrutia Herrada, M.T.; Gomez Garcia, V.M.; Tello Marquina, J. Fusarium wilt on eggplant in Almeria (Spain). Bol. Sanid. Veg. Plagas 2004, 30, 85–92. [Google Scholar]
- Altinok, H.H. First report of fusarium wilt of eggplant caused by Fusarium oxysporum f. sp. melongenae in Turkey. Plant Pathol. 2005, 54, 577–579. [Google Scholar] [CrossRef]
- Van Steekelenburg, N.A.M. Fusarium wilt of eggplant in the Netherlands. Neth. J. Plant Pathol. 1976, 82, 191–192. [Google Scholar] [CrossRef]
- Daunay, M.C.; Lester, R.N.; Laterrot, H. The use of wild species for the genetic improvement of brinjal-eggplant (Solanum melongena) and tomato (Lycopersicon esculentum). In Solanaceae III: Taxonomy, Chemistry, Evolution; Hawkes, J.G., Lester, R.N., Nee, M., Estrada-R., N., Eds.; Royal Botanic Gardens: Richmond, UK, 1991; pp. 389–412. [Google Scholar]
- Rotino, G.L.; Sala, T.; Toppino, L. Eggplant. In Alien Gene Transfer in Crop Plants, Achievements and Impacts; Pratap, A., Kumar, J., Eds.; Springer Science + Business Media, LLC: Berlin, Germany, 2014; Volume 2, Chapter 16; p. 381. [Google Scholar] [CrossRef]
- Abdullaheva, K.; Shifman, I.A. Resistance of eggplant to fusar ium wilt. Sel. Semenovod. 1988, 1, 29–31. [Google Scholar]
- Komochi, S.; Monma, S.; Narikawa, T.; Sakata, Y. Evaluation of resistance to bacterial wilt and verticillium wilt in eggplants (Solanum melongena L.) collected in Malaysia. J. Jpn. Soc. Hortic. Sci. 1996, 65, 81–88. [Google Scholar]
- Monma, S.; Akazawa, S.; Simosaka, K.; Sakata, Y.; Matsunaga, H. ‘Diataro’, a bacterial wilt- and Fusarium wilt-resistant hybrid eggplant for rootstock. Bull. Natl. Res. Inst. Veg. Orn. Plants Tea Ser. A 1997, 12, 73–83. [Google Scholar]
- Mandhare, V.K.; Patil, P.L. Varietal screening and efficacy of fungicides against Fusarium wilt of brinjal. J. Maharashtra Agric. Univ. 1993, 18, 34–36. [Google Scholar]
- Barchi, L.; Toppino, L.; Valentino, D.; Bassolino, L.; Portis, E.; Lanteri, S.; Rotino, G.L. QTL analysis reveals new eggplant loci involved in resistance to fungal wilts. Euphytica 2018, 214, 20. [Google Scholar] [CrossRef]
- Mutlu, N.; Boyacı, F.H.; Göçmen, M.; Abak, K. Development of SRAP, SRAP-RGA, RAPD, and SCAR markers linked with a Fusarium wilt resistance gene in eggplant. Theor. Appl. Genet. 2008, 8, 1303–1312. [Google Scholar] [CrossRef]
- Miyatake, K.; Saito, T.; Negoro, S.; Yamaguchi, H.; Nunome, T.; Ohyama, A.; Fukuoka, H. Detailed mapping of a resistance locus against Fusarium wilt in cultivated eggplant Solanum melongena. Theor. Appl. Genet. 2016, 129, 357–367. [Google Scholar] [CrossRef]
- Daunay, M.C.; Lester, R.N.; Gebhardt, C.; Hennart, J.W.; Jahn, M.; Frary, A.; Doganlar, S. Genetic resources of eggplant (Solanum melongena L.) and allied species: A new challenge for molecular geneticists and eggplant breeders. In Solanaceae V; Advances in Taxonomy and Utilization; Nijmegen University Press: Nijmegen, The Netherlands, 2001; pp. 251–274. [Google Scholar]
- Plazas, M.; Vilanova, S.; Gramazio, P.; Rodríguez-Burruezo, A.; Fita, A.; Herraiz, F.J.; Ranil, R.; Fonseka, R.; Niran, L.; Fonseka, H.; et al. Interspecific hybridization between eggplant and wild relatives from different genepools. J. Am. Soc. Hortic. Sci. 2016, 141, 34–44. [Google Scholar] [CrossRef]
