
Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human and Animal Models
2.2. Primary Cortical Neuronal (CxN) Cell Cultures and Scrapie Infection
2.3. Western Blot Analysis
2.4. Subcellular Fractionation
2.5. Immunohistochemistry and Immunofluorescence
2.6. Image Acquisition and Quantification
2.7. Cell Viability Assay
2.8. DNA Construction
2.9. Mitophagy Assay
2.10. Measurement of Mitochondrial Membrane Potential (ΔΨm) and mtROS
2.11. Determination of Glycolysis and ATP Levels
2.12. Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
2.13. Measurement of Mitochondrial Length
2.14. Caspase Activity Assay
2.15. Statistical Analysis
3. Results
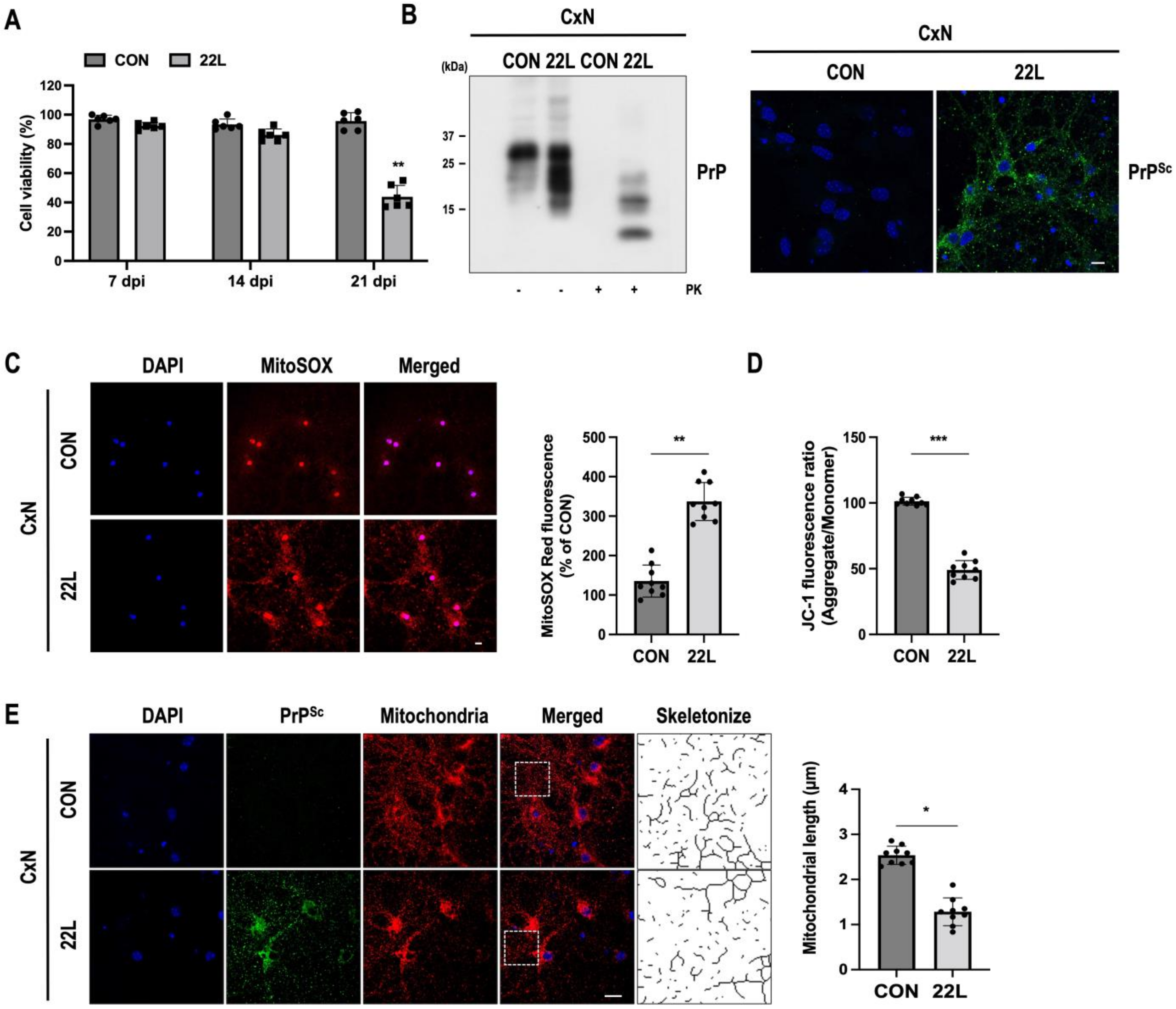
