
Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System

 ,
, 
 ,
,  ,
, 
Abstract
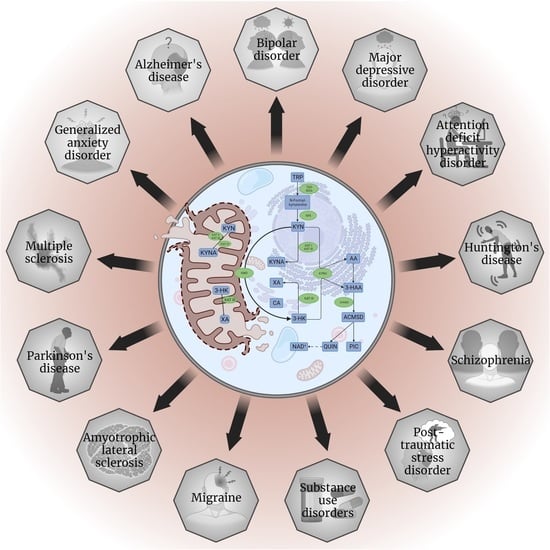
:
1. Introduction
2. Mitochondria in the Central Nervous System
2.1. Mitochondrial Bioenergetics
2.2. Other Mitochondrial Functions
3. The Tryptophan–Kynurenine Metabolic System
3.1. Tryptophan 2,3-Dioxygenase, Indoleamine 2,3-Dioxygenases, and Kynurenine Formamidase
3.2. Kynurenine 3-Monooxygenase
3.3. Kynurenine Aminotransferases
3.4. Kynureninase
3.5. 3-Hydroxyanthranilate 3,4-Dioxygenase, and toward the Tricyclic Carboxylic Cycle
4. Diseases Linked to Mitochondrial Dysfunction
4.1. Primary Mitochondrial Diseases
4.2. Secondary Mitochondrial Dysfunction
4.3. Neurological Disesases Linked to Mitochondrial Dysfunction
4.3.1. Alzheimer’s Disease
4.3.2. Parkinson’s Disease
4.3.3. Multiple Sclerosis
4.3.4. Huntington’s Disease
4.3.5. Amyotrophic Lateral Sclerosis
4.3.6. Migraine
4.4. Psychiatric Disorders Linked to Mitochondrial Dysfunction
4.4.1. Major Depressive Disorder
4.4.2. Generalized Anxiety Disorder
4.4.3. Post-Traumatic Stress Disorder
4.4.4. Bipolar Disorder
4.4.5. Substance Use Disorders
4.4.6. Schizophrenia
4.4.7. Autism Spectrum Disorder
4.4.8. Attention-Deficit Hyperactivity Disorder
5. Conclusions and Future Perspective
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | anthranilic acid |
| Acetyl-CoA | acetyl coenzyme A |
| ACMS | 2-amino-3-carboxymuconate semialdehyde |
| ACMSD | 2-amino-3-carboxymuconate-6-semialdehyde decarboxylase |
| acyl-CoA | acyl coenzyme A |
| AD | Alzheimer’s disease |
| ADHD | attention-deficit hyperactive disorder |
| AHR | aryl hydrocarbon receptor |
| ALS | amyotrophic lateral sclerosis |
| AMPA | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid |
| AMS | 2-aminomuconic-6-semialdehyde |
| AMSD | 2-aminomuconate semialdehyde dehydrogenase |
| Aβ | amyloid beta |
| ASD | autism spectrum disorder |
| ATP | adenosine triphosphate |
| BAC | bacterial artificial chromosome |
| BCG | Bacillus Calmette-Guérin |
| BD | bipolar disorder |
| BPA | bisphenol A |
| BSSG | beta-sitosterol beta-d-glucoside |
| CA | cinnabarinic acid |
| CMS | chronic mild stress |
| C9orf72 | chromosome 9 open reading frame 72 |
| Cys | cysteine |
| EAE | experimental autoimmune/allergic encephalomyelitis |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| FA | Friedreich’s ataxia |
| FADH2 | flavin adenine dinucleotide |
| FVB | Friend leukemia virus B |
| GABA | gamma-aminobutyric acid |
| GAD | generalized anxiety disorder |
| GTP | guanosine triphosphate |
| GPR35 | G-protein-coupled receptor 35 |
| H+ | proton |
| HD | Huntington’s disease |
| 5-HT | serotonin |
| i.c.v. | intracerebroventricular |
| IDO | indoleamine 2,3-dioxygenase |
| IDO1 | indoleamine 2,3-dioxygenase isoform 1 |
| IDO2 | indoleamine 2,3-dioxygenase isoform 2 |
| 3-HAA | 3-hydroxyanthranilic acid |
| 3-HK | 3-hydroxy-L-kynurenine |
| 3-HAO | 3-hydroxyanthranilate oxidase |
| HTT | huntingtin |
| 5-HTT | 5-HT transporter |
| iPSC | induced pluripotent stem cells |
| KAT | kynurenine aminotransferase |
| KFA | kynurenine formamidase |
| KI | knock-in |
| KMO | kynurenine 3-monooxygenase |
| KO | knockout |
| KYN | kynurenine |
| KYNA | Kynurenic acid |
| KYNU | kynureninase |
| MDD | major depressive disorder |
| MELAS | mitochondrial encephalomyopathy with lactic acidosis and stroke-like episodes |
| MPT | mitochondrial permeability transition |
| MS | multiple sclerosis |
| mtDNA | mitochondrial DNA |
| MTF | mitofusin |
| NaAD | nicotinic acid adenine dinucleotide |
| NADH | nicotinamide adenine dinucleotide |
| NaMN | nicotinic acid mononucleotide |
| nDNA | nuclear DNA |
| NDUFS2 | NADH:Ubiquinone Oxidoreductase Core Subunit S2 |
| NMDA | N-methyl-D-aspartic acid |
| NMS | non-motor symptoms |
| OCD | obsessive-compulsive disorder |
| OXPHOS | oxidative phosphorylation |
| parkin | Parkinson juvenile disease protein 2 |
| Pi | inorganic phosphate |
| PINK1 | phosphatase and tensin homolog (PTEN)-induced kinase1 |
| PD | Parkinson’s diseases |
| PFC | prefrontal cortex |
| PIC | picolinic acid |
| PLP | pyridoxal 5’-phosphate |
| PMD | primary mitochondrial disease |
| PMM | primary mitochondrial myopathy |
| PTSD | post-traumatic stress disorder |
| QPRT | quinolinate phosphoribosyltransferase |
| QUIN | quinolinic acid |
| RCS | reactive chemical species |
| SCZ | schizophrenia |
| SNP | single-nucleotide polymorphism |
| SMD | secondary mitochondrial dysfunction |
| SOD1 | superoxide dismutase 1 |
| SSRI | selective serotonin reuptake inhibitor |
| succinyl-CoA | succinyl coenzyme A |
| SUD | substance use disorder |
| TDP-43 | transactive response (TAR) DNA binding protein 43 kDa |
| TDO | tryptophan 2,3-dioxygenase |
| TPH | tryptophan hydroxylase |
| Trp | tryptophan |
| TST | tail suspension test |
| XA | xanthurenic acid |
References
- Fiske, C.H.; Subbarow, Y. Phosphorus Compounds of Muscle and Liver. Science 1929, 70, 381–382. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K. Über die Pyrophosphatfraktion im Muskel. Naturwissenschaften 1929, 17, 624–625. [Google Scholar] [CrossRef]
- Maruyama, K. The Discovery of Adenosine Triphosphate and the Establishment of its Structure. J. Hist. Biol. 1991, 24, 145–154. [Google Scholar] [CrossRef]
- Krebs, H.A.; Johnson, W.A. Metabolism of ketonic acids in animal tissues. Biochem. J. 1937, 31, 645–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedkin, M.; Lehninger, A.L. Esterification of inorganic phosphate coupled to electron transport between dihydrodiphosphopyridine nucleotide and oxygen. J. Biol. Chem. 1949, 178, 611–644. [Google Scholar] [CrossRef]
- Belitser, V.A.; Tsibakova, E.T. About phosphorilation mechanism coupled with respiration. Biokhimiya 1939, 4, 516–534. [Google Scholar]
- Gano, L.B.; Patel, M.; Rho, J.M. Ketogenic diets, mitochondria, and neurological diseases. J. Lipid Res. 2014, 55, 2211–2228. [Google Scholar] [CrossRef] [Green Version]
- Knoop, F. Der Abbau aromatischer Fettsäuren im Tierkörper. Beitr. Chem. Physiol. Pathol. 1904, 6, 150–162. [Google Scholar]
- Klingman, D.; Handler, P. Partial purification and properties of renal glutaminase. J. Biol. Chem. 1958, 232, 369–380. [Google Scholar] [CrossRef]
- Ernster, L.; Ikkos, D.; Luft, R. Enzymatic activities of human skeletal muscle mitochondria: A tool in clinical metabolic research. Nature 1959, 184, 1851–1854. [Google Scholar] [CrossRef]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J., 2nd; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef]
- Bourgeron, T.; Rustin, P.; Chretien, D.; Birch-Machin, M.; Bourgeois, M.; Viegas-Péquignot, E.; Munnich, A.; Rötig, A. Mutation of a nuclear succinate dehydrogenase gene results in mitochondrial respiratory chain deficiency. Nat. Genet. 1995, 11, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Carafoli, E. The interplay of mitochondria with calcium: An historical appraisal. Cell Calcium 2012, 52, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Diogo, C.V.; Yambire, K.F.; Fernández Mosquera, L.; Branco, F.T.; Raimundo, N. Mitochondrial adventures at the organelle society. Biochem. Biophys. Res. Commun. 2018, 500, 87–93. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the Etiological Links behind Neurodegenerative Diseases: Inflammatory Cytokines and Bioactive Kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Tóth, F.; Polyák, H.; Szabó, Á.; Mándi, Y.; Vécsei, L. Immune Influencers in Action: Metabolites and Enzymes of the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 734. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Vécsei, L. Are Kynurenines Accomplices or Principal Villains in Dementia? Maintenance of Kynurenine Metabolism. Molecules 2020, 25, 564. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Vécsei, L. Monitoring the kynurenine system: Concentrations, ratios or what else? Adv. Clin. Exp. Med. 2021, 30, 775–778. [Google Scholar] [CrossRef]
- Stenton, S.L.; Prokisch, H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine 2020, 56, 102784. [Google Scholar] [CrossRef]
- Koene, S.; Wortmann, S.B.; de Vries, M.C.; Jonckheere, A.I.; Morava, E.; de Groot, I.J.; Smeitink, J.A. Developing outcome measures for pediatric mitochondrial disorders: Which complaints and limitations are most burdensome to patients and their parents? Mitochondrion 2013, 13, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Niyazov, D.M.; Kahler, S.G.; Frye, R.E. Primary Mitochondrial Disease and Secondary Mitochondrial Dysfunction: Importance of Distinction for Diagnosis and Treatment. Mol. Syndromol. 2016, 7, 122–137. [Google Scholar] [CrossRef] [Green Version]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative Stress and Mitochondrial Damage in Neurodegenerative Diseases: From Molecular Mechanisms to Targeted Therapies. Oxid. Med. Cell Longev. 2020, 2020, 1270256. [Google Scholar] [CrossRef] [PubMed]
- Rigotto, G.; Basso, E. Mitochondrial Dysfunctions: A Thread Sewing Together Alzheimer’s Disease, Diabetes, and Obesity. Oxid. Med. Cell Longev. 2019, 2019, 7210892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Simoes, I.C.M.; Ren, Z.; Morciano, G.; Perrone, M.; Patalas-Krawczyk, P.; Borchard, S.; Jędrak, P.; Pierzynowska, K.; et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018, 340, 209–344. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [Green Version]
- Daniels, T.E.; Olsen, E.M.; Tyrka, A.R. Stress and Psychiatric Disorders: The Role of Mitochondria. Annu. Rev. Clin. Psychol. 2020, 16, 165–186. [Google Scholar] [CrossRef] [Green Version]
- Raichle, M.E.; Gusnard, D.A. Appraising the brain’s energy budget. Proc. Natl. Acad. Sci. USA 2002, 99, 10237–10239. [Google Scholar] [CrossRef] [Green Version]
- Kay, J.; Weitzman, P.D. Krebs’ Citric Acid Cycle: Half a Century and Still Turning; Biochemical Society: London, UK, 1987; p. 25. [Google Scholar]
- Zhang, H.; Ryu, D.; Wu, Y.; Gariani, K.; Wang, X.; Luan, P.; D’Amico, D.; Ropelle, E.R.; Lutolf, M.P.; Aebersold, R.; et al. NAD+ repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 2016, 352, 1436–1443. [Google Scholar] [CrossRef] [Green Version]
- Stryer, L. Fatty acid metabolism. In Biochemistry, 4th ed.; W.H. Freeman and Company: New York, NY, USA, 1995; pp. 603–628. [Google Scholar]
- Ahmad, M.; Wolberg, A.; Kahwaji, C.I. Biochemistry, Electron Transport Chain. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK526105/ (accessed on 6 July 2022).
- Klingenberg, M. The ADP and ATP transport in mitochondria and its carrier. Biochim. Biophys. Acta 2008, 1778, 1978–2021. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Menazza, S.; Holmström, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.; Zhang, D.; He, X.; Huang, Y.; Shao, H. Transport of Calcium Ions into Mitochondria. Curr. Genom. 2016, 17, 215–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palego, L.; Betti, L.; Rossi, A.; Giannaccini, G. Tryptophan Biochemistry: Structural, Nutritional, Metabolic, and Medical Aspects in Humans. J. Amino Acids 2016, 2016, 8952520. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Gao, Y.; Liu, J.; Huang, Y.; Yin, J.; Feng, Y.; Shi, L.; Meloni, B.P.; Zhang, C.; Zheng, M.; et al. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal. Transduct. Target Ther. 2021, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A. The citric acid cycle and the Szent-Györgyi cycle in pigeon breast muscle. Biochem. J. 1940, 34, 775–779. [Google Scholar] [CrossRef] [Green Version]
- Lipmann, F.; Kaplan, N.O. A common factor in the enzymatic acetylation of sulfanilamide and of choline. J. Biol. Chem. 1946, 162, 743–744. [Google Scholar] [CrossRef]
- Lipmann, F. Development of the Acetylation Problem: A personal Account. Available online: https://www.nobelprize.org/prizes/medicine/1953/lipmann/lecture/ (accessed on 6 July 2022).