- Syfert, M.M.; Castañeda-Álvarez, N.P.; Khoury, C.K.; Särkinen, T.; Sosa, C.C.; Achicanoy, H.A.; Bernau, V.; Prohens, J.; Daunay, M.C.; Knapp, S. Crop wild relatives of the brinjal eggplant (Solanum melongena): Poorly represented in genebanks and many species at risk of extinction. Am. J. Bot. 2016, 103, 635–651. [Google Scholar] [CrossRef]
- Toppino, L.; Prohens, J.; Rotino, G.L.; Plazas, M.; Parisi, M.; Carrizo García, C.; Tripodi, P. Pepper and Eggplant Genetic Resources. In The wild Solanum genomes; Carputo, D., Aversano, R., Ercolano, M.R., Eds.; Springer: Cham, Switzerland, 2021; Chapter 6; ISBN 978-3-030-30343-3. [Google Scholar] [CrossRef]
- Kaushik, P.; Prohens, J.; Vilanova, S.; Gramazio, P.; Plazas, M. Phenotyping of eggplant wild relatives and interspecific hybrids with conventional and phenomics descriptors provides insight for their potential utilization in breeding. Front. Plant Sci. 2016, 7, 677. [Google Scholar] [CrossRef]
- Rizza, F.; Mennella, G.; Collonnier, C.; Sihachakr, D.; Kashyap, V.; Rajam, M.V.; Prestera, M.; Rotino, G.L. Androgenic dihaploids from somatic hybrids between Solanum melongena and S. aethiopicum group gilo as a source of resistance to Fusarium oxysporum f. sp. melongenae. Plant Cell Rep. 2002, 20, 1022–1032. [Google Scholar] [CrossRef]
- Rotino, G.L.; Mennella, G.; Fusari, F.; Vitelli, G.; Tacconi, M.G.; D’Alessandro, A.; Acciarri, N. Towards introgression of resistance to Fusarium oxysporum F. sp. melongenae from Solanum integrifolium into eggplant. In Proceedings of the 11th Eucarpia Meeting on Genetics and Breeding of Capsicum and Eggplant, Antalya, Turkey, 9 April 2001; pp. 303–307. [Google Scholar]
- Rotino, G.L.; Rizza, F.; Mennella, G.; Tacconi, M.G.; Alberti, P.; D’Alessandro, A.; Acciarri, N.; Toppino, L. Production and utilization of sexual “doubled hybrid” between the somatic hybrids S. melongena (+) S. integrifolium and S. melongena (+) S. aethiopicum gr. gilo. In Proceedings of the EUCARPIA, XIIth Meeting on Genetics and Breeding of Capsicum & Eggplant, Noordwijkerhout, The Netherlands, 17–19 May 2004; pp. 203–209. [Google Scholar]
- Rotino, G.L.; Sihachakr, D.; Rizza, F.; Valè, G.; Tacconi, M.G.; Alberti, P.; Mennella, G.; Sabatini, E.; Toppino, L.; D’Alessandro, A.; et al. Current status in production and utilization of dihaploids from somatic hybrids between eggplant (Solanum melongena L.) and its wild relatives. Acta Physiol. Plant. 2005, 27, 723–733. [Google Scholar] [CrossRef]
- Toppino, L.; Valè, G.P.; Rotino, G.L. Inheritance of Fusarium wilt resistance introgressed from Solanum aethiopicum Gilo and Aculeatum groups into cultivated eggplant S. melongena and development of associated PCR-based markers. Mol. Breed. 2008, 222, 237–250. [Google Scholar] [CrossRef]
- Boyaci, F.; Unlu, A.; Abak, K. Screening for resistance to Fusarium wilt of some cultivated eggplants and wild Solanum accessions. In Proceedings of the XXVIII International Horticultural Congress on Science and Horticulture for People IHC2010: International Symposium on New 935, Lisbon, Portugal, 22 August 2010; pp. 23–27. [Google Scholar]