3.1. Scrapie Infection Induces Mitochondrial ROS (mtROS) Production and Reduces Mitochondrial Membrane Potential (ΔΨm)
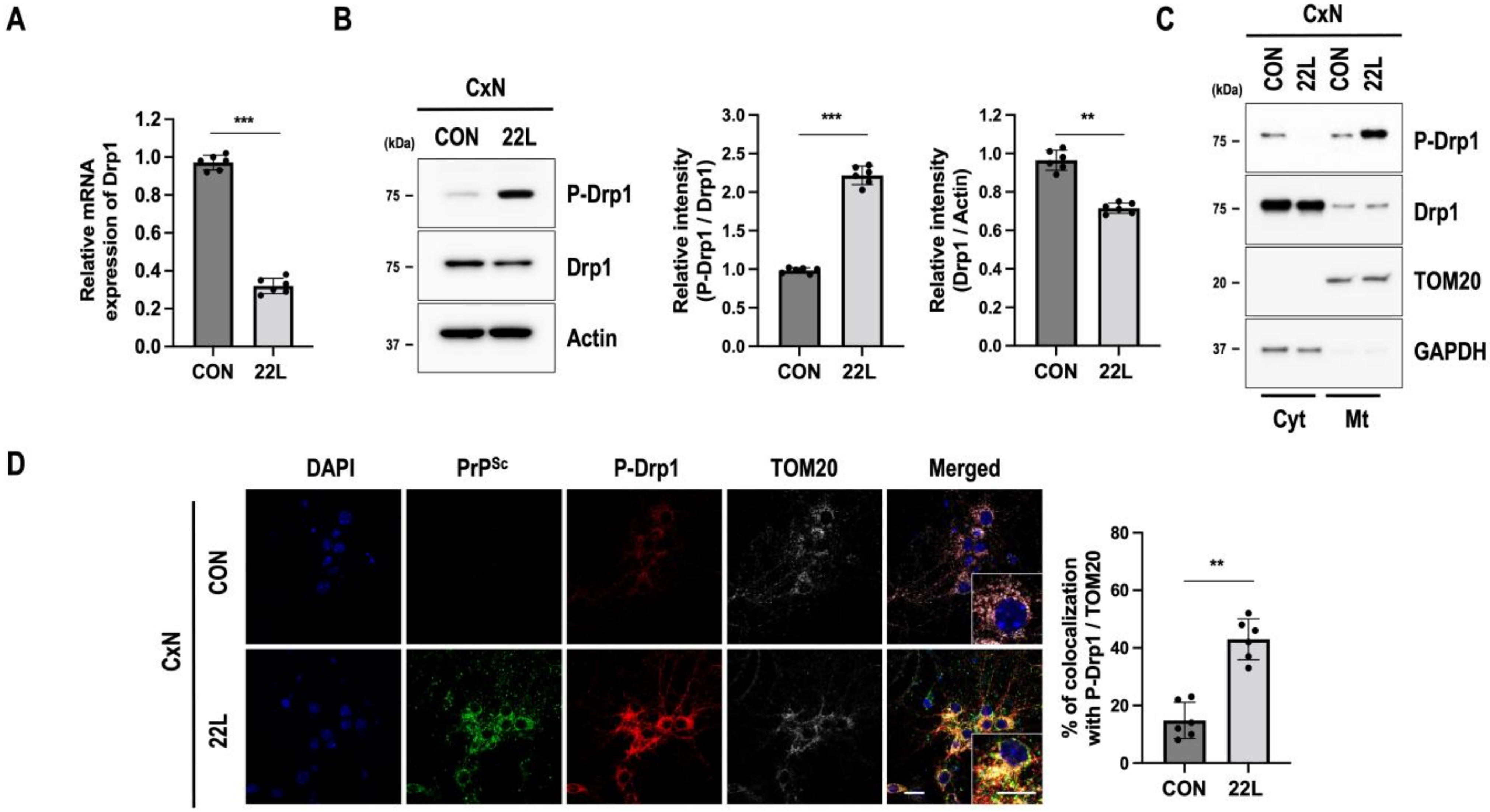
3.2. Scrapie Infection Regulates Drp1-Mediated Mitochondrial Fission
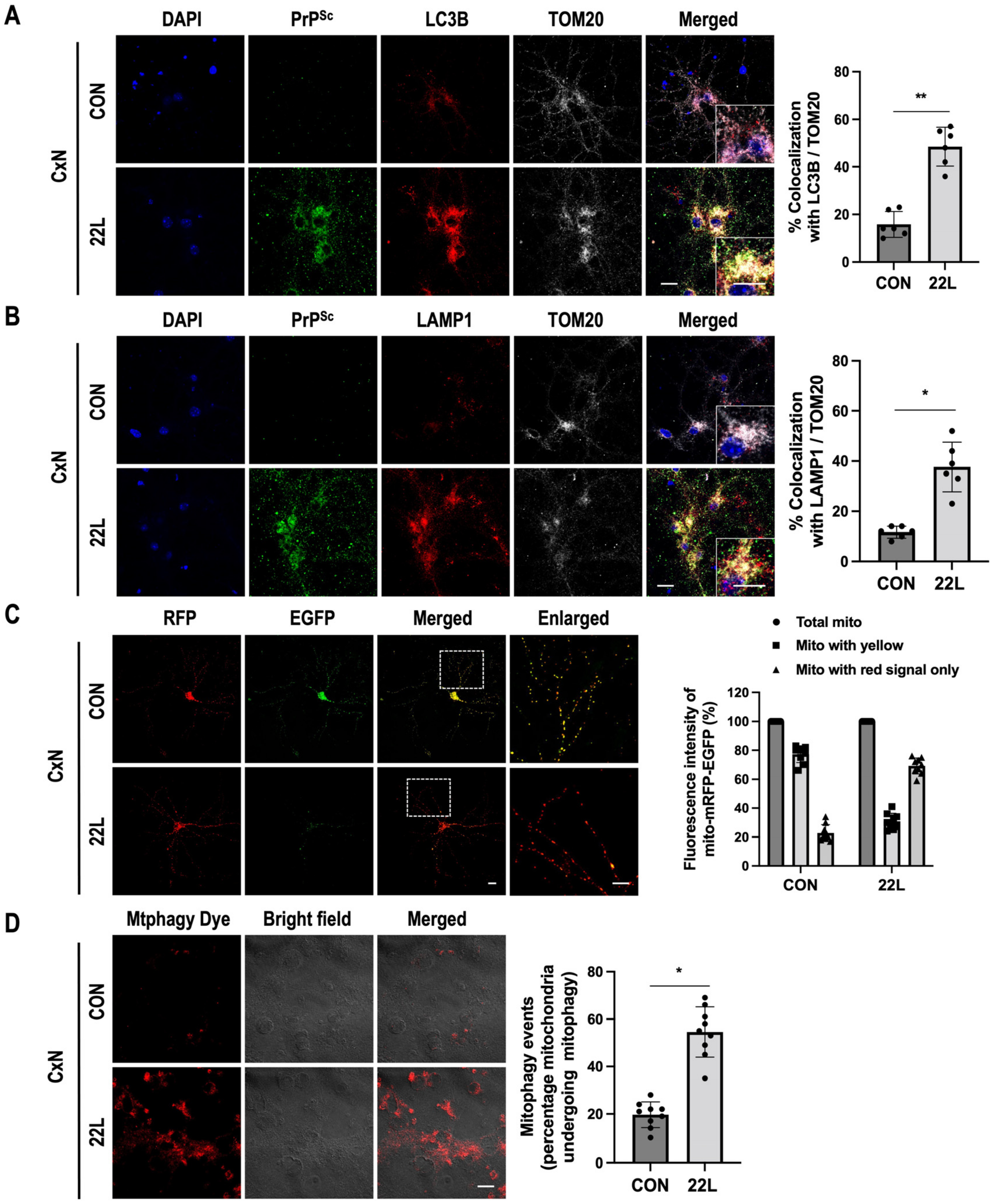
3.3. Scrapie Infection Promotes Mitophagy
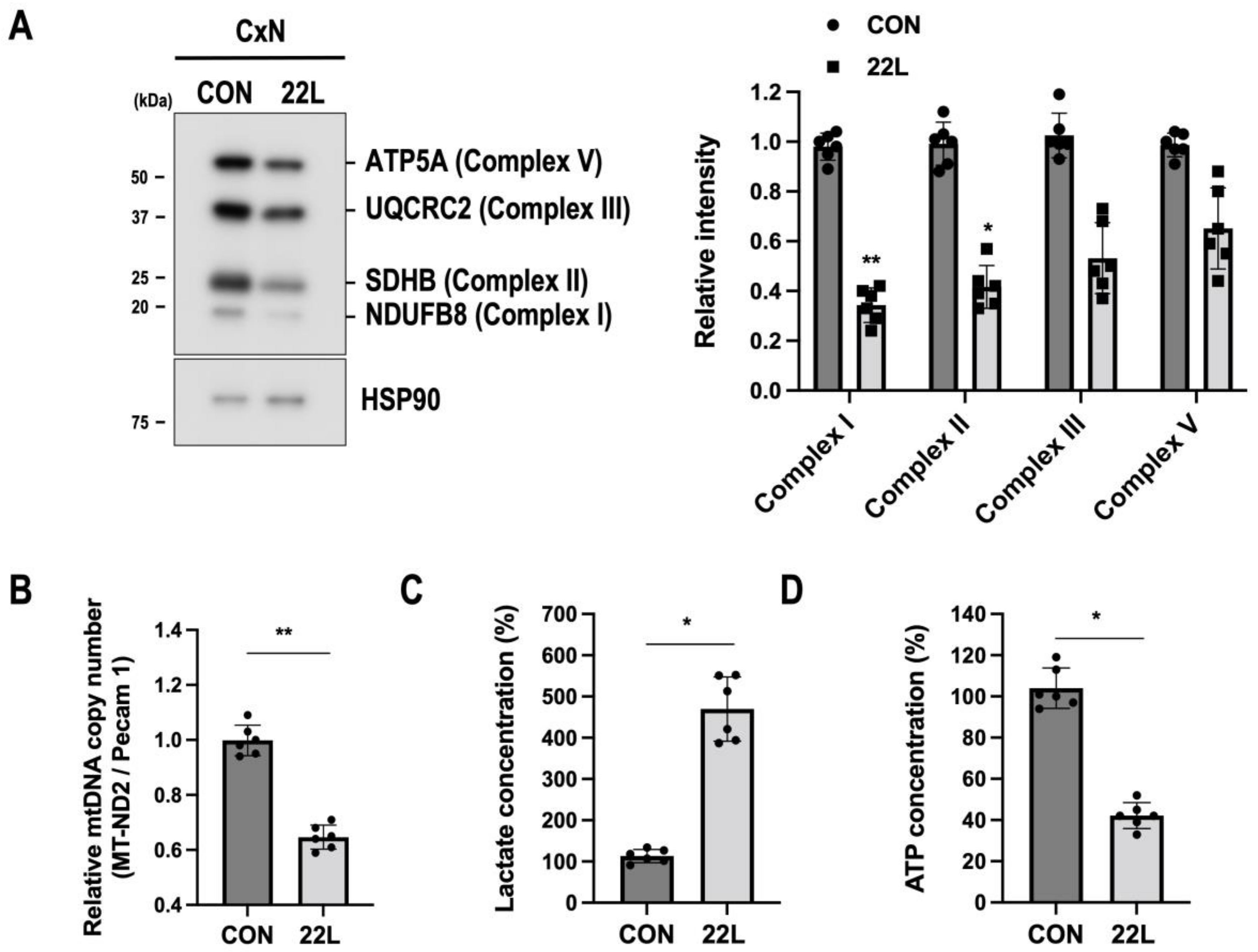
3.4. Scrapie Infection Impairs Mitochondrial Oxidative Phosphorylation (OXPHS) and Alters Glycolytic Metabolism
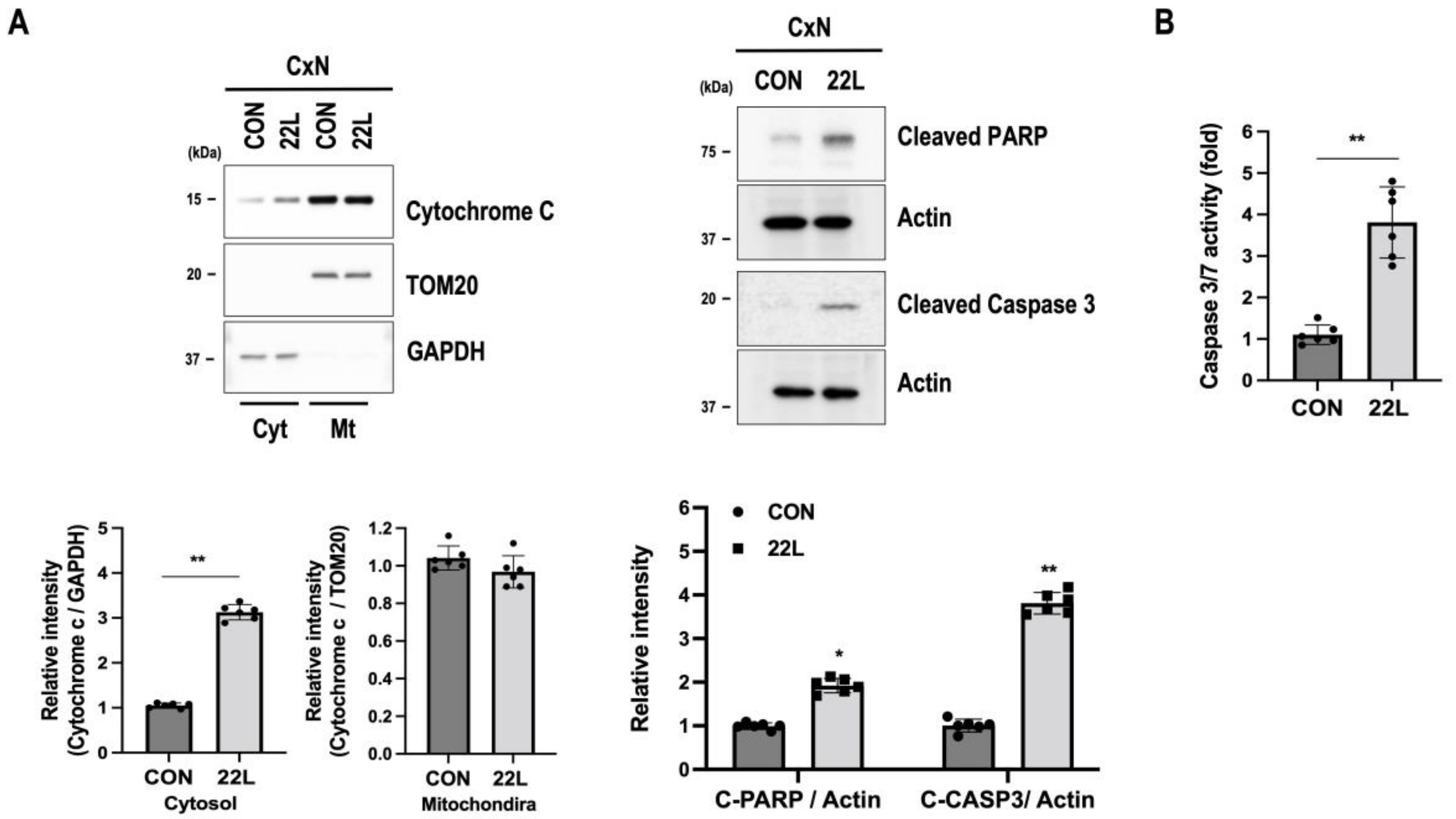
3.5. Scrapie Infection Induces Apoptosis through Mitochondrial Dysfunction
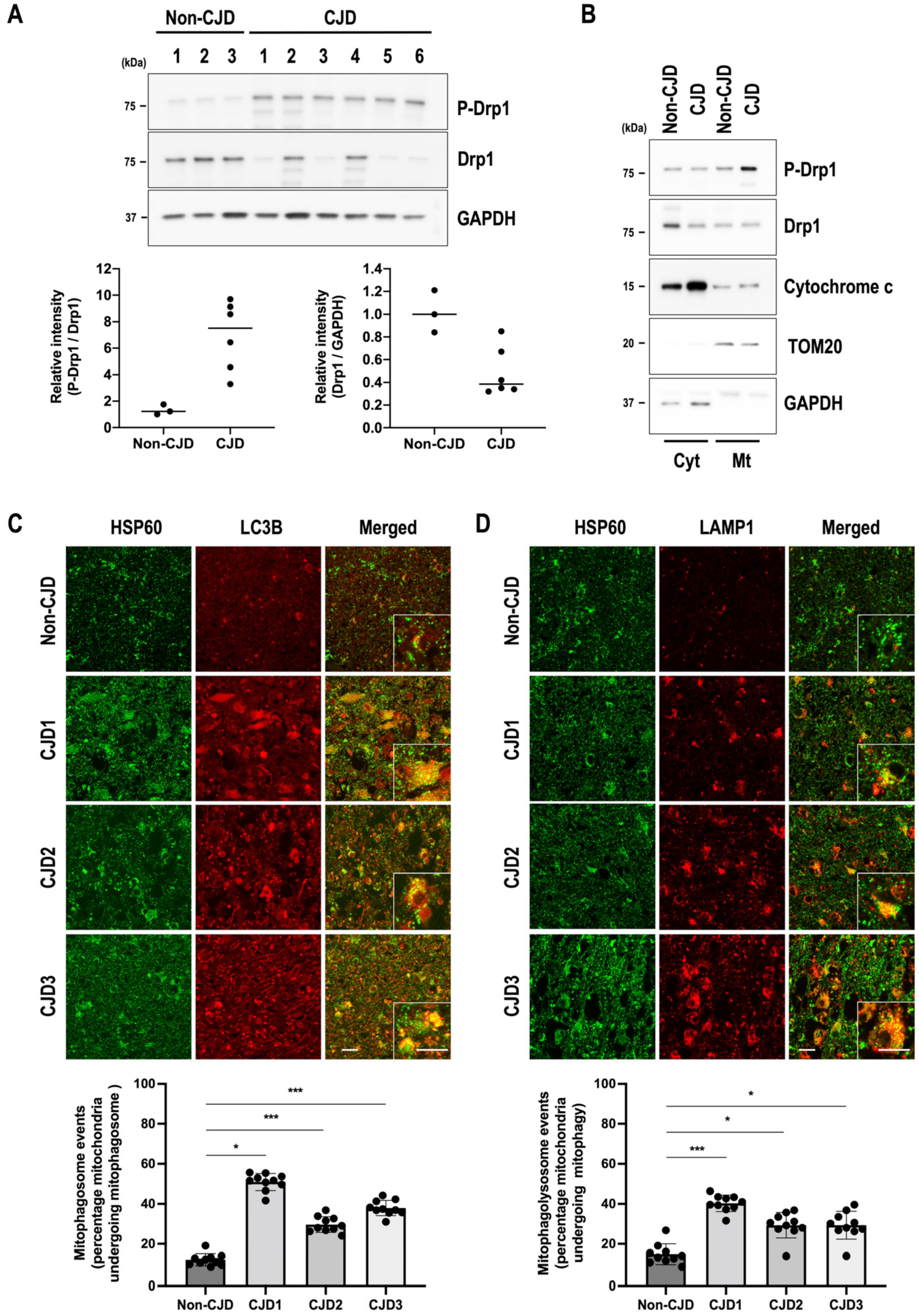
3.6. Mitochondrial Quality Control Is Impaired in sCJD Brains
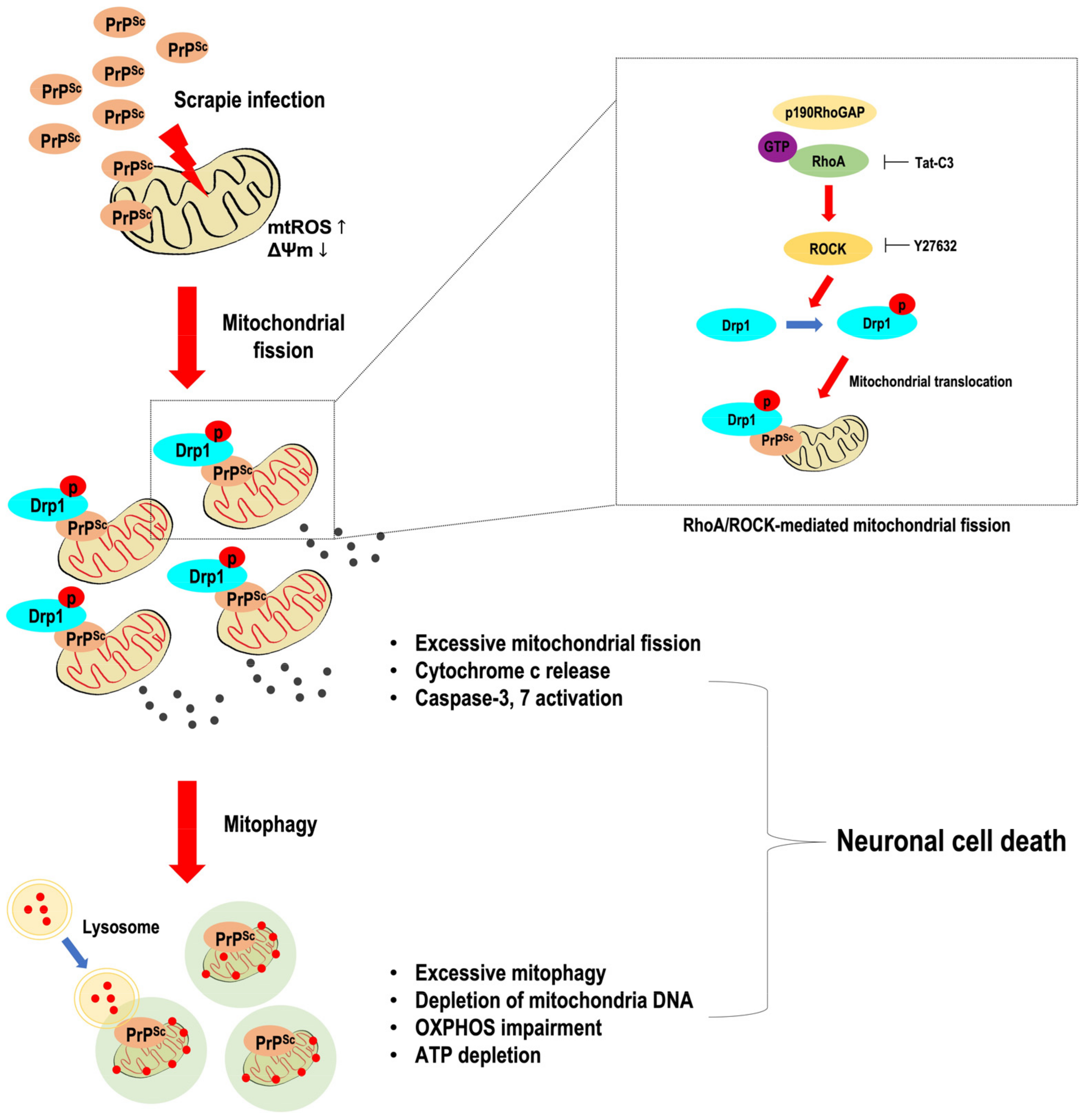