- Romero-Garcia, S.; Prado-Garcia, H. Mitochondrial calcium: Transport and modulation of cellular processes in homeostasis and cancer (Review). Int. J. Oncol. 2019, 54, 1155–1167. [Google Scholar] [CrossRef] [Green Version]
- Xia, M.; Zhang, Y.; Jin, K.; Lu, Z.; Zeng, Z.; Xiong, W. Communication between mitochondria and other organelles: A brand-new perspective on mitochondria in cancer. Cell Biosci. 2019, 9, 27. [Google Scholar] [CrossRef] [Green Version]
- Soledad, R.B.; Charles, S.; Samarjit, D. The secret messages between mitochondria and nucleus in muscle cell biology. Arch. Biochem. Biophys. 2019, 666, 52–62. [Google Scholar] [CrossRef]
- Yeo, A.J.; Chong, K.L.; Gatei, M.; Zou, D.; Stewart, R.; Withey, S.; Wolvetang, E.; Parton, R.G.; Brown, A.D.; Kastan, M.B.; et al. Impaired endoplasmic reticulum-mitochondrial signaling in ataxia-telangiectasia. iScience 2020, 24, 101972. [Google Scholar] [CrossRef]
- Todkar, K.; Ilamathi, H.S.; Germain, M. Mitochondria and Lysosomes: Discovering Bonds. Front. Cell Dev. Biol. 2017, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demarquoy, J.; Le Borgne, F. Crosstalk between mitochondria and peroxisomes. World J. Biol. Chem. 2015, 6, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noguchi, M.; Kasahara, A. Mitochondrial dynamics coordinate cell differentiation. Biochem. Biophys. Res. Commun. 2018, 27, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Hales, K.G. Mitochondrial Fusion and Division. Nat. Educ. 2010, 3, 12. [Google Scholar]
- Thomas, R.L.; Gustafsson, A.B. Mitochondrial autophagy—An essential quality control mechanism for myocardial homeostasis. Circ. J. 2013, 77, 2449–2454. [Google Scholar] [CrossRef] [Green Version]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial Interactome: A Focus on Antiviral Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100. [Google Scholar] [CrossRef]
- Barik, S. The Uniqueness of Tryptophan in Biology: Properties, Metabolism, Interactions and Localization in Proteins. Int. J. Mol. Sci. 2020, 21, 8776. [Google Scholar] [CrossRef] [PubMed]
- Van Donkelaar, E.L.; Blokland, A.; Ferrington, L.; Kelly, P.A.; Steinbusch, H.W.; Prickaerts, J. Mechanism of acute tryptophan depletion: Is it only serotonin? Mol. Psychiatry 2011, 16, 695–713. [Google Scholar] [CrossRef]
- Blankfield, A. A Brief Historic Overview of Clinical Disorders Associated with Tryptophan: The Relevance to Chronic Fatigue Syndrome (CFS) and Fibromyalgia (FM). Int. J. Tryptophan. Res. 2012, 5, 27–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marx, W.; McGuinness, A.J.; Rocks, T.; Ruusunen, A.; Cleminson, J.; Walker, A.J.; Gomes-da-Costa, S.; Lane, M.; Sanches, M.; Diaz, A.P.; et al. The kynurenine pathway in major depressive disorder, bipolar disorder, and schizophrenia: A meta-analysis of 101 studies. Mol. Psychiatry 2021, 26, 4158–4178. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for Peripheral Biomarkers in Neurodegenerative Diseases: The Tryptophan-Kynurenine Metabolic Pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef] [PubMed]
- Jamshed, L.; Debnath, A.; Jamshed, S.; Wish, J.V.; Raine, J.C.; Tomy, G.T.; Thomas, P.J.; Holloway, A.C. An Emerging Cross-Species Marker for Organismal Health: Tryptophan-Kynurenine Pathway. Int. J. Mol. Sci. 2022, 23, 6300. [Google Scholar] [CrossRef] [PubMed]
- UniProtKB—P48775 (T23O_HUMAN). Available online: https://www.uniprot.org/uniprot/P48775 (accessed on 6 July 2022).
- Comings, D.E. Clinical and molecular genetics of ADHD and Tourette syndrome. Two related polygenic disorders. Ann. N. Y. Acad. Sci. 2001, 931, 50–83. [Google Scholar] [CrossRef]
- Nabi, R.; Serajee, F.J.; Chugani, D.C.; Zhong, H.; Huq, A.H. Association of tryptophan 2,3 dioxygenase gene polymorphism with autism. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2004, 125B, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kanai, M.; Funakoshi, H.; Takahashi, H.; Hayakawa, T.; Mizuno, S.; Matsumoto, K.; Nakamura, T. Tryptophan 2,3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol. Brain 2009, 27, 8. [Google Scholar] [CrossRef] [Green Version]
- Too, L.K.; Li, K.M.; Suarna, C.; Maghzal, G.J.; Stocker, R.; McGregor, I.S.; Hunt, N.H. Behavioral and cognitive data in mice with different tryptophan-metabolizing enzymes knocked out. Data Brief 2016, 6, 275–287. [Google Scholar] [CrossRef]
- Too, L.K.; Li, K.M.; Suarna, C.; Maghzal, G.J.; Stocker, R.; McGregor, I.S.; Hunt, N.H. Deletion of TDO2, IDO-1 and IDO-2 differentially affects mouse behavior and cognitive function. Behav. Brain Res. 2016, 1, 102–117. [Google Scholar] [CrossRef]
- Hattori, S.; Takao, K.; Funakoshi, H.; Miyakawa, T. Comprehensive behavioral analysis of tryptophan 2,3-dioxygenase (Tdo2) knockout mice. Neuropsychopharmacol. Rep. 2018, 38, 52–60. [Google Scholar] [CrossRef] [Green Version]
- UniProtKB—P14902 (I23O1_HUMAN). Available online: https://www.uniprot.org/uniprot/P14902 (accessed on 6 July 2022).
- UniProtKB—Q6ZQW0 (I23O2_HUMAN). Available online: https://www.uniprot.org/uniprot/Q6ZQW0 (accessed on 6 July 2022).
- Kim, H.; Chen, L.; Lim, G.; Sung, B.; Wang, S.; McCabe, M.F.; Rusanescu, G.; Yang, L.; Tian, Y.; Mao, J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J. Clin. Investig. 2012, 122, 2940–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, J.C.; Lawson, M.A.; André, C.; Briley, E.M.; Szegedi, S.S.; Lestage, J.; Castanon, N.; Herkenham, M.; Dantzer, R.; Kelley, K.W. Induction of IDO by bacille Calmette-Guérin is responsible for development of murine depressive-like behavior. J. Immunol. 2009, 182, 3202–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, N.; Hattori, S.; Shoji, H.; Funakoshi, H.; Miyakawa, T. Comprehensive behavioral analysis of indoleamine 2,3-dioxygenase knockout mice. Neuropsychopharmacol. Rep. 2018, 38, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, A.K.; Simon, J.S.; Gustafson, E.L.; Noviello, S.; Cubells, J.F.; Epstein, M.P.; Devlin, D.J.; Qiu, P.; Albrecht, J.K.; Brass, C.A.; et al. Association of a polymorphism in the indoleamine-2,3-dioxygenase gene and interferon-α-induced depression in patients with chronic hepatitis C. Mol. Psychiatry 2012, 17, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutler, J.A.; Rush, A.J.; McMahon, F.J.; Laje, G. Common genetic variation in the indoleamine-2,3-dioxygenase genes and antidepressant treatment outcome in major depressive disorder. J. Psychopharmacol. 2012, 26, 360–367. [Google Scholar] [CrossRef]
- UniProtKB—Q63HM1 (KFA_HUMAN). Available online: https://www.uniprot.org/uniprot/Q63HM1 (accessed on 6 July 2022).
- Ramírez Ortega, D.; Ugalde Muñiz, P.E.; Blanco Ayala, T.; Vázquez Cervantes, G.I.; Lugo Huitrón, R.; Pineda, B.; González Esquivel, D.F.; Pérez de la Cruz, G.; Pedraza Chaverrí, J.; Sánchez Chapul, L.; et al. On the Antioxidant Properties of L-Kynurenine: An Efficient ROS Scavenger and Enhancer of Rat Brain Antioxidant Defense. Antioxidants 2022, 11, 31. [Google Scholar] [CrossRef]
- UniProtKB—O15229 (KMO_HUMAN). Available online: https://www.uniprot.org/uniprot/O15229 (accessed on 6 July 2022).
- Mor, A.; Tankiewicz-Kwedlo, A.; Krupa, A.; Pawlak, D. Role of Kynurenine Pathway in Oxidative Stress during Neurodegenerative Disorders. Cells 2021, 10, 1603. [Google Scholar] [CrossRef]
- Giorgini, F.; Huang, S.Y.; Sathyasaikumar, K.V.; Notarangelo, F.M.; Thomas, M.A.; Tararina, M.; Wu, H.Q.; Schwarcz, R.; Muchowski, P.J. Targeted deletion of kynurenine 3-monooxygenase in mice: A new tool for studying kynurenine pathway metabolism in periphery and brain. J. Biol. Chem. 2013, 288, 36554–36566. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, S.; Pocivavsek, A.; Repici, M.; Liu, X.C.; Imbeault, S.; Maddison, D.C.; Thomas, M.A.R.; Smalley, J.L.; Larsson, M.K.; Muchowski, P.J.; et al. Adaptive and Behavioral Changes in Kynurenine 3-Monooxygenase Knockout Mice: Relevance to Psychotic Disorders. Biol. Psychiatry 2017, 82, 756–765. [Google Scholar] [CrossRef] [Green Version]
- Holtze, M.; Saetre, P.; Engberg, G.; Schwieler, L.; Werge, T.; Andreassen, O.A.; Hall, H.; Terenius, L.; Agartz, I.; Jönsson, E.G.; et al. Kynurenine 3-monooxygenase polymorphisms: Relevance for kynurenic acid synthesis in patients with schizophrenia and healthy controls. J. Psychiatry Neurosci. 2012, 37, 53–57. [Google Scholar] [CrossRef] [Green Version]
- Wonodi, I.; McMahon, R.P.; Krishna, N.; Mitchell, B.D.; Liu, J.; Glassman, M.; Hong, L.E.; Gold, J.M. Influence of kynurenine 3-monooxygenase (KMO) gene polymorphism on cognitive function in schizophrenia. Schizophr. Res. 2014, 160, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Cai, T.; Tagle, D.A.; Robinson, H.; Li, J. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci. Rep. 2008, 28, 205–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- UniProtKB—Q16773 (KAT1_HUMAN). Available online: https://www.uniprot.org/uniprot/Q16773 (accessed on 6 July 2022).
- UniProtKB—Q4W5N8 (Q4W5N8_HUMAN). Available online: https://www.uniprot.org/uniprot/Q4W5N8 (accessed on 6 July 2022).
- UniProtKB—Q6YP21 (KAT3_HUMAN). Available online: https://www.uniprot.org/uniprot/Q6YP21 (accessed on 6 July 2022).
- UniProtKB—P00505 (AATM_HUMAN). Available online: https://www.uniprot.org/uniprot/P00505 (accessed on 6 July 2022).
- Okada, K.; Angkawidjaja, C.; Koga, Y.; Takano, K.; Kanaya, S. Characteristic features of kynurenine aminotransferase allosterically regulated by (alpha)-ketoglutarate in cooperation with kynurenine. PLoS ONE 2012, 7, e40307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herédi, J.; Berkó, A.M.; Jankovics, F.; Iwamori, T.; Iwamori, N.; Ono, E.; Horváth, S.; Kis, Z.; Toldi, J.; Vécsei, L.; et al. Astrocytic and neuronal localization of kynurenine aminotransferase-2 in the adult mouse brain. Brain Struct. Funct. 2017, 222, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Prescott, C.; Weeks, A.M.; Staley, K.J.; Partin, K.M. Kynurenic acid has a dual action on AMPA receptor responses. Neurosci. Lett. 2006, 402, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Rózsa, E.; Robotka, H.; Vécsei, L.; Toldi, J. The Janus-face kynurenic acid. J. Neural. Transm. 2008, 115, 1087–1091. [Google Scholar] [CrossRef]
- Stone, T.W. Does kynurenic acid act on nicotinic receptors? An assessment of the evidence. J. Neurochem. 2020, 152, 627–649. [Google Scholar] [CrossRef] [Green Version]
- Copeland, C.S.; Neale, S.A.; Salt, T.E. Actions of Xanthurenic acid, a putative endogenous Group II metabotropic glutamate receptor agonist, on sensory transmission in the thalamus. Neuropharmacology 2013, 66, 133–142. [Google Scholar] [CrossRef]
- Bartlett, R.D.; Esslinger, C.S.; Thompson, C.M.; Bridges, R.J. Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles. Neuropharmacology 1998, 37, 839–846. [Google Scholar] [CrossRef]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Cavallari, M.; Zappulla, C.; Ulivieri, M.; Napoletano, F.; Capi, M.; Corigliano, V.; et al. Xanthurenic Acid Activates mGlu2/3 Metabotropic Glutamate Receptors and is a Potential Trait Marker for Schizophrenia. Sci. Rep. 2015, 8, 17799. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Di Prospero, N.A.; Sapko, M.T.; Cai, T.; Chen, A.; Melendez-Ferro, M.; Du, F.; Whetsell, W.O., Jr.; Guidetti, P.; Schwarcz, R.; et al. Biochemical and phenotypic abnormalities in kynurenine aminotransferase II-deficient mice. Mol. Cell. Biol. 2004, 24, 6919–6930. [Google Scholar] [CrossRef] [Green Version]
- Martos, D.; Tuka, B.; Tanaka, M.; Vécsei, L.; Telegdy, G. Memory Enhancement with Kynurenic Acid and Its Mechanisms in Neurotransmission. Biomedicines 2022, 10, 849. [Google Scholar] [CrossRef]
- Tanaka, M.; Bohár, Z.; Martos, D.; Telegdy, G.; Vécsei, L. Antidepressant-like effects of kynurenic acid in a modified forced swim test. Pharmacol. Rep. 2020, 72, 449–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Szabó, Á.; Lőrinczi, B.; Szatmári, I.; Fülöp, F.; Vécsei, L. Antidepressant-like Effects of Kynurenic Acid Analogues. In Proceedings of the 1st International Electronic Conference on Biomedicine, Online, 1–26 June 2021. [Google Scholar] [CrossRef]
- Potter, M.C.; Elmer, G.I.; Bergeron, R.; Albuquerque, E.X.; Guidetti, P.; Wu, H.Q.; Schwarcz, R. Reduction of endogenous kynurenic acid formation enhances extracellular glutamate, hippocampal plasticity, and cognitive behavior. Neuropsychopharmacology 2010, 35, 1734–1742. [Google Scholar] [CrossRef] [PubMed]
- Vécsei, L.; (University of Szeged, Szeged, Hungary). Personal communication, 2020.
- UniProtKB—Q16719 (KYNU_HUMAN). Available online: https://www.uniprot.org/uniprot/Q16719 (accessed on 6 July 2022).