- Boyaci, H.F.; Unlu, A.; Abak, K. Genetic analysis of resistance to wilt caused by Fusarium (Fusarium oxysporum melongenae) in eggplant (Solanum melongena). Indian J. Agric. Sci. 2011, 81, 812–815. [Google Scholar]
- Michelmore, R.W.; Paran, I.; Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef]
- Barchi, L.; Lanteri, S.; Portis, E.; Valè, G.; Volante, A.; Pulcini, L.; Ciriaci, T.; Acciarri, N.; Barbierato, V.; Toppino, L.; et al. A RAD Tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS ONE 2012, 7, e43740. [Google Scholar] [CrossRef]
- Barchi, L.; Pietrella, M.; Venturini, L.; Minio, A.; Toppino, L.; Acquadro, A.; Andolfo, G.; Aprea, G.; Avanzato, C.; Bassolino, L.; et al. A chromosome-anchored eggplant genome sequence reveals key events in Solanaceae evolution. Sci. Rep. 2019, 9, 11769. [Google Scholar] [CrossRef]
- Portis, E.; Barchi, L.; Toppino, L.; Lanteri, S.; Acciarri, N.; Felicioni, N.; Fusari, F.; Barbierato, V.; Cericola, F.; Valè, G.; et al. QTL mapping in eggplant reveals clusters of yield-related loci and orthology with the tomato genome. PLoS ONE 2014, 9, e89499. [Google Scholar] [CrossRef]
- Fukuoka, H.; Miyatake, K.; Nunome, T.; Negoro, S.; Shirasawa, K.; Isobe, S.; Asamizu, E.; Yamaguchi, H.; Ohyama, A. Development of gene-based markers and construction of an integrated linkage map in eggplant by using Solanum orthologous (SOL) gene sets. Theor. Appl. Genet. 2012, 125, 47–56. [Google Scholar] [CrossRef]
- Hirakawa, H.; Shirasawa, K.; Miyatake, K.; Nunome, T.; Negoro, S.; Ohyama, A.; Tamaguchi, H.; Sato, S.; Isobe, S.; Tabata, S.; et al. Draft genome sequence of eggplant (Solanum melongena L.): The representative Solanum species indigenous to the Old World. DNA Res. 2014, 21, 649–660. [Google Scholar] [CrossRef]
- Barbierato, V.; Toppino, L.; Rinaldi, P.; Sala, T.; Bassolino, L.; Valè, G.; Ferrarini, A.; Delledonne, M.; Bagnaresi, P.; Rotino, G.L. Phenotype and gene expression analyses of the Rfo-sa1 resistant aubergine interaction with Fusarium oxysporum f. sp. melongenae and Verticillium dahliae. Plant Pathol. 2016, 5, 1297–1309. [Google Scholar] [CrossRef]
- Toppino, L.; Barchi, L.; Mercati, F.; Acciarri, N.; Perrone, D.; Martina, M.; Gattolin, S.; Sala, T.; Fadda, S.; Mauceri, A.; et al. A New Intra-Specific and High-Resolution Genetic Map of Eggplant Based on a RIL Population, and Location of QTLs Related to Plant Anthocyanin Pigmentation and Seed Vigour. Genes 2020, 11, 745. [Google Scholar] [CrossRef]
- Florio, F.E.; Gattolin, S.; Toppino, L.; Bassolino, L.; Fibiani, M.; Lo Scalzo, R.; Rotino, G.L. A SmelAAT Acyltransferase Variant Causes a Major Difference in Eggplant (Solanum melongena L.) Peel Anthocyanin Composition. Int. J. Mol. Sci. 2021, 22, 9174. [Google Scholar] [CrossRef]
- Sulli, M.; Barchi, L.; Toppino, L.; Diretto, G.; Sala, T.; Lanteri, S.; Rotino, G.L.; Giuliano, G. An Eggplant Recombinant Inbred Population Allows the Discovery of Metabolic QTLs Controlling Fruit Nutritional Quality. Front. Plant Sci. 2021, 12, 638195. [Google Scholar] [CrossRef]
- Barchi, L.; Portis, E.; Toppino, L.; Rotino, G.L. Molecular Mapping, QTL Identification, and GWA Analysis. In The Eggplant Genome; Springer International Publishing: Cham, Switzerland, 2019; pp. 41–54. [Google Scholar]