- Issa, F.; Kirch, D.G.; Gerhardt, G.A.; Bartko, J.J.; Suddath, R.L.; Freedman, R.; Wyatt, R.J. A Multidimensional Approach to Analysis of Cerebrospinal Fluid Biogenic Amines in Schizophrenia: II. Correlations with Psychopathology. Psychiatry Res. 1994, 52, 251–258. [Google Scholar] [CrossRef]
- Badawy, A.A. Kynurenine Pathway of Tryptophan Metabolism: Regulatory and Functional Aspects. Int. J. Tryptophan Res. 2017, 15, 1178646917691938. [Google Scholar] [CrossRef] [Green Version]
- Bala, S.; Kamboj, S.; Saini, V.; Prasad, D.N. Anti-inflammatory, analgesic evaluation and molecular docking studies of N-phenyl anthranilic acid-based 1,3,4-oxadiazole analogues. J. Chem. 2013, 2013, 412053. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.M.M.A.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 10, 377, 544–552. [Google Scholar] [CrossRef]
- Zhang, Y.; Shen, J.; He, X.; Zhang, K.; Wu, S.; Xiao, B.; Zhou, X.; Phillips, R.S.; Gao, P.; Jeunemaitre, X.; et al. A rare variant at the KYNU gene is associated with kynureninase activity and essential hypertension in the Han Chinese population. Circ. Cardiovasc. Genet. 2011, 4, 687–694. [Google Scholar] [CrossRef] [Green Version]
- Farooq, R.K.; Asghar, K.; Kanwal, S.; Zulqernain, A. Role of inflammatory cytokines in depression: Focus on interleukin-1β. Biomed. Rep. 2017, 6, 15–20. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Qu, P.; Sun, Y.; Li, Z.; Liu, L.; Yang, L. Association between increased inflammatory cytokine expression in the lateral habenular nucleus and depressive-like behavior induced by unpredictable chronic stress in rats. Exp. Neurol. 2022, 349, 113964. [Google Scholar] [CrossRef] [PubMed]
- Hepsomali, P.; Coxon, C. Inflammation and diet: Focus on mental and cognitive health. Adv. Clin. Exp. Med. 2022. [published online ahead of print, 2022 Aug 11]. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhou, L.J.; Wang, J.; Li, D.; Ren, W.J.; Peng, J.; Wei, X.; Xu, T.; Xin, W.J.; Pang, R.P.; et al. TNF-α Differentially Regulates Synaptic Plasticity in the Hippocampus and Spinal Cord by Microglia-Dependent Mechanisms after Peripheral Nerve Injury. J. Neurosci. 2017, 37, 871–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boes, A.D.; Kelly, M.S.; Trapp, N.T.; Stern, A.P.; Press, D.Z.; Pascual-Leone, A. Noninvasive Brain Stimulation: Challenges and Opportunities for a New Clinical Specialty. J. Neuropsychiatry Clin. Neurosci. 2018, 30, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Thayer, J.F. Functional interplay between central and autonomic nervous systems in human fear conditioning. Trends Neurosci. 2022, 45, 504–506. [Google Scholar] [CrossRef]
- Battaglia, S.; Orsolini, S.; Borgomaneri, S.; Barbieri, R.; Diciotti, S.; di Pellegrino, G. Characterizing cardiac autonomic dynamics of fear learning in humans. Psychophysiology 2022, e14122. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Garofalo, S.; Tortora, F.; Avenanti, A.; di Pellegrino, G. State-Dependent TMS over Prefrontal Cortex Disrupts Fear-Memory Reconsolidation and Prevents the Return of Fear. Curr. Biol. 2020, 30, 3672–3679.e4. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories are not written in stone: Re-writing fear memories by means of non-invasive brain stimulation and optogenetic manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Avenanti, A.; Pellegrino, G.D. Don’t Hurt Me No More: State-dependent Transcranial Magnetic Stimulation for the treatment of specific phobia. J. Affect Disord. 2021, 286, 78–79. [Google Scholar] [CrossRef]
- Gonzalez-Escamilla, G.; Dörfel, D.; Becke, M.; Trefz, J.; Bonanno, G.A.; Groppa, S. Associating Flexible Regulation of Emotional Expression with Psychopathological Symptoms. Front. Behav. Neurosci. 2022, 16, 924305. [Google Scholar] [CrossRef]
- UniProtKB—P46952 (3HAO_HUMAN). Available online: https://www.uniprot.org/uniprot/P46952 (accessed on 6 July 2022).
- Lugo-Huitrón, R.; Ugalde Muñiz, P.; Pineda, B.; Pedraza-Chaverrí, J.; Ríos, C.; Pérez-de la Cruz, V. Quinolinic acid: An endogenous neurotoxin with multiple targets. Oxid. Med. Cell. Longev. 2013, 2013, 104024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majláth, Z.; Toldi, J.; Vécsei, L. The potential role of kynurenines in Alzheimer’s disease: Pathomechanism and therapeutic possibilities by influencing the glutamate receptors. J. Neural. Transm. 2014, 121, 881–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Egashira, N.; Masuda, S. Recent Topics on The Mechanisms of Immunosuppressive Therapy-Related Neurotoxicities. Int. J. Mol. Sci. 2019, 20, 3210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, P.F.; Tonin, A.; da Costa Ferreira, G.; Rosa, R.B.; Latini, A.; Balestro, F.; Perry, M.L.; Wannmacher, C.M.; de Souza Wyse, A.T.; Wajner, M. In vitro effect of quinolinic acid on energy metabolism in brain of young rats. Neurosci. Res. 2007, 57, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Essa, M.M.; de Paula Martins, R.; Lovejoy, D.B.; Bilgin, A.A.; Waly, M.I.; Al-Farsi, Y.M.; Al-Sharbati, M.; Al-Shaffae, M.A.; Guillemin, G.J. Altered kynurenine pathway metabolism in autism: Implication for immune-induced glutamatergic activity. Autism Res. 2016, 9, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Ulivieri, M.; Wierońska, J.M.; Lionetto, L.; Martinello, K.; Cieslik, P.; Chocyk, A.; Curto, M.; Di Menna, L.; Iacovelli, L.; Traficante, A.; et al. The Trace Kynurenine, Cinnabarinic Acid, Displays Potent Antipsychotic-Like Activity in Mice and Its Levels Are Reduced in the Prefrontal Cortex of Individuals Affected by Schizophrenia. Schizophr. Bull. 2020, 46, 1471–1481. [Google Scholar] [CrossRef]
- Lesiewska, N.; Borkowska, A.; Junik, R.; Kamińska, A.; Jaracz, K.; Bieliński, M. Consequences of diabetes and pre-diabetes and the role of biochemical parameters of carbohydrate metabolism for the functioning of the prefrontal cortex in obese patients. Front. Biosci. 2022, 27, 76. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Serio, G.; Battaglia, S. Please, don’t do it! Fifteen years of progress of non-invasive brain stimulation in action inhibition. Cortex 2020, 132, 404–422. [Google Scholar] [CrossRef]
- Battaglia, S.; Cardellicchio, P.; Di Fazio, C.; Nazzi, C.; Fracasso, A.; Borgomaneri, S. The Influence of Vicarious Fear-Learning in “Infecting” Reactive Action Inhibition. Front. Behav. Neurosci. 2022, 16, 946263. [Google Scholar] [CrossRef]
- Sellitto, M.; Terenzi, D.; Starita, F.; di Pellegrino, G.; Battaglia, S. The Cost of Imagined Actions in a Reward-Valuation Task. Brain Sci. 2022, 12, 582. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2022, 27, 784–786. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, S.; Timmermann, C.; Battaglia, S.; Maier, M.E.; di Pellegrino, G. Mediofrontal Negativity Signals Unexpected Timing of Salient Outcomes. J. Cogn. Neurosci. 2017, 29, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, O.L.; Chuan-Peng, H.; Salas, C.E.; Yuen, K.S.L. Obliviate! Reviewing Neural Fundamentals of Intentional Forgetting from a Meta-Analytic Perspective. Biomedicines 2022, 10, 1555. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G.; Starita, F. Revaluing the Role of vmPFC in the Acquisition of Pavlovian Threat Conditioning in Humans. J. Neurosci. 2020, 40, 8491–8500. [Google Scholar] [CrossRef]
- Battaglia, S.; Serio, G.; Scarpazza, C.; D’Ausilio, A.; Borgomaneri, S. Frozen in (e)motion: How reactive motor inhibition is influenced by the emotional content of stimuli in healthy and psychiatric populations. Behav. Res. Ther. 2021, 146, 103963. [Google Scholar] [CrossRef]
- Milad, M.R.; Pitman, R.K.; Ellis, C.B.; Gold, A.L.; Shin, L.M.; Lasko, N.B.; Zeidan, M.A.; Handwerger, K.; Orr, S.P.; Rauch, S.L. Neurobiological basis of failure to recall extinction memory in posttraumatic stress disorder. Biol. Psychiatry 2009, 66, 1075–1082. [Google Scholar] [CrossRef]
- Holt, D.J.; Lebron-Milad, K.; Milad, M.R.; Rauch, S.L.; Pitman, R.K.; Orr, S.P.; Cassidy, B.S.; Walsh, J.P.; Goff, D.C. Extinction memory is impaired in schizophrenia. Biol. Psychiatry 2009, 65, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Fabius, J.H.; Moravkova, K.; Fracasso, A.; Borgomaneri, S. The Neurobiological Correlates of Gaze Perception in Healthy Individuals and Neurologic Patients. Biomedicines 2022, 10, 627. [Google Scholar] [CrossRef]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef]
- Santuy, A.; Turégano-López, M.; Rodríguez, J.R.; Alonso-Nanclares, L.; DeFelipe, J.; Merchán-Pérez, A. A Quantitative Study on the Distribution of Mitochondria in the Neuropil of the Juvenile Rat Somatosensory Cortex. Cereb. Cortex 2018, 28, 3673–3684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.; Hirabayashi, Y.; Kwon, S.K.; Lewis, T.L., Jr.; Polleux, F. Emerging roles of mitochondria in synaptic transmission and neurodegeneration. Curr. Opin. Physiol. 2018, 3, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Huang, M.; Shang, D.; Yan, X.; Zhao, B.; Zhang, X. Mitochondrial Behavior in Axon Degeneration and Regeneration. Front. Aging Neurosci. 2021, 13, 650038. [Google Scholar] [CrossRef] [PubMed]
- Carelli, V.; Chan, D.C. Mitochondrial DNA: Impacting central and peripheral nervous systems. Neuron 2014, 84, 1126–1142. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.; Ahmad, K.; Sue, C.M. The broadening spectrum of mitochondrial disease: Shifts in the diagnostic paradigm. Biochim. Biophys. Acta 2014, 1840, 1360–1367. [Google Scholar] [CrossRef] [Green Version]
- Frye, R.E. Mitochondrial Dysfunction in Autism Spectrum Disorder: Unique Abnormalities and Targeted Treatments. Semin. Pediatr. Neurol. 2020, 35, 100829. [Google Scholar] [CrossRef]
- Scaglia, F. The role of mitochondrial dysfunction in psychiatric disease. Dev. Disabil. Res. Rev. 2010, 16, 136–143. [Google Scholar] [CrossRef]
- Khan, N.A.; Govindaraj, P.; Meena, A.K.; Thangaraj, K. Mitochondrial disorders: Challenges in diagnosis & treatment. Indian J. Med. Res. 2015, 141, 13–26. [Google Scholar] [CrossRef]
- Falk, M.J.; Sondheimer, N. Mitochondrial genetic diseases. Curr. Opin. Pediatr. 2010, 22, 711–716. [Google Scholar] [CrossRef] [Green Version]
- Almannai, M.; El-Hattab, A.W.; Ali, M.; Soler-Alfonso, C.; Scaglia, F. Clinical trials in mitochondrial disorders, an update. Mol. Genet. Metab. 2020, 131, 1–13. [Google Scholar] [CrossRef]
- McCormick, E.M.; Zolkipli-Cunningham, Z.; Falk, M.J. Mitochondrial disease genetics update: Recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr. Opin. Pediatr. 2018, 30, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Kanungo, S.; Morton, J.; Neelakantan, M.; Ching, K.; Saeedian, J.; Goldstein, A. Mitochondrial disorders. Ann. Transl. Med. 2018, 6, 475. [Google Scholar] [CrossRef] [PubMed]
- Montano, V.; Gruosso, F.; Simoncini, C.; Siciliano, G.; Mancuso, M. Clinical features of mtDNA-related syndromes in adulthood. Arch. Biochem. Biophys. 2021, 697, 108689. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.C.; Lee, H.F.; Yue, C.T.; Chi, C.S. Clinical Characteristics of Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Episodes. Life 2021, 11, 1111. [Google Scholar] [CrossRef]
- Tetsuka, S.; Ogawa, T.; Hashimoto, R.; Kato, H. Clinical features, pathogenesis, and management of stroke-like episodes due to MELAS. Metab. Brain Dis. 2021, 36, 2181–2193. [Google Scholar] [CrossRef]
- Schlieben, L.D.; Prokisch, H. The Dimensions of Primary Mitochondrial Disorders. Front. Cell Dev. Biol. 2020, 8, 600079. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef] [Green Version]
- Beekman, M.; Dowling, D.K.; Aanen, D.K. The costs of being male: Are there sex-specific effects of uniparental mitochondrial inheritance? Philos. Trans. R. Soc. Lond. Bol. Sci. 2014, 369, 20130440. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [Green Version]
- Ruhoy, I.S.; Saneto, R.P. The genetics of Leigh syndrome and its implications for clinical practice and risk management. Appl. Clin. Genet. 2014, 7, 221–234. [Google Scholar] [CrossRef] [Green Version]
- Thompson Legault, J.; Strittmatter, L.; Tardif, J.; Sharma, R.; Tremblay-Vaillancourt, V.; Aubut, C.; Boucher, G.; Clish, C.B.; Cyr, D.; Daneault, C.; et al. A Metabolic Signature of Mitochondrial Dysfunction Revealed through a Monogenic Form of Leigh Syndrome. Cell Rep. 2015, 13, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Sundaramurthy, S.; SelvaKumar, A.; Ching, J.; Dharani, V.; Sarangapani, S.; Yu-Wai-Man, P. Leber hereditary optic neuropathy-new insights and old challenges. Graefes. Arch. Clin. Exp. Ophthalmol. 2021, 259, 2461–2472. [Google Scholar] [CrossRef] [PubMed]
- Stenton, S.L.; Sheremet, N.L.; Catarino, C.B.; Andreeva, N.A.; Assouline, Z.; Barboni, P.; Barel, O.; Berutti, R.; Bychkov, I.; Caporali, L.; et al. Impaired complex I repair causes recessive Leber’s hereditary optic neuropathy. J. Clin. Investig. 2021, 131, e138267. [Google Scholar] [CrossRef]
- Klivenyi, P.; Karg, E.; Rozsa, C.; Horvath, R.; Komoly, S.; Nemeth, I.; Turi, S.; Vécsei, L. alpha-Tocopherol/lipid ratio in blood is decreased in patients with Leber’s hereditary optic neuropathy and asymptomatic carriers of the 11,778 mtDNA mutation. J. Neurol. Neurosurg. Psychiatry 2001, 70, 359–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khajuria, K.; Khajuria, V.; Sawhney, V. Secondary Mitochondrial Dysfunction. Int. J. Pharm. Pharm. Sci. 2021, 13, 14–19. [Google Scholar] [CrossRef]
- Barahona, A.J.; Bursac, Z.; Veledar, E.; Lucchini, R.; Tieu, K.; Richardson, J.R. Relationship of Blood and Urinary Manganese Levels with Cognitive Function in Elderly Individuals in the United States by Race/Ethnicity, NHANES 2011–2014. Toxics 2022, 10, 191. [Google Scholar] [CrossRef]
- Jopowicz, A.; Wiśniowska, J.; Tarnacka, B. Cognitive and Physical Intervention in Metals’ Dysfunction and Neurodegeneration. Brain Sci. 2022, 12, 345. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial diseases of the brain. Free Radic. Biol. Med. 2013, 63, 1–29. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue “Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry”. Biomedicines 2021, 9, 517. [Google Scholar] [CrossRef]
- Tanaka, M.; Vécsei, L. Editorial of Special Issue ‘Dissecting Neurological and Neuropsychiatric Diseases: Neurodegeneration and Neuroprotection’. Int. J. Mol. Sci. 2022, 23, 6991. [Google Scholar] [CrossRef] [PubMed]
- Telegdy, G.; Tanaka, M.; Schally, A.V. Effects of the LHRH antagonist Cetrorelix on the brain function in mice. Neuropeptides 2009, 43, 229–234. [Google Scholar] [CrossRef]
- Telegdy, G.; Tanaka, M.; Schally, A.V. Effects of the growth hormone-releasing hormone (GH-RH) antagonist on brain functions in mice. Behav. Brain Res. 2011, 224, 155–1558. [Google Scholar] [CrossRef]
- Palotai, M.; Telegdy, G.; Tanaka, M.; Bagosi, Z.; Jászberényi, M. Neuropeptide AF induces anxiety-like and antidepressant-like behavior in mice. Behav. Brain Res. 2014, 274, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Kádár, K.; Tóth, G.; Telegdy, G. Antidepressant-like effects of urocortin 3 fragments. Brain Res. Bull. 2011, 84, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Mirchandani-Duque, M.; Barbancho, M.A.; López-Salas, A.; Alvarez-Contino, J.E.; García-Casares, N.; Fuxe, K.; Borroto-Escuela, D.O.; Narváez, M. Galanin and Neuropeptide Y Interaction Enhances Proliferation of Granule Precursor Cells and Expression of Neuroprotective Factors in the Rat Hippocampus with Consequent Augmented Spatial Memory. Biomedicines 2022, 10, 1297. [Google Scholar] [CrossRef]
- Simon, C.; Soga, T.; Ahemad, N.; Bhuvanendran, S.; Parhar, I. Kisspeptin-10 Rescues Cholinergic Differentiated SHSY-5Y Cells from α-Synuclein-Induced Toxicity In Vitro. Int. J. Mol. Sci. 2022, 23, 5193. [Google Scholar] [CrossRef] [PubMed]
- Naito, M.; Iwakoshi-Ukena, E.; Moriwaki, S.; Narimatsu, Y.; Kato, M.; Furumitsu, M.; Miyamoto, Y.; Esumi, S.; Ukena, K. Immunohistochemical Analysis of Neurotransmitters in Neurosecretory Protein GL-Producing Neurons of the Mouse Hypothalamus. Biomedicines 2022, 10, 454. [Google Scholar] [CrossRef]
- Dias, F.L.; Silva, R.M.; Moraes, E.N.; Caramelli, P. Clinical and autonomic profile of patients with Alzheimer’s disease and mixed dementia patients. Rev. Assoc. Médica Bras. 2013, 59, 435–441. [Google Scholar] [CrossRef] [Green Version]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef]
- Orso, B.; Lorenzini, L.; Arnaldi, D.; Girtler, N.; Brugnolo, A.; Doglione, E.; Mattioli, P.; Biassoni, E.; Massa, F.; Peira, E.; et al. The Role of Hub and Spoke Regions in Theory of Mind in Early Alzheimer’s Disease and Frontotemporal Dementia. Biomedicines 2022, 10, 544. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. CSH Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [Green Version]
- Dai, M.H.; Zheng, H.; Zeng, L.D.; Zhang, Y. The genes associated with early-onset Alzheimer’s disease. Oncotarget 2017, 9, 15132–15143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rönnbäck, A.; Pavlov, P.F.; Mansory, M.; Gonze, P.; Marlière, N.; Winblad, B.; Graff, C.; Behbahani, H. Mitochondrial dysfunction in a transgenic mouse model expressing human amyloid precursor protein (APP) with the Arctic mutation. J. Neurochem. 2016, 136, 497–502. [Google Scholar] [CrossRef] [Green Version]
- Dixit, S.; Fessel, J.P.; Harrison, F.E. Mitochondrial dysfunction in the APP/PSEN1 mouse model of Alzheimer’s disease and a novel protective role for ascorbate. Free Radic. Biol. Med. 2017, 112, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Mehla, J.; Lacoursiere, S.G.; Lapointe, V.; McNaughton, B.L.; Sutherland, R.J.; McDonald, R.J.; Mohajerani, M.H. Age-dependent behavioral and biochemical characterization of single APP knock-in mouse (APPNL-G-F/NL-G-F) model of Alzheimer’s disease. Neurobiol. Aging 2019, 75, 25–37. [Google Scholar] [CrossRef]
- Guillemin, G.J.; Brew, B.J.; Noonan, C.E.; Takikawa, O.; Cullen, K.M. Indoleamine 2,3 hippocampus dioxygenase and quinolinic acid immunoreactivity in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2005, 31, 395–404. [Google Scholar] [CrossRef]
- Bonda, D.; Mailankot, M.; Stone, J.G.; Garrett, M.R.; Staniszewska, M.; Castellani, R.J.; Siedlak, S.L.; Zhu, X.; Lee, H.; Perry, G.; et al. Indoleamine 2,3-dioxygenase and 3-hydroxykynurenine modifications are found in the neuropathology of Alzheimer’s disease. Redox Rep. 2010, 15, 161–168. [Google Scholar] [CrossRef] [Green Version]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef]
- Almulla, A.F.; Supasitthumrong, T.; Amrapala, A.; Tunvirachaisakul, C.; Jaleel, A.K.A.; Oxenkrug, G.; Al-Hakeim, H.K.; Maes, M. The Tryptophan Catabolite or Kynurenine Pathway in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Alzheimer’s Dis. 2022; in press. [Google Scholar] [CrossRef]
- Schwarcz, M.J.; Guillemin, G.J.; Teipel, S.J.; Buerger, K.; Hampel, H. Increased 3-hydroxykynurenine serum concentrations differentiate Alzheimer’s disease patients from controls. Eur. Arch. Psychiatry Clin. Neurosci. 2013, 263, 345. [Google Scholar] [CrossRef]
- Jacobs, K.; Lim, C.K.; Blennow, K.; Zetterberg, H.; Chatterjee, P.; Martins, R.N.; Brew, B.; Guillemin, G.; Lovejoy, D. Correlation between plasma and CSF concentrations of kynurenine pathway metabolites in Alzheimer’s disease and relationship to amyloid-β and tau. Neurobiol. Aging 2019, 80, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef]
- Park, A.; Stacy, M. Non-motor symptoms in Parkinson’s disease. J. Neurol. 2009, 256 (Suppl. S3), 293–298. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Ellena, G.; Battaglia, S.; Làdavas, E. The spatial effect of fearful faces in the autonomic response. Exp. Brain. Res. 2020, 238, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Borgomaneri, S.; Vitale, F.; Battaglia, S.; Avenanti, A. Early Right Motor Cortex Response to Happy and Fearful Facial Expressions: A TMS Motor-Evoked Potential Study. Brain Sci. 2021, 11, 1203. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Moisoi, N.; Fedele, V.; Edwards, J.; Martins, L.M. Loss of PINK1 enhances neurodegeneration in a mouse model of Parkinson’s disease triggered by mitochondrial stress. Neuropharmacology 2014, 77, 350–357. [Google Scholar] [CrossRef]
- Chia, S.J.; Tan, E.-K.; Chao, Y.-X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [Green Version]
- Kee, T.R.; Espinoza Gonzalez, P.; Wehinger, J.L.; Bukhari, M.Z.; Ermekbaeva, A.; Sista, A.; Kotsiviras, P.; Liu, T.; Kang, D.E.; Woo, J.A. Mitochondrial CHCHD2: Disease-Associated Mutations, Physiological Functions, and Current Animal Models. Front. Aging Neurosci. 2021, 13, 660843. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.W.; Choi, W.S.; Sorscher, N.; Park, H.J.; Tronche, F.; Palmiter, R.D.; Xia, Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 2015, 36, 2617–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latchoumycandane, C.; Anantharam, V.; Jin, H.; Kanthasamy, A.; Kanthasamy, A. Dopaminergic neurotoxicant 6-OHDA induces oxidative damage through proteolytic activation of PKCδ in cell culture and animal models of Parkinson’s disease. Toxicol. Appl. Pharmacol. 2011, 256, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vécsei, L. Kynurenine metabolism in plasma and in red blood cells in Parkinson’s disease. J. Neurol. Sci. 2005, 239, 31–35. [Google Scholar] [CrossRef]
- Lewitt, P.A.; Li, J.; Lu, M.; Beach, T.G.; Adler, C.H.; Guo, L. Arizona Parkinson’s Disease Consortium. 3-hydroxykynurenine and other Parkinson’s disease biomarkers discovered by metabolomic analysis. Mov. Disord. 2013, 28, 1653–1660. [Google Scholar] [CrossRef] [PubMed]
- Török, N.; Maszlag-Török, R.; Molnár, K.; Szolnoki, Z.; Somogyvári, F.; Boda, K.; Tanaka, M.; Klivényi, P.; Vécsei, L. Single Nucleotide Polymorphisms of Indoleamine 2,3-Dioxygenase 1 Influenced the Age Onset of Parkinson’s Disease. Preprints 2020, 2020090470. [Google Scholar] [CrossRef]
- Boeschoten, R.E.; Braamse, A.M.J.; Beekman, A.T.F.; Cuijpers, P.; van Oppen, P.; Dekker, J.; Uitdehaag, B.M.J. Prevalence of Depression and Anxiety in Multiple Sclerosis: A Systematic Review and Meta-Analysis. J. Neurol. Sci. 2017, 372, 331–341. [Google Scholar] [CrossRef]
- Huang, W.J.; Chen, W.W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barcelos, I.P.D.; Troxell, R.M.; Graves, J.S. Mitochondrial Dysfunction and Multiple Sclerosis. Biology 2019, 8, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Vécsei, L. Monitoring the Redox Status in Multiple Sclerosis. Biomedicines 2020, 8, 406. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; De Rosa, V.; Pucino, V.; Formisano, L.; Matarese, G. Animal models of Multiple Sclerosis. Eur. J. Pharmacol. 2015, 759, 182–191. [Google Scholar] [CrossRef]
- Pukoli, D.; Polyák, H.; Rajda, C.; Vécsei, L. Kynurenines and Neurofilament Light Chain in Multiple Sclerosis. Front. Neurosci. 2021, 15, 658202. [Google Scholar] [CrossRef]
- Qi, X.; Lewin, A.S.; Sun, L.; Hauswirth, W.W.; Guy, J. Mitochondrial protein nitration primes neurodegeneration in experimental autoimmune encephalomyelitis. J. Biol. Chem. 2006, 281, 31950–31962. [Google Scholar] [CrossRef]
- Nikić, I.; Merkler, D.; Sorbara, C.; Brinkoetter, M.; Kreutzfeldt, M.; Bareyre, F.M.; Brück, W.; Bishop, D.; Misgeld, T.; Kerschensteiner, M. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat. Med. 2011, 17, 495–499. [Google Scholar] [CrossRef]
- Sadeghian, M.; Mastrolia, V.; Rezaei Haddad, A.; Mosley, A.; Mullali, G.; Schiza, D.; Sajic, M.; Hargreaves, I.; Heales, S.; Duchen, M.R.; et al. Mitochondrial dysfunction is an important cause of neurological deficits in an inflammatory model of multiple sclerosis. Sci. Rep. 2016, 14, 33249. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Asano, M.; Kurono, C. Mechanism of the formation of megamitochondria induced by copper-chelating agents. II. Isolation and some properties of megamitochondria from the cuprizone-treated mouse liver. Acta Pathol. Jpn. 1975, 25, 39–49. [Google Scholar]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; Lenoël, I.; Stys, P.K.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef]
- Praet, J.; Guglielmetti, C.; Berneman, Z.; Van der Linden, A.; Ponsaerts, P. Cellular and molecular neuropathology of the cuprizone mouse model: Clinical relevance for multiple sclerosis. Neurosci. Biobehav. Rev. 2014, 47, 485–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flatmark, T.; Kryvi, H.; Tangerås, A. Induction of megamitochondria by cuprizone (biscyclohexanone oxaldihydrazone). Evidence for an inhibition of the mitochondrial division process. Eur. J. Cell Biol. 1980, 23, 141–148. [Google Scholar] [PubMed]
- Kozin, M.; Kulakova, O.; Kiselev, I.; Baulina, N.; Boyko, A.; Favorova, O. Mitonuclear interactions influence multiple sclerosis risk. Gene 2020, 758, 144962. [Google Scholar] [CrossRef]
- Polyák, H.; Cseh, E.K.; Bohár, Z.; Rajda, C.; Zádori, D.; Klivényi, P.; Toldi, J.; Vécsei, L. Cuprizone markedly decreases kynurenic acid levels in the rodent brain tissue and plasma. Heliyon 2021, 7, e06124. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.K.; Bilgin, A.; Lovejoy, D.B.; Tan, V.; Bustamante, S.; Taylor, B.V.; Bessede, A.; Brew, B.J.; Guillemin, G.J. Kynurenine pathway metabolomics predicts and provides mechanistic insight into multiple sclerosis progression. Sci. Rep. 2017, 7, 41473. [Google Scholar] [CrossRef]
- Rajda, C.; Galla, Z.; Polyák, H.; Maróti, Z.; Babarczy, K.; Pukoli, D.; Vécsei, L. Cerebrospinal Fluid Neurofilament Light Chain Is Associated with Kynurenine Pathway Metabolite Changes in Multiple Sclerosis. Int. J. Mol. Sci. 2020, 21, 2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aeinehband, S.; Brenner, P.; Ståhl, S.; Bhat, M.; Fidock, M.D.; Khademi, M.; Olsson, T.; Engberg, G.; Jokinen, J.; Erhardt, S.; et al. Cerebrospinal fluid kynurenines in multiple sclerosis; relation to disease course and neurocognitive symptoms. Brain Behav. Immun. 2016, 51, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartai, Z.; Klivenyi, P.; Janaky, T.; Penke, B.; Dux, L.; Vécsei, L. Kynurenine metabolism in multiple sclerosis. Acta Neurol. Scand. 2005, 112, 93–96. [Google Scholar] [CrossRef]
- Reiner, A.; Dragatsis, I.; Dietrich, P. Genetics and neurophatology of huntington’s disease. Int. Rev. Neurobiol. 2011, 98, 325–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangiarini, L.; Sathasivam, K.; Seller, M.; Cozens, B.; Harper, A.; Hetherington, C.; Lawton, M.; Trottier, Y.; Lehrach, H.; Davies, S.W.; et al. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell 1996, 87, 493–506. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Di Pardo, A.; Maglione, V.; Schwarzer, C.; Squitieri, F. Mitochondrial Respiration Changes in R6/2 Huntington’s Disease Model Mice during Aging in a Brain Region Specific Manner. Int. J. Mol. Sci. 2020, 21, 5412. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.; Shirasaki, D.I.; Cepeda, C.; André, V.M.; Wilburn, B.; Lu, X.H.; Tao, J.; Yamazaki, I.; Li, S.H.; Sun, Y.E.; et al. Full-length human mutant huntingtin with a stable polyglutamine repeat can elicit progressive and selective neuropathogenesis in BACHD mice. J. Neurosci. 2008, 28, 6182–6195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menalled, L.B. Knock-in mouse models of Huntington’s disease. NeuroRx 2005, 2, 465–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Choo, Y.S.; Johnson, G.V.; MacDonald, M.; Detloff, P.J.; Lesort, M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum. Mol. Genet. 2004, 13, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Kaye, J.; Reisine, T.; Finkbeiner, S. Huntington’s disease mouse models: Unraveling the pathology caused by CAG repeat expansion. Fac. Rev. 2021, 10, 77. [Google Scholar] [CrossRef]
- Widner, B.; Leblhuber, F.; Walli, J.; Tilz, G.P.; Demel, U.; Fuchs, D. Degradation of tryptophan in neurodegenerative disorders. Adv. Exp. Med. Biol. 1999, 467, 133–138. [Google Scholar] [CrossRef]
- Guidetti, P.; Luthi-Carter, R.E.; Augood, S.J.; Schwarcz, R. Neostriatal and cortical quinolinate levels are increased in early grade Huntington’s disease. Neurobiol. Dis. 2004, 17, 455–461. [Google Scholar] [CrossRef]
- Pearson, S.J.; Reynolds, G.P. Increased brain concentrations of a neurotoxin, 3-hydroxykynurenine, in Huntington’s disease. Neurosci. Lett. 1992, 144, 199–201. [Google Scholar] [CrossRef]
- Iłzecka, J.; Kocki, T.; Stelmasiak, Z.; Turski, W.A. Endogenous protectant kynurenic acid in amyotrophic lateral sclerosis. Acta Neurol. Scand. 2003, 107, 412–418. [Google Scholar] [CrossRef]