- Barchi, L.; Rabanus-Wallace, M.T.; Prohens, J.; Toppino, L.; Padmarasu, S.; Portis, E.; Rotino, G.L.; Stein, N.; Lanteri, S.; Giuliano, G. Improved genome assembly and pan-genome provide key insights into eggplant domestication and breeding. Plant J. 2021, 107, 579–596. [Google Scholar] [CrossRef]
- Toppino, L.; Barchi, L.; LoScalzo, R.; Palazzolo, E.; Francese, G.; Fibiani, M.; D’Alessandro, A.; Papa, V.; Laudicina, V.A.; Sabatino, L.; et al. Mapping Quantitative Trait Loci Affecting Biochemical and Morphological Fruit Properties in Eggplant (Solanum melongena L.). Front. Plant Sci. 2016, 7, 256. [Google Scholar] [CrossRef]
- Cappelli, C.; Stravato, V.M.; Rotino, G.L.; Buonaurio, R. Sources of resistance among Solanum spp. to an Italian isolate of Fusarium oxysporum f. sp. melongenae. In EUCARPIA, Proceedings of the 9th Meeting of Genetics and Breeding of Capsicum and Eggplant; Andràsfalvi, A., Moòr, A., Zatykò, L., Eds.; SINCOP: Budapest, Hungary, 1995; pp. 221–224. [Google Scholar]
- Sall, J.; Stephens, M.L.; Lehman, A.; Loring, S. JMP Start Statistics: A Guide to Statistics and Data Analysis Using JMP; Sas Institute: Cary, NC, USA, 2017. [Google Scholar]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef]
- Lander, E.S.; Botstein, D. Mapping mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 1989, 121, 185–199. [Google Scholar] [CrossRef]
- Churchill, G.A.; Doerge, R.W. Empirical threshold values for quantitative trait mapping. Genetics 1994, 138, 963–971. [Google Scholar] [CrossRef]
- Sen, S.; Churchill, G.A. A statistical framework for quantitative trait mapping. Genetics 2001, 159, 371–387. [Google Scholar] [CrossRef]
- Darvasi, A.; Soller, M. A simple method to calculate resolving power and confidence interval of QTL map location. Behav. Genet. 1997, 27, 125–132. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118, iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 2047X–2217X. [Google Scholar] [CrossRef]
- Hoff, K.J.; Stanke, M. Predicting genes in single genomes with AUGUSTUS. Curr. Protoc. Bioinform. 2019, 65, e57. [Google Scholar] [CrossRef]
- Barbierato, V.; Sala, T.; Rinaldi, P.; Bassolino, L.; Barchi, L.; Rotino, G.L.; Toppino, L. A spiking strategy facilitates housekeeping selection for RT-qPCR analysis under different biotic stresses in eggplant. Protoplasma 2017, 254, 2215–2223. [Google Scholar] [CrossRef]
- Gramazio, P.; Prohens, J.; Plazas, M.; Andújar, I.; Herraiz, F.J.; Castillo, E.; Knapp, S.; Meyer, R.S.; Vilanova, S. Location of chlorogenic acid biosynthesis pathway and polyphenol oxidase genes in a new interspecific anchored linkage map of eggplant. BMC Plant Biol. 2014, 14, 350. [Google Scholar] [CrossRef]
- Song, B.; Song, Y.; Fu, Y.; Kizito, E.B.; Kamenya, S.N.; Kabod, P.N.; Liu, H.; Muthemba, S.; Kariba, R.; Njuguna, J.; et al. Draft genome sequence of Solanum aethiopicum provides insights into disease resistance, drought tolerance, and the evolution of the genome. GigaScience 2019, 8, giz115. [Google Scholar] [CrossRef]