- Beal, M.F.; Matson, W.R.; Swartz, K.J.; Gamache, P.H.; Bird, E.D. Kynurenine pathway measurements in Huntington’s disease striatum: Evidence for reduced formation of kynurenic acid. J. Neurochem. 1990, 55, 1327–1339. [Google Scholar] [CrossRef] [PubMed]
- Forrest, C.M.; Mackay, G.M.; Stoy, N.; Spiden, S.L.; Taylor, R.; Stone, T.W.; Darlington, L.G. Blood levels of kynurenines, interleukin-23 and soluble human leucocyte antigen-G at different stages of Huntington’s disease. J. Neurochem. 2010, 112, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Gubbay, S.; Kahana, E.; Zilber, N.; Cooper, G.; Pintov, S.; Leibowitz, Y. Amyotrophic lateral sclerosis. A study of its presentation and prognosis. J. Neurol. 1985, 232, 295–300. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef] [PubMed]
- Morrice, J.R.; Gregory-Evans, C.Y.; Shaw, C.A. Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regen. Res. 2018, 13, 2050–2054. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar] [CrossRef] [Green Version]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014, 23, 1413–1424. [Google Scholar] [CrossRef] [Green Version]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef]
- Wang, W.; Li, L.; Lin, W.L.; Dickson, D.W.; Petrucelli, L.; Zhang, T.; Wang, X. The ALS disease-associated mutant TDP-43 impairs mitochondrial dynamics and function in motor neurons. Hum. Mol. Genet. 2013, 22, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, S.; Yadav, A.; Tiwari, S.K.; Seth, B.; Chauhan, L.K.; Khare, P.; Ray, R.S.; Chaturvedi, R.K. Dynamin-related Protein 1 Inhibition Mitigates Bisphenol A-mediated Alterations in Mitochondrial Dynamics and Neural Stem Cell Proliferation and Differentiation. J. Biol. Chem. 2016, 291, 15923–15939. [Google Scholar] [CrossRef] [Green Version]
- Panov, A.; Kubalik, N.; Brooks, B.R.; Shaw, C.A. In vitro effects of cholesterol β-D-glucoside, cholesterol and cycad phytosterol glucosides on respiration and reactive oxygen species generation in brain mitochondria. J. Membr. Biol. 2010, 237, 71–77. [Google Scholar] [CrossRef]
- Chen, Y.; Stankovic, R.; Cullen, K.; Meininger, V.; Garner, B.; Coggan, S.; Grant, R.; Brew, B.J.; Guillemin, G.J. The kynurenine pathway and inflammation in amyotrophic lateral sclerosis. Neurotox. Res. 2010, 18, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Sparaco, M.; Feleppa, M.; Lipton, R.B.; Rapoport, A.M.; Bigal, M.E. Mitochondrial dysfunction and migraine: Evidence and hypotheses. Cephalalgia 2006, 26, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Montagna, P.; Sacquegna, T.; Martinelli, P.; Cortelli, P.; Bresolin, N.; Moggio, M.; Baldrati, A.; Riva, R.; Lugaresi, E. Mitochondrial Abnormalities in Migraine. Preliminary Findings. Headache J. Head Face Pain 1988, 28, 477–480. [Google Scholar] [CrossRef] [PubMed]
- Uncini, A.; Lodi, R.; Di Muzio, A.; Silvestri, G.; Servidei, S.; Lugaresi, A.; Iotti, S.; Zaniol, P.; Barbiroli, B. Abnormal brain and muscle energy metabolism shown by 31P-MRS in familial hemiplegic migraine. J. Neurol. Sci. 1995, 129, 214–222. [Google Scholar] [CrossRef]
- Skinhøj, E. Hemodynamic Studies within the Brain during Migraine. Arch. Neurol. 1973, 29, 95–98. [Google Scholar] [CrossRef]
- Okada, H.; Araga, S.; Takeshima, T.; Nakashima, K. Plasma lactic acid and pyruvic acid levels in migraine and tension-type headache. Headache 1998, 38, 39–42. [Google Scholar] [CrossRef]
- Littlewood, J.; Glover, V.; Sandler, M.; Peatfield, R.; Petty, R.; Clifford Rose, F. Low platelet monoamine oxidase activity in headache: No correlation with phenolsulphotransferase, succinate dehydrogenase, platelet preparation method or smoking. J. Neurol. Neurosurg. Psychiatry 1984, 47, 338–343. [Google Scholar] [CrossRef] [Green Version]
- Sangiorgi, S.; Mochi, M.; Riva, R.; Cortelli, P.; Monari, L.; Pierangeli, G.; Montagna, P. Abnormal platelet mitochondrial function in patients affected by migraine with and without aura. Cephalalgia 1994, 14, 21–23. [Google Scholar] [CrossRef]
- Barbiroli, B.; Montagna, P.; Cortelli, P.; Funicello, R.; Iotti, S.; Monari, L.; Pierangeli, G.; Zaniol, P.; Lugaresi, E. Abnormal brain and muscle energy metabolism shown by 31P magnetic resonance spectroscopy in patients affected by migraine with aura. Neurology 1992, 42, 1209–1214. [Google Scholar] [CrossRef]
- Schulz, U.G.; Blamire, A.M.; Corkill, R.G.; Davies, P.; Styles, P.; Rothwell, P.M. Association between cortical metabolite levels and clinical manifestations of migrainous aura: An MR-spectroscopy study. Brain 2007, 130 Pt 12, 3102–3110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyngoudt, H.; Achten, E.; Paemeleire, K. Magnetic resonance spectroscopy in migraine: What have we learned so far? Cephalalgia 2012, 32, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, E.C.; Lisicki, M.; Fischer, D.; Sándor, P.S.; Schoenen, J. The metabolic face of migraine—From pathophysiology to treatment. Nat. Rev. Neurol. 2019, 15, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Reyngoudt, H.; Paemeleire, K.; Descamps, B.; De Deene, Y.; Achten, E. 31P-MRS demonstrates a reduction in high-energy phosphates in the occipital lobe of migraine without aura patients. Cephalalgia 2011, 31, 1243–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Guan, X.; Chen, K.; Jin, S.; Wang, C.; Yan, L.; Shi, Z.; Zhang, X.; Chen, L.; Wan, Q. Abnormal mitochondrial dynamics and impaired mitochondrial biogenesis in trigeminal ganglion neurons in a rat model of migraine. Neurosci. Lett. 2017, 636, 127–133. [Google Scholar] [CrossRef]
- Fried, N.T.; Moffat, C.; Seifert, E.L.; Oshinsky, M.L. Functional mitochondrial analysis in acute brain sections from adult rats reveals mitochondrial dysfunction in a rat model of migraine. Am. J. Physiol. Cell Physiol. 2014, 307, C1017–C1030. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Liu, Y.; Chen, N.; Zhang, Y.; Song, G.; Zhang, Z. Valproate Attenuates Nitroglycerin-Induced Trigeminovascular Activation by Preserving Mitochondrial Function in a Rat Model of Migraine. Med. Sci. Monit. 2016, 22, 3229–3237. [Google Scholar] [CrossRef] [Green Version]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Perugino, F.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered serum levels of kynurenine metabolites in patients affected by cluster headache. J. Headache Pain 2015, 17, 27. [Google Scholar] [CrossRef] [Green Version]
- Curto, M.; Lionetto, L.; Negro, A.; Capi, M.; Fazio, F.; Giamberardino, M.A.; Simmaco, M.; Nicoletti, F.; Martelletti, P. Altered kynurenine pathway metabolites in serum of chronic migraine patients. J. Headache Pain 2015, 17, 47. [Google Scholar] [CrossRef] [Green Version]
- Tuka, B.; Nyári, A.; Cseh, E.K.; Körtési, T.; Veréb, D.; Tömösi, F.; Kecskeméti, G.; Janáky, T.; Tajti, J.; Vécsei, L. Clinical relevance of depressed kynurenine pathway in episodic migraine patients: Potential prognostic markers in the peripheral plasma during the interictal period. J. Headache Pain 2021, 22, 60. [Google Scholar] [CrossRef]
- Nagy-Grócz, G.; Tar, L.; Bohár, Z.; Fejes-Szabó, A.; Laborc, K.F.; Spekker, E.; Vécsei, L.; Párdutz, Á. The modulatory effect of anandamide on nitroglycerin-induced sensitization in the trigeminal system of the rat. Cephalalgia 2016, 36, 849–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spekker, E.; Tanaka, M.; Szabó, Á.; Vécsei, L. Neurogenic Inflammation: The Participant in Migraine and Recent Advancements in Translational Research. Biomedicines 2022, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Török, N.; Vécsei, L. Are 5-HT1 receptor agonists effective anti-migraine drugs? Expert. Opin. Pharmacother. 2021, 22, 1221–1225. [Google Scholar] [CrossRef]
- Tanaka, M.; Török, N.; Tóth, F.; Szabó, Á.; Vécsei, L. Co-Players in Chronic Pain: Neuroinflammation and the Tryptophan-Kynurenine Metabolic Pathway. Biomedicines 2021, 9, 897. [Google Scholar] [CrossRef]
- Ciapała, K.; Mika, J.; Rojewska, E. The Kynurenine Pathway as a Potential Target for Neuropathic Pain Therapy Design: From Basic Research to Clinical Perspectives. Int. J. Mol. Sci. 2021, 22, 11055. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, F.; Candido, K.D.; Knezevic, N.N. The Role of the Kynurenine Signaling Pathway in Different Chronic Pain Conditions and Potential Use of Therapeutic Agents. Int. J. Mol. Sci. 2020, 21, 6045. [Google Scholar] [CrossRef]
- Gecse, K.; Édes, A.E.; Nagy, T.; Demeter, A.K.; Virág, D.; Király, M.; Dalmadi Kiss, B.; Ludányi, K.; Környei, Z.; Denes, A.; et al. Citalopram Neuroendocrine Challenge Shows Altered Tryptophan and Kynurenine Metabolism in Migraine. Cells 2022, 11, 2258. [Google Scholar] [CrossRef]
- Fila, M.; Chojnacki, J.; Pawlowska, E.; Szczepanska, J.; Chojnacki, C.; Blasiak, J. Kynurenine Pathway of Tryptophan Metabolism in Migraine and Functional Gastrointestinal Disorders. Int. J. Mol. Sci. 2021, 22, 10134. [Google Scholar] [CrossRef]
- Tanaka, M.; Schally, A.V.; Telegdy, G. Neurotransmission of the antidepressant-like effects of the growth hormone-releasing hormone antagonist MZ-4-71. Behav. Brain Res. 2012, 228, 388–391. [Google Scholar] [CrossRef]
- Tanaka, M.; Telegdy, G. Neurotransmissions of antidepressant-like effects of neuromedin U-23 in mice. Behav. Brain Res. 2014, 259, 196–199. [Google Scholar] [CrossRef]
- Castillo-Mariqueo, L.; Giménez-Llort, L. Impact of Behavioral Assessment and Re-Test as Functional Trainings That Modify Survival, Anxiety and Functional Profile (Physical Endurance and Motor Learning) of Old Male and Female 3xTg-AD Mice and NTg Mice with Normal Aging. Biomedicines 2022, 10, 973. [Google Scholar] [CrossRef]
- Tanaka, M.; Telegdy, G. Antidepressant-like Effects of Neuropeptide SF (NPSF). arXiv 2020, arXiv:200513256. [Google Scholar] [CrossRef]
- Gutiérrez-Rojas, L.; Porras-Segovia, A.; Dunne, H.; Andrade-González, N.; Cervilla, J.A. Prevalence and correlates of major depressive disorder: A systematic review. Braz. J. Psychiatry 2020, 42, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Gładka, A.; Zatoński, T.; Rymaszewska, J. Association between the long-term exposure to air pollution and depression. Adv. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Chen, C. Recent advances in the study of the comorbidity of depressive and anxiety disorders. Adv. Clin. Exp. Med. 2022, 31, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Carrera-González, M.; Cantón-Habas, V.; Rich-Ruiz, M. Aging, depression and dementia: The inflammatory process. Adv. Clin. Exp. Med. 2022, 31, 469–473. [Google Scholar] [CrossRef]
- Tanaka, M. Crosstalk between Depression, Anxiety, and Dementia: Comorbidity in Behavioral Neurology and Neuropsychiatry, 1st ed.; MDPI: Basel, Switzerland, 2022; pp. 1–266. [Google Scholar]
- Delgado, P.L. Depression: The case for a monoamine deficiency. J. Clin. Psychiatry 2000, 61 (Suppl. S6), 7–11. [Google Scholar]
- Balogh, L.; Tanaka, M.; Török, N.; Vécsei, L.; Taguchi, S. Crosstalk between Existential Phenomenological Psychotherapy and Neurological Sciences in Mood and Anxiety Disorders. Biomedicines 2021, 9, 340. [Google Scholar] [CrossRef]
- Wohleb, E.; Franklin, T.; Iwata, M.; Duman, R.S. Integrating neuroimmune systems in the neurobiology of depression. Nat. Rev. Neurosci. 2016, 17, 497–511. [Google Scholar] [CrossRef]
- Adeel, M.; Chen, C.-C.; Lin, B.-S.; Chen, H.-C.; Liou, J.-C.; Li, Y.-T.; Peng, C.-W. Safety of Special Waveform of Transcranial Electrical Stimulation (TES): In Vivo Assessment. Int. J. Mol. Sci. 2022, 23, 6850. [Google Scholar] [CrossRef]
- Taliaz, D.; Spinrad, A.; Barzilay, R.; Barnett-Itzhaki, Z.; Averbuch, D.; Teltsh, O.; Schurr, R.; Darki-Morag, S.; Lerer, B. Optimizing prediction of response to antidepressant medications using machine learning and integrated genetic, clinical, and demographic data. Transl. Psychiatry 2021, 11, 381. [Google Scholar] [CrossRef] [PubMed]
- Gamaro, G.D.; Streck, E.L.; Matté, C.; Prediger, M.E.; Wyse, A.T.; Dalmaz, C. Reduction of hippocampal Na+, K+-ATPase activity in rats subjected to an experimental model of depression. Neurochem. Res. 2003, 28, 1339–1344. [Google Scholar] [CrossRef] [PubMed]
- Gong, Y.; Chai, Y.; Ding, J.H.; Sun, X.L.; Hu, G. Chronic mild stress damages mitochondrial ultrastructure and function in mouse brain. Neurosci. Lett. 2011, 488, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Jin, Y.; Li, L.; Sun, S.; Cheng, S.; Zhang, S.; Zhang, Y.; Svenningsson, P. Exercise prevents raphe nucleus mitochondrial overactivity in a rat depression model. Physiol. Behav. 2014, 132, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Trowbridge, S.; Narboux-Nême, N.; Gaspar, P. Genetic models of serotonin (5-HT) depletion: What do they tell us about the developmental role of 5-HT? Anat. Rec. 2011, 294, 1615–1623. [Google Scholar] [CrossRef]
- Crane, J.D.; Palanivel, R.; Mottillo, E.P.; Bujak, A.L.; Wang, H.; Ford, R.J.; Collins, A.; Blümer, R.M.; Fullerton, M.D.; Yabut, J.M.; et al. Inhibiting peripheral serotonin synthesis reduces obesity and metabolic dysfunction by promoting brown adipose tissue thermogenesis. Nat. Med. 2015, 21, 166–172. [Google Scholar] [CrossRef] [Green Version]
- Mosienko, V.; Bert, B.; Beis, D.; Matthes, S.; Fink, H.; Bader, M.; Alenina, N. Exaggerated aggression and decreased anxiety in mice deficient in brain serotonin. Transl. Psychiatry 2012, 2, e122. [Google Scholar] [CrossRef] [Green Version]
- Gutknecht, L.; Popp, S.; Waider, J.; Sommerlandt, F.M.; Göppner, C.; Post, A.; Reif, A.; van den Hove, D.; Strekalova, T.; Schmitt, A.; et al. Interaction of brain 5-HT synthesis deficiency, chronic stress and sex differentially impact emotional behavior in Tph2 knockout mice. Psychopharmacology 2015, 232, 2429–2441. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Timberlake, M.A., 2nd; Prall, K.; Dwivedi, Y. The recent progress in animal models of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 99–109. [Google Scholar] [CrossRef]