- Fradin, E.F.; Zhang, Z.; Juarez Ayala, J.C.; Castroverde, C.D.; Nazar, R.N.; Robb, J.; Liu, C.M.; Thomma, B.P. Genetic dissection of Verticillium wilt resistance mediated by tomato Ve1. Plant Physiol. 2009, 150, 320–332. [Google Scholar] [CrossRef]
- King, M.D.; Fountain, C.; Dakhlallah, D.; Bearman, P.S. Estimated autism risk, older reproductive age, and parameterization response. Am. J. Public Health 2010, 100, 390. [Google Scholar] [CrossRef]
- Tiwari, V.; Singh, R.; Pandey, A.K. Efficacy of some antagonistic fungi and botanicals against Fusarium solani causing damping-off disease in eggplant (Solanum Melongena L.). J. Pure Appl. Microbiol. 2017, 43, 1–2. [Google Scholar]
- Tiwari, V.; Singh, R.; Rizvi, G.; Pandey, A.K. Antagonistic potential of some rhizospheric fungi against damping-off pathogen (Fusarium solani) of eggplant, Solanum melongena. Biochem. Cell. Arch. 2018, 18, 459–464. [Google Scholar]
- Li, W.; Deng, Y.; Ning, Y.; He, Z.; Wang, G.L. Exploiting broad-spectrum disease resistance in crops: From molecular dissection to breeding. Annu. Rev. Plant Biol. 2020, 71, 575–603. [Google Scholar] [CrossRef]
- Deng, Y.; Ning, Y.; Yang, D.L.; Zhai, K.; Wang, G.L.; He, Z. Molecular basis of disease resistance and perspectives on breeding strategies for resistance improvement in crops. Mol. Plant 2020, 13, 1402–1419. [Google Scholar] [CrossRef]
- Gururani, M.A.; Venkatesh, J.; Upadhyaya, C.P.; Nookaraju, A.; Pandey, S.K.; Park, S.W. Plant disease resistance genes: Current status and future directions. Physiol. Mol. Plant Pathol. 2012, 78, 51–65. [Google Scholar] [CrossRef]
- Le Nguyen, K.; Grondin, A.; Courtois, B.; Gantet, P. Next-generation sequencing accelerates crop gene discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef]
- Clevenger, J.; Chu, Y.; Chavarro, C.; Botton, S.; Culbreath, A.; Isleib, T.G.; Holbrook, C.C.; Ozias-Akins, P. Mapping late leaf spot resistance in peanut (Arachis hypogaea) using QTL-seq reveals markers for marker-assisted selection. Front. Plant Sci. 2018, 9, 83. [Google Scholar] [CrossRef]
- Kayam, G.; Brand, Y.; Faigenboim-Doron, A.; Patil, A.; Hedvat, I.; Hovav, R. Fine-mapping the branching habit trait in cultivated peanut by combining bulked segregant analysis and high-throughput sequencing. Front. Plant Sci. 2017, 8, 467. [Google Scholar] [CrossRef]
- Klein, H.; Xiao, Y.; Conklin, P.A.; Govindarajulu, R.; Kelly, J.A.; Scanlon, M.J.; Whipple, C.J.; Bartlett, M. Bulked-segregant analysis coupled to whole genome sequencing (BSA-Seq) for rapid gene cloning in maize. G3 Genes Genomes Genet. 2018, 8, 3583–3592. [Google Scholar] [CrossRef]
- Vogel, G.; LaPlant, K.E.; Mazourek, M.; Gore, M.A.; Smart, C.D. A combined BSA-Seq and linkage mapping approach identifies genomic regions associated with Phytophthora root and crown rot resistance in squash. Theor. Appl. Genet. 2021, 134, 1015–1031. [Google Scholar] [CrossRef]
- Zou, C.; Wang, P.; Xu, Y. Bulked sample analysis in genetics, genomics and crop improvement. Plant Biotechnol. J. 2016, 14, 1941–1955. [Google Scholar] [CrossRef]
- Park, T.H.; Gros, J.; Sikkema, A.; Vleeshouwers, V.G.; Muskens, M.; Allefs, S.; Jacobsen, E.; Visser, R.G.; van der Vossen, E.A. The late blight resistance locus Rpi-blb3 from Solanum bulbocastanum belongs to a major late blight R gene cluster on chromosome 4 of potato. Mol. Plant-Microbe Interact. 2005, 18, 722–729. [Google Scholar] [CrossRef]