- Beaulieu, J.M.; Zhang, X.; Rodriguiz, R.M.; Sotnikova, T.D.; Cools, M.J.; Wetsel, W.C.; Gainetdinov, R.R.; Caron, M.G. Role of GSK3 beta in behavioral abnormalities induced by serotonin deficiency. Proc. Natl. Acad. Sci. USA 2008, 105, 1333–1338. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, S.; Fujii, T.; Koga, N.; Hori, H.; Teraishi, T.; Hattori, K.; Noda, T.; Higuchi, T.; Motohashi, N.; Kunugi, H. Plasma L-tryptophan concentration in major depressive disorder: New data and meta-analysis. J. Clin. Psychiatry 2014, 75, e906–e915. [Google Scholar] [CrossRef] [PubMed]
- Ogyu, K.; Kubo, K.; Noda, Y.; Iwata, Y.; Tsugawa, S.; Omura, Y.; Wada, M.; Tarumi, R.; Plitman, E.; Moriguchi, S.; et al. Kynurenine pathway in depression: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2018, 90, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Török, N.; Vécsei, L. Novel Pharmaceutical Approaches in Dementia. In NeuroPsychopharmacotherapy; Riederer, P., Laux, G., Nagatsu, T., Le, W., Riederer, C., Eds.; Springer: Cham, Switzerland, 2021. [Google Scholar] [CrossRef]
- Hunt, C.; Macedo e Cordeiro, T.; Suchting, R.; de Dios, C.; Cuellar Leal, V.A.; Soares, J.C.; Dantzer, R.; Teixeira, A.L.; Selvaraj, S. Effect of immune activation on the kynurenine pathway and depression symptoms—A systematic review and meta-analysis. Neurosci. Biobeha. Rev. 2020, 118, 514. [Google Scholar] [CrossRef] [PubMed]
- Erabi, H.; Okada, G.; Shibasaki, C.; Setoyama, D.; Kang, D.; Takamura, M.; Yoshino, A.; Fuchikami, M.; Kurata, A.; Kato, T.A.; et al. Kynurenic acid is a potential overlapped biomarker between diagnosis and treatment response for depression from metabolome analysis. Sci. Rep. 2020, 10, 16822. [Google Scholar] [CrossRef] [PubMed]
- Carrillo-Mora, P.; Pérez-De la Cruz, V.; Estrada-Cortés, B.; Toussaint-González, P.; Martínez-Cortéz, J.A.; Rodríguez-Barragán, M.; Quinzaños-Fresnedo, J.; Rangel-Caballero, F.; Gamboa-Coria, G.; Sánchez-Vázquez, I.; et al. Serum Kynurenines Correlate with Depressive Symptoms and Disability in Poststroke Patients: A Cross-sectional Study. Neurorehabilit. Neural Repair 2020, 34, 154596832095367. [Google Scholar] [CrossRef]
- Ruscio, A.M.; Hallion, L.S.; Lim, C.C.W.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Alonso, J.; Andrade, L.H.; Borges, G.; Bromet, E.J.; Bunting, B.; et al. Cross-sectional Comparison of the Epidemiology of DSM-5 Generalized Anxiety Disorder Across the Globe. JAMA Psychiatry 2017, 74, 465–475. [Google Scholar] [CrossRef] [Green Version]
- Martin, E.I.; Ressler, K.J.; Binder, E.; Nemeroff, C.B. The neurobiology of anxiety disorders: Brain imaging, genetics, and psychoneuroendocrinology. Psychiatr. Clin. N. Am. 2009, 32, 549–575. [Google Scholar] [CrossRef] [Green Version]
- Strawn, J.R.; Geracioti, L.; Rajdev, N.; Clemenza, K.; Levine, A. Pharmacotherapy for generalized anxiety disorder in adult and pediatric patients: An evidence-based treatment review. Expert. Opin. Pharmacother. 2018, 19, 1057–1070. [Google Scholar] [CrossRef]
- Bandelow, B.; Michaelis, S.; Wedekind, D. Treatment of anxiety disorders. Dialogues Clin. Neurosci. 2017, 19(2), 93–107. [Google Scholar] [CrossRef]
- Chen, A.P.; Chen, L.; Kim, T.A.; Xiong, Q. Integrating the Roles of Midbrain Dopamine Circuits in Behavior and Neuropsychiatric Disease. Biomedicines 2021, 9, 647. [Google Scholar] [CrossRef]
- Gebara, E.; Zanoletti, O.; Ghosal, S.; Grosse, J.; Schneider, B.L.; Knott, G.; Astori, S.; Sandi, C. Mitofusin-2 in the Nucleus Accumbens Regulates Anxiety and Depression-like Behaviors Through Mitochondrial and Neuronal Actions. Biol. Psychiatry 2021, 89, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Hollis, F.; van der Kooij, M.A.; Zanoletti, O.; Lozano, L.; Cantó, C.; Sandi, C. Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 2015, 112, 15486–15491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlikov, A.B.; Prakhye, I.B.; Ryzov, I.V. Kynurenine in blood plasma and DST in patients with endogenous anxiety and endogenous depression. Biol. Psychiatry 1994, 36, 97–102. [Google Scholar] [CrossRef]
- Altmaier, E.; Emeny, R.T.; Krumsiek, J.; Lacruz, M.E.; Lukaschek, K.; Häfner, S.; Kastenmüller, G.; Römisch-Margl, W.; Prehn, C.; Mohney, R.P.; et al. Metabolomic profiles in individuals with negative affectivity and social inhibition: A population-based study of Type D personality. Psychoneuroendocrinology 2013, 38, 1299–1309. [Google Scholar] [CrossRef]
- National Institute of Mental Health. Transforming the Understanding and Treatment of Mental Illnesses. Post-Traumatic Stress Disorder. Available online: https://www.nimh.nih.gov/health/publications/post-traumatic-stress-disorder-ptsd (accessed on 15 June 2022).
- Sareen, J.; Stein, M.B.; Friedman, M. Posttraumatic Stress Disorder in Adults: Epidemiology, Pathophysiology, Clinical Manifestations, Course, Assessment, and Diagnosis. UpToDate 2022. Available online: https://www.uptodate.com/contents/posttraumatic-stress-disorder-in-adults-epidemiology-pathophysiology-clinical-manifestations-course-assessment-and-diagnosis/print#:~:text=PTSD%20prevalence%20%E2%80%94%20The%20lifetime%20prevalence,percent%20%5B6%2C7%5D (accessed on 15 June 2022).
- Bedard-Gilligan, M.; Zoellner, L.A.; Feeny, N.C. Is Trauma Memory Special? Trauma Narrative Fragmentation in PTSD: Effects of Treatment and Response. Clin. Psychol. Sci. 2017, 5, 212–225. [Google Scholar] [CrossRef]
- Ehret, M. Treatment of posttraumatic stress disorder: Focus on pharmacotherapy. Ment. Health Clin. 2019, 9, 373–382. [Google Scholar] [CrossRef]
- Richter-Levin, G.; Stork, O.; Schmidt, M.V. Animal models of PTSD: A challenge to be met. Mol. Psychiatry 2019, 24, 1135–1156. [Google Scholar] [CrossRef] [Green Version]
- Sabbagh, J.J.; O’Leary, J.C., 3rd; Blair, L.J.; Klengel, T.; Nordhues, B.A.; Fontaine, S.N.; Binder, E.B.; Dickey, C.A. Age-associated epigenetic upregulation of the FKBP5 gene selectively impairs stress resiliency. PLoS ONE 2014, 9, e107241. [Google Scholar] [CrossRef]
- King, S.B.; Lezak, K.R.; O’Reilly, M.; Toufexis, D.J.; Falls, W.A.; Braas, K.; May, V.; Hammack, S.E. The Effects of Prior Stress on Anxiety-Like Responding to Intra-BNST Pituitary Adenylate Cyclase Activating Polypeptide in Male and Female Rats. Neuropsychopharmacology 2017, 42, 1679–1687. [Google Scholar] [CrossRef]
- Otto, C.; Martin, M.; Wolfer, D.P.; Lipp, H.P.; Maldonado, R.; Schütz, G. Altered emotional behavior in PACAP-type-I-receptor-deficient mice. Brain Res. Mol. Brain Res. 2001, 92, 78–84. [Google Scholar] [CrossRef]
- Hill, J.L.; Hardy, N.F.; Jimenez, D.V.; Maynard, K.R.; Kardian, A.S.; Pollock, C.J.; Schloesser, R.J.; Martinowich, K. Loss of promoter IV-driven BDNF expression impacts oscillatory activity during sleep, sensory information processing and fear regulation. Transl. Psychiatry 2016, 6, e873. [Google Scholar] [CrossRef] [PubMed]
- Soliman, F.; Glatt, C.E.; Bath, K.G.; Levita, L.; Jones, R.M.; Pattwell, S.S.; Jing, D.; Tottenham, N.; Amso, D.; Somerville, L.H.; et al. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 2010, 327, 863–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klemenhagen, K.C.; Gordon, J.A.; David, D.J.; Hen, R.; Gross, C.T. Increased fear response to contextual cues in mice lacking the 5-HT1A receptor. Neuropsychopharmacology 2006, 31, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellman, C.L.; Izquierdo, A.; Garrett, J.E.; Martin, K.P.; Carroll, J.; Millstein, R.; Lesch, K.P.; Murphy, D.L.; Holmes, A. Impaired stress-coping and fear extinction and abnormal corticolimbic morphology in serotonin transporter knock-out mice. J. Neurosci. 2007, 27, 684–691. [Google Scholar] [CrossRef]
- O’Tuathaigh, C.M.; Clarke, G.; Walsh, J.; Desbonnet, L.; Petit, E.; O’Leary, C.; Tighe, O.; Clarke, N.; Karayiorgou, M.; Gogos, J.A.; et al. Genetic vs. pharmacological inactivation of COMT influences cannabinoid-induced expression of schizophrenia-related phenotypes. Int. J. Neuropsychopharmacol. 2012, 15, 1331–1342. [Google Scholar] [CrossRef] [Green Version]
- Bergado-Acosta, J.R.; Sangha, S.; Narayanan, R.T.; Obata, K.; Pape, H.C.; Stork, O. Critical role of the 65-kDa isoform of glutamic acid decarboxylase in consolidation and generalization of Pavlovian fear memory. Learn. Mem. 2008, 15, 163–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangha, S.; Narayanan, R.T.; Bergado-Acosta, J.R.; Stork, O.; Seidenbecher, T.; Pape, H.C. Deficiency of the 65 kDa isoform of glutamic acid decarboxylase impairs extinction of cued but not contextual fear memory. J. Neurosci. 2009, 29, 15713–15720. [Google Scholar] [CrossRef] [PubMed]
- Shaban, H.; Humeau, Y.; Herry, C.; Cassasus, G.; Shigemoto, R.; Ciocchi, S.; Barbieri, S.; van der Putten, H.; Kaupmann, K.; Bettler, B.; et al. Generalization of amygdala LTP and conditioned fear in the absence of presynaptic inhibition. Nat. Neurosci. 2006, 9, 1028–1035. [Google Scholar] [CrossRef]
- Fride, E.; Suris, R.; Weidenfeld, J.; Mechoulam, R. Differential response to acute and repeated stress in cannabinoid CB1 receptor knockout newborn and adult mice. Behav. Pharmacol. 2005, 16, 431–440. [Google Scholar] [CrossRef]
- Preston, G.; Emmerzaal, T.; Kirdar, F.; Schrader, L.; Henckens, M.; Morava, E.; Kozicz, T. Cerebellar mitochondrial dysfunction and concomitant multi-system fatty acid oxidation defects are sufficient to discriminate PTSD-like and resilient male mice. Brain Behav. Immun. Health 2020, 6, 100104. [Google Scholar] [CrossRef]
- Jia, Y.; Han, Y.; Wang, X.; Han, F. Role of apoptosis in the Post-traumatic stress disorder model-single prolonged stressed rats. Psychoneuroendocrinology 2018, 95, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Gómez, P.; Ballestín, R.; Gil de Biedma-Elduayen, L.; Vidal, R.; Ferrer-Pérez, C.; Reguilón, M.D.; O’Shea, E.; Miñarro, J.; Colado, M.I.; Rodríguez-Arias, M. Decreased kynurenine pathway potentiate resilience to social defeat effect on cocaine reward. Neuropharmacology 2021, 197, 108753. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Cheng, P.-Y.; Hsiao, M.; Liu, Y.-P. Effects of RU486 in Treatment of Traumatic Stress-Induced Glucocorticoid Dysregulation and Fear-Related Abnormalities: Early versus Late Intervention. Int. J. Mol. Sci. 2022, 23, 5494. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.D.; Lee, S.; Yoon, S. Inflammation in Post-Traumatic Stress Disorder (PTSD): A Review of Potential Correlates of PTSD with a Neurological Perspective. Antioxidants 2020, 9, 107. [Google Scholar] [CrossRef] [Green Version]
- Merikangas, K.R.; Jin, R.; He, J.P.; Kessler, R.C.; Lee, S.; Sampson, N.A.; Viana, M.C.; Andrade, L.H.; Hu, C.; Karam, E.G.; et al. Prevalence and correlates of bipolar spectrum disorder in the world mental health survey initiative. Arch. Gen. Psychiatry 2011, 68, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Drevets, W.C.; Price, J.L.; Furey, M.L. Brain structural and functional abnormalities in mood disorders: Implications for neurocircuitry models of depression. Brain Struct. Funct. 2008, 213, 93–118. [Google Scholar] [CrossRef] [Green Version]
- Womer, F.Y.; Kalmar, J.H.; Wang, F.; Blumberg, H.P. A Ventral Prefrontal-Amygdala Neural System in Bipolar Disorder: A View from Neuroimaging Research. Acta Neuropsychiatr. 2009, 21, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Cao, B.; Passos, I.C.; Mwangi, B.; Amaral-Silva, H.; Tannous, J.; Wu, M.J.; Zunta-Soares, G.B.; Soares, J.C. Hippocampal subfield volumes in mood disorders. Mol. Psychiatry 2017, 22, 1352–1358. [Google Scholar] [CrossRef] [Green Version]
- Brady, R.O., Jr.; McCarthy, J.M.; Prescot, A.P.; Jensen, J.E.; Cooper, A.J.; Cohen, B.M.; Renshaw, P.F.; Ongür, D. Brain gamma-aminobutyric acid (GABA) abnormalities in bipolar disorder. Bipolar Disord. 2013, 15, 434–439. [Google Scholar] [CrossRef]
- Beyer, D.K.E.; Freund, N. Animal models for bipolar disorder: From bedside to the cage. Int. J. Bipolar Disord. 2017, 5, 35. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, M.; Nierenberg, A.A.; Østergaard, S.D. Face and predictive validity of the ClockΔ19 mouse as an animal model for bipolar disorder: A systematic review. Mol. Psychiatry 2018, 23, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Van Enkhuizen, J.; Minassian, A.; Young, J.W. Further evidence for ClockΔ19 mice as a model for bipolar disorder mania using cross-species tests of exploration and sensorimotor gating. Behav. Brain Res. 2013, 249, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, T.; Takata, A.; Kato, T.M.; Kubota-Sakashita, M.; Sawada, T.; Kakita, A.; Mizukami, H.; Kaneda, D.; Ozawa, K.; Kato, T. Depression-like episodes in mice harboring mtDNA deletions in paraventricular thalamus. Mol. Psychiatry 2016, 21, 39–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T. Neurobiological basis of bipolar disorder: Mitochondrial dysfunction hypothesis and beyond. Schizophr. Res. 2017, 187, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giménez-Palomo, A.; Dodd, S.; Anmella, G.; Carvalho, A.F.; Scaini, G.; Quevedo, J.; Pacchiarotti, I.; Vieta, E.; Berk, M. The Role of Mitochondria in Mood Disorders: From Physiology to Pathophysiology and to Treatment. Front. Psychiatry 2021, 12, 546801. [Google Scholar] [CrossRef] [PubMed]
- Birner, A.; Platzer, M.; Bengesser, S.A.; Dalkner, N.; Fellendorf, F.T.; Queissner, R.; Pilz, R.; Rauch, P.; Maget, A.; Hamm, C.; et al. Increased breakdown of kynurenine towards its neurotoxic branch in bipolar disorder. PLoS ONE 2017, 12, e0172699. [Google Scholar] [CrossRef]
- Arnone, D.; Saraykar, S.; Salem, H.; Teixeira, A.L.; Dantzer, R.; Selvaraj, S. Role of Kynurenine pathway and its metabolites in mood disorders: A systematic review and meta-analysis of clinical studies. Neurosci. Biobehav. Rev. 2018, 92, 477–485. [Google Scholar] [CrossRef]
- Hiles, S.A.; Baker, A.L.; de Malmanche, T.; Attia, J. A meta-analysis of differences in IL-6 and IL-10 between people with and without depression: Exploring the causes of heterogeneity. Brain Behav. Immun. 2012, 26, 1180–1188. [Google Scholar] [CrossRef]
- Substance Abuse and Mental Health Services Administration (US); Office of the Surgeon General (US). Chapter 2, the Neurobiology of Substance Use, Misuse, and Addiction. Facing Addiction in America: The Surgeon General’s Report on Alcohol, Drugs, and Health [Internet]; US Department of Health and Human Services: Washington, DC, USA, 2016. Available online: https://www.ncbi.nlm.nih.gov/books/NBK424849/ (accessed on 15 June 2022).
- National Institute on Drug Abuse. Advancing Addiction Science. Available online: https://www.drugabuse.gov/publications/media-guide/science-drug-use-addiction-basics (accessed on 15 June 2022).
- Uhl, G.R.; Koob, G.F.; Cable, J. The neurobiology of addiction. Ann. N. Y. Acad. Sci. 2019, 451, 5–28. [Google Scholar] [CrossRef]
- Calarco, C.A.; Fox, M.E.; Van Terheyden, S.; Turner, M.D.; Alipio, J.B.; Chandra, R.; Lobo, M.K. Mitochondria-Related Nuclear Gene Expression in the Nucleus Accumbens and Blood Mitochondrial Copy Number After Developmental Fentanyl Exposure in Adolescent Male and Female C57BL/6 Mice. Front. Psychiatry 2021, 12, 737389. [Google Scholar] [CrossRef]
- Morales-Puerto, N.; Giménez-Gómez, P.; Pérez-Hernández, M.; Abuin-Martínez, C.; Gil de Biedma-Elduayen, L.; Vidal, R.; Gutiérrez-López, M.D.; O’Shea, E.; Colado, M.I. Addiction and the kynurenine pathway: A new dancing couple? Pharmacol. Ther. 2021, 223, 107807. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, F.; Mestre-Pintó, J.I.; Gómez-Gómez, À.; Martinez-Sanvisens, D.; Rodríguez-Minguela, R.; Papaseit, E.; Pérez-Mañá, C.; Langohr, K.; Valverde, O.; Pozo, Ó.J.; et al. The Tryptophan System in Cocaine-Induced Depression. J. Clin. Med. 2020, 9, 4103. [Google Scholar] [CrossRef] [PubMed]
- Salazar de Pablo, G.; Woods, S.W.; Drymonitou, G.; de Diego, H.; Fusar-Poli, P. Prevalence of Individuals at Clinical High-Risk of Psychosis in the General Population and Clinical Samples: Systematic Review and Meta-Analysis. Brain Sci. 2021, 11, 1544. [Google Scholar] [CrossRef] [PubMed]
- Gaebler, A.J.; Finner-Prével, M.; Sudar, F.P.; Langer, F.H.; Keskin, F.; Gebel, A.; Zweerings, J.; Mathiak, K. The Interplay between Vitamin D, Exposure of Anticholinergic Antipsychotics and Cognition in Schizophrenia. Biomedicines 2022, 10, 1096. [Google Scholar] [CrossRef] [PubMed]
- Nyatega, C.O.; Qiang, L.; Adamu, M.J.; Younis, A.; Kawuwa, H.B. Altered Dynamic Functional Connectivity of Cuneus in Schizophrenia Patients: A Resting-State fMRI Study. Appl. Sci. 2021, 11, 11392. [Google Scholar] [CrossRef]
- Tanaka, M.; Spekker, E.; Szabó, Á.; Polyák, H.; Vécsei, L. Modelling the neurodevelopmental pathogenesis in neuropsychiatric disorders. Bioactive kynurenines and their analogues as neuroprotective agents—In celebration of 80th birthday of Professor Peter Riederer. J. Neural. Transm. 2022, 129, 627–642. [Google Scholar] [CrossRef]
- Panov, G. Dissociative Model in Patients with Resistant Schizophrenia. Front. Psychiatry 2022, 13, 845493. [Google Scholar] [CrossRef]
- Avan, R.; Sahebnasagh, A.; Hashemi, J.; Monajati, M.; Faramarzi, F.; Henney, N.C.; Montecucco, F.; Jamialahmadi, T.; Sahebkar, A. Update on Statin Treatment in Patients with Neuropsychiatric Disorders. Life 2021, 11, 1365. [Google Scholar] [CrossRef]
- Correia, B.S.B.; Nani, J.V.; Waladares Ricardo, R.; Stanisic, D.; Costa, T.B.B.C.; Hayashi, M.A.F.; Tasic, L. Effects of Psychostimulants and Antipsychotics on Serum Lipids in an Animal Model for Schizophrenia. Biomedicines 2021, 9, 235. [Google Scholar] [CrossRef]
- Rog, J.; Błażewicz, A.; Juchnowicz, D.; Ludwiczuk, A.; Stelmach, E.; Kozioł, M.; Karakula, M.; Niziński, P.; Karakula-Juchnowicz, H. The Role of GPR120 Receptor in Essential Fatty Acids Metabolism in Schizophrenia. Biomedicines 2020, 8, 243. [Google Scholar] [CrossRef]
- Dahoun, T.; Trossbach, S.V.; Brandon, N.J.; Korth, C.; Howes, O.D. The impact of Disrupted-in-Schizophrenia 1 (DISC1) on the dopaminergic system: A systematic review. Transl. Psychiatry 2017, 7, e1015. [Google Scholar] [CrossRef] [PubMed]
- Norkett, R.; Modi, S.; Birsa, N.; Atkin, T.A.; Ivankovic, D.; Pathania, M.; Trossbach, S.V.; Korth, C.; Hirst, W.D.; Kittler, J.T. DISC1-dependent Regulation of Mitochondrial Dynamics Controls the Morphogenesis of Complex Neuronal Dendrites. J. Biol. Chem. 2016, 291, 613–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kvajo, M.; McKellar, H.; Drew, L.J.; Lepagnol-Bestel, A.M.; Xiao, L.; Levy, R.J.; Blazeski, R.; Arguello, P.A.; Lacefield, C.O.; Mason, C.A.; et al. Altered axonal targeting and short-term plasticity in the hippocampus of Disc1 mutant mice. Proc. Natl. Acad. Sci. USA 2011, 108, E1349–E1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niigaki, S.T.; Peres, F.F.; Ferreira, L.; Libanio, T.; Gouvea, D.A.; Levin, R.; Almeida, V.; Silva, N.D.; Diana, M.C.; Suiama, M.A.; et al. Young spontaneously hypertensive rats (SHRs) display prodromal schizophrenia-like behavioral abnormalities. Prog. Neuropsychopharmacol. Biol. Psychiatry 2019, 2, 169–176. [Google Scholar] [CrossRef]
- Park, C.; Park, S.K. Molecular links between mitochondrial dysfunctions and schizophrenia. Mol. Cells 2012, 33, 105–110. [Google Scholar] [CrossRef] [Green Version]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef]
- Okusaga, O.; Fuchs, D.; Reeves, G.; Giegling, I.; Hartmann, A.M.; Konte, B.; Friedl, M.; Groer, M.; Cook, T.B.; Stearns-Yoder, K.A.; et al. Kynurenine and Tryptophan Levels in Patients with Schizophrenia and Elevated Antigliadin Immunoglobulin G Antibodies. Psychosom. Med. 2016, 78, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.K.; Miller, B.J. Meta-analysis of Cerebrospinal Fluid Cytokine and Tryptophan Catabolite Alterations in Psychiatric Patients: Comparisons Between Schizophrenia, Bipolar Disorder, and Depression. Schizophr. Bull. 2018, 44, 75–83. [Google Scholar] [CrossRef] [Green Version]
- Faras, H.; Al Ateeqi, N.; Tidmarsh, L. Autism spectrum disorders. Ann. Saudi. Med. 2010, 30, 295–300. [Google Scholar] [CrossRef]
- Candini, M.; Battaglia, S.; Benassi, M.; di Pellegrino, G.; Frassinetti, F. The physiological correlates of inter-personal space. Sci. Rep. 2021, 11, 2611. [Google Scholar] [CrossRef]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Thabault, M.; Turpin, V.; Maisterrena, A.; Jaber, M.; Egloff, M.; Galvan, L. Cerebellar and Striatal Implications in Autism Spectrum Disorders: From Clinical Observations to Animal Models. Int. J. Mol. Sci. 2022, 23, 2294. [Google Scholar] [CrossRef]
- Agarwala, S.; Ramachandra, N.B. Role of CNTNAP2 in autism manifestation outlines the regulation of signaling between neurons at the synapse. Egypt. J. Med. Hum. Genet. 2021, 22, 22. [Google Scholar] [CrossRef]
- Yardeni, T.; Cristancho, A.G.; McCoy, A.J.; Schaefer, P.M.; McManus, M.J.; Marsh, E.D.; Wallace, D.C. An mtDNA mutant mouse demonstrates that mitochondrial deficiency can result in autism endophenotypes. Proc. Natl. Acad. Sci. USA 2021, 118, e2021429118. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.A.; Lin, Y.-K.; Lai, J.-H.; Lo, Y.-C.; Yang, Y.-C.S.H.; Ye, S.-Y.; Lee, C.-J.; Wang, C.-C.; Chiang, Y.-H.; Tseng, S.-H. Maternal Immune Activation Causes Social Behavior Deficits and Hypomyelination in Male Rat Offspring with an Autism-Like Microbiota Profile. Brain Sci. 2021, 11, 1085. [Google Scholar] [CrossRef]