- Bittner-Eddy, P.D.; Crute, I.R.; Holub, E.B.; Beynon, J.L. RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica. Plant J. 2000, 21, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Toppino, L.; Mennella, G.; Rizza, F.; D’Alessandro, A.; Sihachakr, D.; Rotino, G.L. ISSR and Isozyme Characterization of androgenetic dihaploids reveals tetrasomic inheritance in tetraploid somatic hybrids between Solanum melongena and Solanum aethiopicum group gilo. J. Hered. 2008, 99, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Barchi, L.; Lanteri, S.; Portis, E.; Stagel, A.; Valè, G.; Toppino, L.; Rotino, G.L. Segregation distortion and linkage analysis in eggplant (Solanum melongena L.). Genome 2010, 53, 805–815. [Google Scholar] [CrossRef] [PubMed]
- Hammond-Kosack, K.E.; Jones, J.D. Plant disease resistance genes. Annu. Rev. Plant Biol. 1997, 48, 575–607. [Google Scholar] [CrossRef]
- Vossen, J.H.; van Arkel, G.; Bergervoet, M.; Jo, K.R.; Jacobsen, E.; Visser, R.G. The Solanum demissum R8 late blight resistance gene is an Sw-5 homologue that has been deployed worldwide in late blight resistant varieties. Theor. Appl. Genet. 2016, 129, 1785–1796. [Google Scholar] [CrossRef]







| Gene Abbreviation | Oligo Sequences for RT-qPCR |
|---|---|
| RES_1 | RES_1_FW 5′_TGGCAGAATCTCCACAACCT_3′ RES_1_RV 5′_GATGATGAAGGACTGCTCGC_3′ |
| RES_2 | RES_2_FW 5′_ACCAGCACTGATCTGTCTCC_3′ RES_2_RV 5′_TATGACCGGTCCCTTTTCCC_3′ |
| RES_3 | RES_3_FW 5′_AGTACAAGGGAAGCCGTGAG_3′ RES_3_RV 5′_GAGCAGCATCAGATCAGCAC_3′ |
| RES_4 | RES_4_FW 5’_ACGGAACTAGAGCGACAACA_3′ RES_4_RV 5’_TAGCCTTGCCTCTATCCTGC_3′ |
| RES_5 | RES_5_FW 5′_CCGCCAATTCACTGCGTAG_3′ RES_5_RV 5′_TTGTATCCTCCTCCTCGCTG_3′ |
| RES_6 | RES_6_FW 5′_TTTGAGCTGTTGGGCCAATC_3′ RES_6_RV 5′_CCGTGGTGCATTATAGCCAC_3′ |
| RES_7 | RES_7_FW 5′_AATGGGAAGTGCAGTGGAGA_3′ RES_7_RV 5′_GGGGAAGTTGGCAGCATAAG_3′ |
| RES_8 | RES_8_FW 5′_ACCAGGTTAAGTTACAGCTCTGA_3′ RES_8_RV 5′_ACCCCTTTCCAGACACATCA_3′ |
| Disease Score (Mean ± SD) | Skewness | SE | Kurtosis | SE | Heritability | Transgr. vs. ‘305E40’ | Transgr. vs. ‘67/3’ | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ‘305E40’ | ‘67/3’ | S. aeth | ‘Tal1/1’ | F1 | RIL Population | |||||||
| 100 ± 0 | 60 ± 0 | 100 ± 0 | 0 ± 0 | 100 ± 0 | 59.8 ± 42.0 | −0.455 | 0.193 | −1.502 | 0.384 | 0.98 | 66 | |
| QTL Name | Chr | cM | LOD | PVE (%) | Add. Eff. | CI (cM) | |
|---|---|---|---|---|---|---|---|
| Start | End | ||||||
| FomCH02 | 2 | 222.7 | 29.03 | 46.47 | −30.9 | 221.66 | 223.74 |
| FomCH11 | 11 | 88.5 | 18.98 | 25.72 | 14.61 | 86.61 | 90.39 |
| Interaction 2*11 | 7.42 | 8.37 | 13.15 | ||||
| Diff. Enriched Region | Physical Interval in V3 | Number of Annotated Genes | Number of Resistance Genes | Gene ID According to Gene Annotation by BARCHI et al. [35] |
|---|---|---|---|---|
| A | 65,030–65,120 Mb | 2 | 2 | SMEL_011g374890.1 Similar to At1g58602: Probable disease resistance protein At1g58602 (A. thaliana) |
| SMEL_011g374900.1 Similar to RPP13: Disease resistance protein RPP13 (A. thaliana) | ||||
| B | 65,532–65,553 Mb | 2 | 2 | SMEL_011g375310.1 Similar to RPP13: Disease resistance protein RPP13 (A. thaliana) |
| SMEL_011g375320. 1 Similar to RPP13: Disease resistance protein RPP13 (A. thaliana) | ||||
| C | 66,920–67,557 Mb | 31 | 0 | |
| D | 68,093–68,171 Mb | 7 | 3 | SMEL_011g376860.1 Similar to XA21: Receptor kinase-like protein Xa21 (O. Sativa subsp. Indica) |
| SMEL_011g376900.1 Similar to FLS2: LRR receptor-like serine/threonine-protein kinase FLS2 (A. thaliana) | ||||
| SMEL_011g376910.1 Similar to FLS2: LRR receptor-like serine/threonine-protein kinase FLS2 (A. thaliana) | ||||
| E | 69,366–69,410 Mb | 7 | 2 | SMEL_011g377340.1 Similar to R1A: Late blight resistance protein R1-A (S. demissum) |
| SMEL_011g377380.1 Similar to R1C-3: Putative late blight resistance protein homolog R1C-3 (S. demissum) |
| Expression Level | ||||||||
|---|---|---|---|---|---|---|---|---|
| Diff. Region | Gene ID | SnpEff | Roots | Expanded Leaves | Open Flowers | Fruits 2–4 cm | Fruit Stage B | Fruit Stage C |
| A | SMEL_011g374890.1 | Mod | 0 | 0 | 0 | 0 | 0 | 0 |
| A | SMEL_011g374900.1 | High | 0.716154 | 12.2523 | 0 | 8.27647 | 3.05559 | 2.95479 |
| A+ | SMEL_011g374910.1 | High | 0 | 5.38543 | 0 | 4.35919 | 4.00833 | 16.8497 |
| A+ | SMEL_011g374920.1 | High | 26.9772 | 25.087 | 31.93 | 9.58168 | 16.8583 | 50.3871 |
| B | SMEL_011g375310.1 | High | 0 | 0 | 0 | 0 | 0.221595 | 0.474865 |
| B | SMEL_011g375320.1 | Mod | 0 | 0 | 0 | 0 | 0.636017 | 0 |
| D | SMEL_011g376860.1 | High | 0 | 0 | 0 | 0 | 0 | 0 |
| D | SMEL_011g376900.1 | High | 0 | 0 | 0 | 0 | 0 | 0 |
| D | SMEL_011g376910.1 | High | 0 | 0 | 0 | 0 | 0 | 0 |
| E | SMEL_011g377340.1 | High | 1.31895 | 0 | 0 | 0.761885 | 1.12618 | 0 |
| E | SMEL_011g377380.1 | ND | 0 | 0 | 0 | 0 | 0 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tassone, M.R.; Bagnaresi, P.; Desiderio, F.; Bassolino, L.; Barchi, L.; Florio, F.E.; Sunseri, F.; Sirangelo, T.M.; Rotino, G.L.; Toppino, L. A Genomic BSAseq Approach for the Characterization of QTLs Underlying Resistance to Fusarium oxysporum in Eggplant. Cells 2022, 11, 2548. https://doi.org/10.3390/cells11162548
Tassone MR, Bagnaresi P, Desiderio F, Bassolino L, Barchi L, Florio FE, Sunseri F, Sirangelo TM, Rotino GL, Toppino L. A Genomic BSAseq Approach for the Characterization of QTLs Underlying Resistance to Fusarium oxysporum in Eggplant. Cells. 2022; 11(16):2548. https://doi.org/10.3390/cells11162548
Chicago/Turabian StyleTassone, Maria Rosaria, Paolo Bagnaresi, Francesca Desiderio, Laura Bassolino, Lorenzo Barchi, Francesco Elia Florio, Francesco Sunseri, Tiziana Maria Sirangelo, Giuseppe Leonardo Rotino, and Laura Toppino. 2022. "A Genomic BSAseq Approach for the Characterization of QTLs Underlying Resistance to Fusarium oxysporum in Eggplant" Cells 11, no. 16: 2548. https://doi.org/10.3390/cells11162548
APA StyleTassone, M. R., Bagnaresi, P., Desiderio, F., Bassolino, L., Barchi, L., Florio, F. E., Sunseri, F., Sirangelo, T. M., Rotino, G. L., & Toppino, L. (2022). A Genomic BSAseq Approach for the Characterization of QTLs Underlying Resistance to Fusarium oxysporum in Eggplant. Cells, 11(16), 2548. https://doi.org/10.3390/cells11162548