- Abuaish, S.; Al-Otaibi, N.M.; Abujamel, T.S.; Alzahrani, S.A.; Alotaibi, S.M.; AlShawakir, Y.A.; Aabed, K.; El-Ansary, A. Fecal Transplant and Bifidobacterium Treatments Modulate Gut Clostridium Bacteria and Rescue Social Impairment and Hippocampal BDNF Expression in a Rodent Model of Autism. Brain Sci. 2021, 11, 1038. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Cakir, J.; Rose, S.; Delhey, L.; Bennuri, S.C.; Tippett, M.; Palmer, R.F.; Austin, C.; Curtin, P.; Arora, M. Early life metal exposure dysregulates cellular bioenergetics in children with regressive autism spectrum disorder. Transl. Psychiatry 2020, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Bryn, V.; Verkerk, R.; Skjeldal, O.H.; Saugstad, O.D.; Ormstad, H. Kynurenine Pathway in Autism Spectrum Disorders in Children. Neuropsychobiology 2017, 76, 82–88. [Google Scholar] [CrossRef]
- Mahone, E.M.; Denckla, M.B. Attention-Deficit/Hyperactivity Disorder: A Historical Neuropsychological Perspective. J. Int. Neuropsychol. Soc. 2017, 23, 916–929. [Google Scholar] [CrossRef] [Green Version]
- Kyaga, S.; Landén, M.; Boman, M.; Hultman, C.M.; Långström, N.; Lichtenstein, P. Mental illness, suicide and creativity: 40-year prospective total population study. J. Psychiatr. Res. 2013, 47, 83–90. [Google Scholar] [CrossRef]
- Saccaro, L.F.; Schilliger, Z.; Perroud, N.; Piguet, C. Inflammation, Anxiety, and Stress in Attention-Deficit/Hyperactivity Disorder. Biomedicines 2021, 9, 1313. [Google Scholar] [CrossRef] [PubMed]
- Murakami, Y.; Imamura, Y.; Saito, K.; Sakai, D.; Motoyama, J. Altered kynurenine pathway metabolites in a mouse model of human attention-deficit hyperactivity/autism spectrum disorders: A potential new biological diagnostic marker. Sci. Rep. 2019, 9, 13182. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, T.I.; Landaas, E.T.; Hegvik, T.A.; Ulvik, A.; Halmøy, A.; Ueland, P.M.; Haavik, J. Serum concentrations of kynurenines in adult patients with attention-deficit hyperactivity disorder (ADHD): A case-control study. Behav. Brain Funct. 2015, 11, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, M.; McEwen, B.S. Psychological Stress and Mitochondria: A Systematic Review. Psychosom. Med. 2018, 80, 141–153. [Google Scholar] [CrossRef] [Green Version]
- Jiang, S.; Postovit, L.; Cattaneo, A.; Binder, E.B.; Aitchison, K.J. Epigenetic Modifications in Stress Response Genes Associated with Childhood Trauma. Front. Psychiatry 2019, 10, 808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.I.; Lee, S.Y.; Park, M.; Kim, S.Y.; Kim, J.W.; Kim, S.A.; Kim, B.N. Peripheral Mitochondrial DNA Copy Number is Increased in Korean Attention-Deficit Hyperactivity Disorder Patients. Front. Psychiatr. 2019, 10, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]



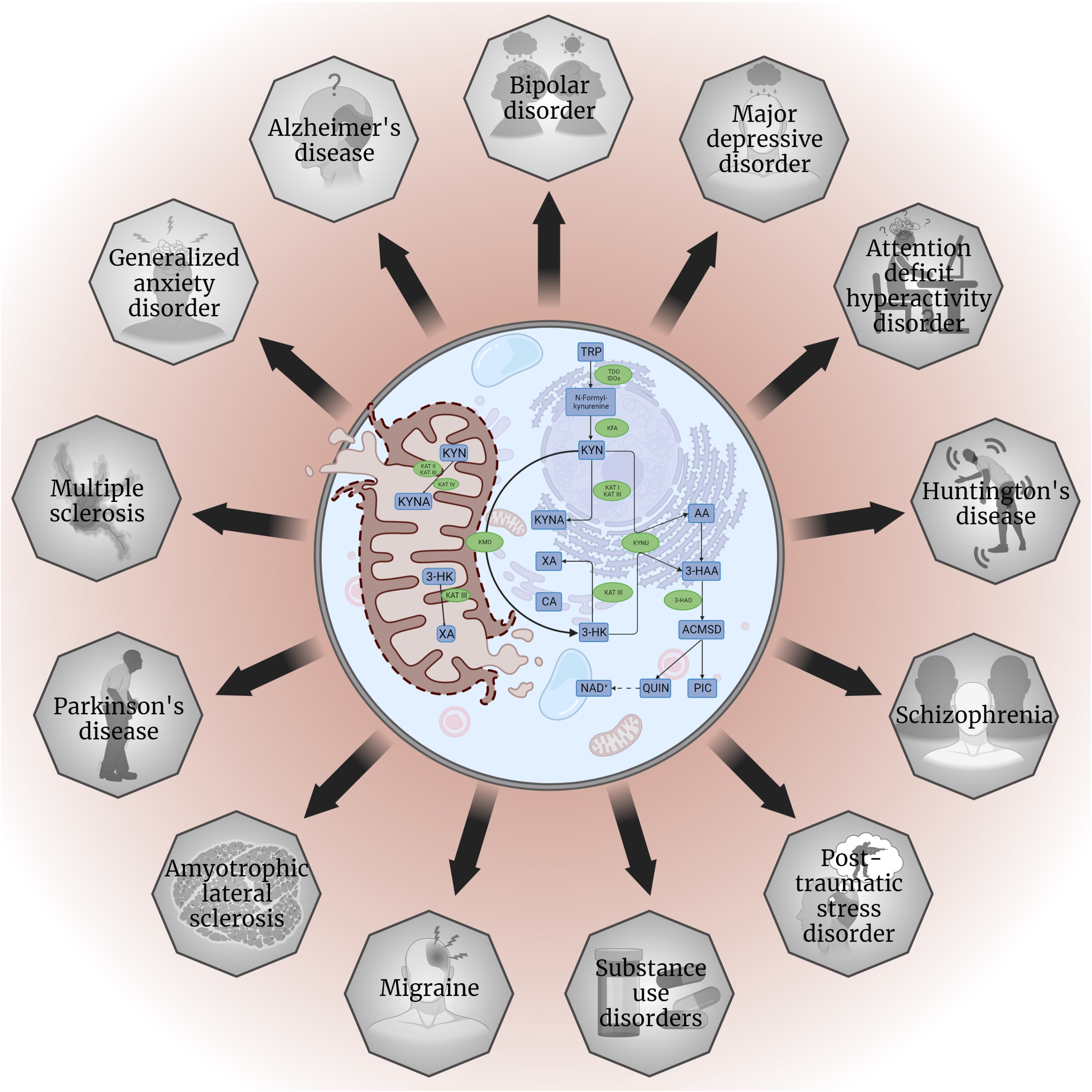{kind=link}
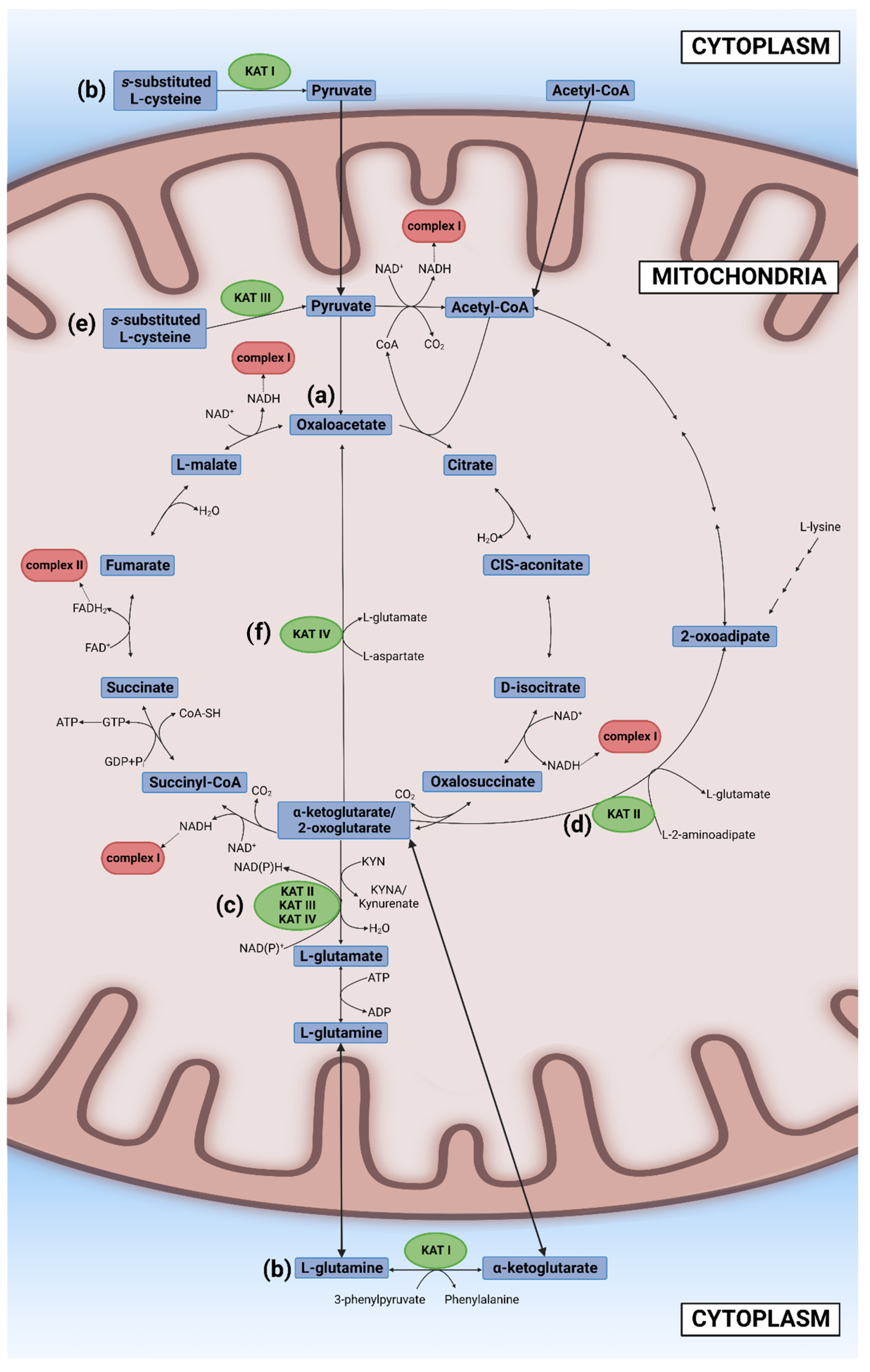{kind=link}
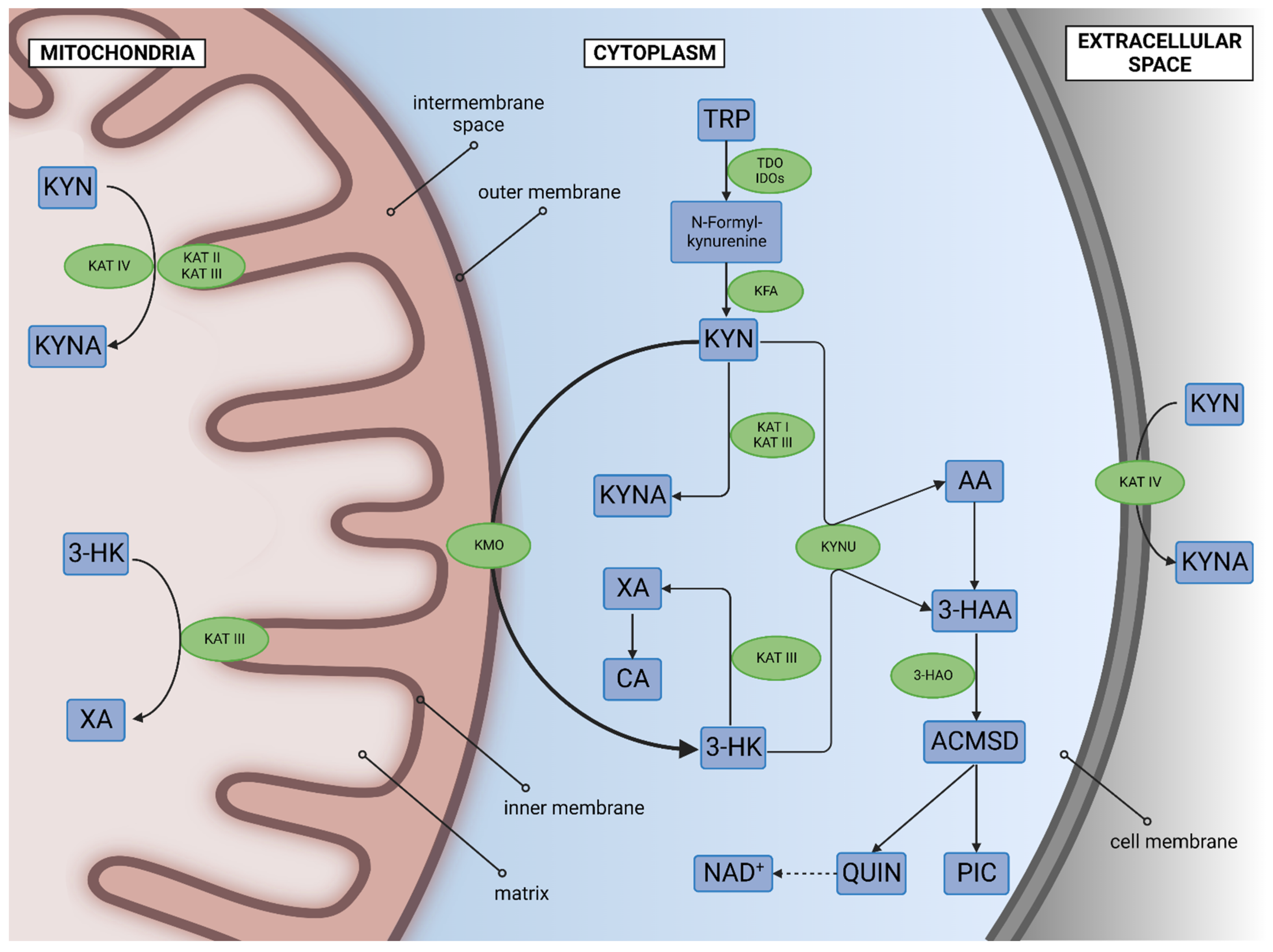{kind=link}
| Enzymes | Genes | Substrates | Products | Locations | Transgenic Models | Animal Traits | Human Gene Variants |
|---|---|---|---|---|---|---|---|
| TDO | tdo2 | L-Trp | N-formyl-L-kynurenine | Cytosol | tdo2−/− |
|
|
| IDO1 | ido1 | L-Trp, D-Trp | N-formyl-L-kynurenine | Cytosol | ido1−/− |
|
|
| IDO2 | ido2 | L-Trp | N-formyl-L-kynurenine | Cytosol | Ido2−/− | - |
|
| KFA | afmid | N-formyl-L-kynurenine | L-KYN | Cytosol | - | - | - |
| KMO | kmo | L-KYN | 3-HK | Mitochondria (outer membrane) | kmo−/− |
|
|
| KAT I (Kynurenine--oxoglutarate transaminase 1) | kyat1 | L-KYN S-substituted L-Cys 3-phenylpyruvate L-glutamine | KYNA thiol, NH4, pyruvate 2-oxoglutarate L-phenylalanine | Cytosol | - | - | - |
| KAT II (kynurenine/α-aminoadipate aminotransferase) (KAT/AadAT) | aadat | L-KYN α-ketoglutarate 2-oxoglutarate | KYNA L-glutamate L-glutamate | Inner membrane of mitochondria | aadat−/− (aka kat2−/−) |
| - |
| KAT III (kynurenine--oxoglutarate transaminase 3) | kyat3 | L-KYN α-ketoglutarate 3-HK glyoxylate glyoxylate L-KYN S-substituted L-Cys H2O | KYNA L-glutamate Glycine H2O XA Glycine H2O KYNA Thiol NH4+ pyruvate | Cytosol inner membrane of mitochondria | - | - | - |
| KAT IV (aspartate aminotransferase, mAspAT) | got2 | L-KYN α-ketoglutarate 2-oxoglutarate L-aspartate | KYNA L-glutamate L-glutamate oxaloacetate | Matrix of mitochondria plasma membrane | - | - | - |
| KYNU | kynu | L-KYN L-alanine 3-HK | AA 3-HAA (3-arylcarbonyl)-alanine | Cytosol | kynu−/− | - |
|
| 3-HAO | haao | 3-HAA | ACMS | Cytosol | - | - |
|
| Neurological Diseases | Preclinical Models | Mitochondrial Involvement | Findings in Kynurenines |
|---|---|---|---|
| Alzheimer’s disease | >170 genetic models (APP, PSEN-1, PSEN-2) | - |
|
| 3xTg-AD |
| ||
| TgAPParc |
| ||
| APPSWE | - | ||
| PSEN1dE9 | - | ||
| SVCT2+/− | - | ||
| human Aβ-KI | - | ||
| Parkinson’s disease | PINK1 Parkin Parkinson disease protein 7 | - - - |
|
| CHCHD2 |
| ||
| complex I Park model |
| ||
| methyl-4-phenyl-1,2,3,6-tetrahydropyridine | - | ||
| Rotenone | - | ||
| 6-hydroxydopamine | - | ||
| Multiple sclerosis | experimental autoimmune/allergic encephalomyelitis (EAE) |
|
|
| Theiler’s murine encephalomyelitis virus-induced chronic demyelination | - | ||
| cuprizone-induced demyelination |
| ||
| Huntington’s disease | R6/1 | - |
|
| R6/2 | - | ||
| HTT+97CAG-CAA repeats | - | ||
| KI (endogenous Hdh promoter) | - | ||
| HdhQ111KI |
| ||
| Amyotrophic lateral sclerosis | FVB-C9orf72 BAC | - |
|
| Cu/Zn SOD1-G93A | - | ||
| TDP43-Q331K | - | ||
| iPSC model of C9orf72-associated ALS |
| ||
| SOD1 G93A | - | ||
| BPA |
| ||
| BSSG | - | ||
| Migraine | inflammatory soup |
|
|
| nitroglycerin-induced trigeminovascular activation | - |
| Psychiatric Diseases | Preclinical Models | Mitochondrial Involvement | Findings in Kynurenines |
|---|---|---|---|
| Major depressive disorder | CMS |
|
|
| TST |
| ||
| FST | - | ||
| Tph1−/− | - | ||
| Tph2−/− | - | ||
| Tph1/Tph2−/− | - | ||
| TPH2 variant (R439H) KI | - | ||
| Generalized anxiety disorder | outbred Wistar rats |
|
|
| social hierarchy |
| ||
| Post-traumatic stress disorder | FKBP5−/− | - | - |
| PAC1R−/− | - | ||
| 5-HT1AR−/− | - | ||
| COMT−/− | - | ||
| GAD6−/− | - | ||
| GABAB1a−/− | - | ||
| CB1R−/− | - | ||
| single prolonged stress model |
| ||
| Bipolar disorder | ClockΔ19 | - |
|
| dominant negative mutant of mtDNA Polg1 | - | ||
| - |
| ||
| Substance use disorder | - |
|
|
| Schizophrenia | DISC1 |
|
|
| hypertensive rats | - | ||
| Autism spectrum disorder | ND6P25LKI | - |
|
| Shank3Δc/Δc | - | ||
| Cntnap2 KO | - | ||
| ADGRL3−/− | - | ||
| valproate | - | ||
| polyinosinic–polycytidylic acid |
| ||
| Attention-deficit hyperactivity disorder | Ptchd1−/− | - |
|
| - |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, M.; Szabó, Á.; Spekker, E.; Polyák, H.; Tóth, F.; Vécsei, L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells 2022, 11, 2607. https://doi.org/10.3390/cells11162607
Tanaka M, Szabó Á, Spekker E, Polyák H, Tóth F, Vécsei L. Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells. 2022; 11(16):2607. https://doi.org/10.3390/cells11162607
Chicago/Turabian StyleTanaka, Masaru, Ágnes Szabó, Eleonóra Spekker, Helga Polyák, Fanni Tóth, and László Vécsei. 2022. "Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System" Cells 11, no. 16: 2607. https://doi.org/10.3390/cells11162607
APA StyleTanaka, M., Szabó, Á., Spekker, E., Polyák, H., Tóth, F., & Vécsei, L. (2022). Mitochondrial Impairment: A Common Motif in Neuropsychiatric Presentation? The Link to the Tryptophan–Kynurenine Metabolic System. Cells, 11(16), 2607. https://doi.org/10.3390/cells11162607