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aguzzi, A.; Heikenwalder, M.; Polymenidou, M. Insights into prion strains and neurotoxicity. Nat. Rev. Mol. Cell Biol. 2007, 8, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Woerman, A.L.; Berry, D.B.; Prusiner, S.B. Bioassays and Inactivation of Prions. Cold Spring Harb. Perspect. Biol. 2017, 9, a023499. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.L.; Ironside, J.W. Neuropathology of Human Prion Diseases. Prog. Mol. Biol. Transl. Sci. 2017, 150, 319–339. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Lee, H.G.; Choi, J.K.; Kim, J.I.; Choi, E.K.; Carp, R.I.; Kim, Y.S. The cellular prion protein (PrPC) prevents apoptotic neuronal cell death and mitochondrial dysfunction induced by serum deprivation. Brain Res. Mol. Brain Res. 2004, 124, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, B.H.; Park, S.J.; Jin, J.K.; Jeon, Y.C.; Wen, G.Y.; Shin, H.Y.; Carp, R.I.; Kim, Y.S. Association of endothelial nitric oxide synthase and mitochondrial dysfunction in the hippocampus of scrapie-infected mice. Hippocampus 2011, 21, 319–333. [Google Scholar] [CrossRef] [PubMed]
- Faris, R.; Moore, R.A.; Ward, A.; Sturdevant, D.E.; Priola, S.A. Mitochondrial Respiration Is Impaired during Late-Stage Hamster Prion Infection. J. Virol. 2017, 91, e00524-17. [Google Scholar] [CrossRef]
- Han, S.; Jeong, Y.Y.; Sheshadri, P.; Cai, Q. Mitophagy coordination with retrograde transport ensures the integrity of synaptic mitochondria. Autophagy 2020, 16, 1925–1927. [Google Scholar] [CrossRef]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 30. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45. [Google Scholar] [CrossRef]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Westermann, B. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol. 2010, 11, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Chang, C.R.; Blackstone, C. Drp1 phosphorylation and mitochondrial regulation. EMBO Rep. 2007, 8, 1088–1089. [Google Scholar] [CrossRef] [PubMed]
- Higuchi-Sanabria, R.; Frankino, P.A.; Paul, J.W., 3rd; Tronnes, S.U.; Dillin, A. A Futile Battle? Protein Quality Control and the Stress of Aging. Dev. Cell 2018, 44, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Syed, G.H.; Kim, S.J.; Siddiqui, A. Mitochondrial dynamics and viral infections: A close nexus. Biochim. Biophys. Acta 2015, 1853, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, M.W.; Haynes, C.M. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015, 13, 22. [Google Scholar] [CrossRef]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef]
- Evans, C.S.; Holzbaur, E.L.F. Quality Control in Neurons: Mitophagy and Other Selective Autophagy Mechanisms. J. Mol. Biol. 2020, 432, 240–260. [Google Scholar] [CrossRef]
- Sung, K.; Jimenez-Sanchez, M. Autophagy in Astrocytes and its Implications in Neurodegeneration. J. Mol. Biol. 2020, 432, 2605–2621. [Google Scholar] [CrossRef]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.S.; Choi, Y.G.; Shin, H.Y.; Oh, J.M.; Park, J.H.; Kim, J.I.; Carp, R.I.; Choi, E.K.; Kim, Y.S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014, 448, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Ansoleaga, B.; Garcia-Esparcia, P.; Llorens, F.; Hernandez-Ortega, K.; Carmona Tech, M.; Antonio Del Rio, J.; Zerr, I.; Ferrer, I. Altered Mitochondria, Protein Synthesis Machinery, and Purine Metabolism Are Molecular Contributors to the Pathogenesis of Creutzfeldt-Jakob Disease. J. Neuropathol. Exp. Neurol. 2016, 75, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Flones, I.H.; Ricken, G.; Klotz, S.; Lang, A.; Strobel, T.; Dolle, C.; Kovacs, G.G.; Tzoulis, C. Mitochondrial respiratory chain deficiency correlates with the severity of neuropathology in sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. Commun. 2020, 8, 50. [Google Scholar] [CrossRef] [PubMed]
- Hannaoui, S.; Maatouk, L.; Privat, N.; Levavasseur, E.; Faucheux, B.A.; Haik, S. Prion propagation and toxicity occur in vitro with two-phase kinetics specific to strain and neuronal type. J. Virol. 2013, 87, 2535–2548. [Google Scholar] [CrossRef]
- Choi, J.K.; Park, S.J.; Jun, Y.C.; Oh, J.M.; Jeong, B.H.; Lee, H.P.; Park, S.N.; Carp, R.I.; Kim, Y.S. Generation of monoclonal antibody recognized by the GXXXG motif (glycine zipper) of prion protein. Hybridoma 2006, 25, 271–277. [Google Scholar] [CrossRef]
- Jang, B.; Shin, H.Y.; Choi, J.K.; Nguyen, D.P.T.; Jeong, B.H.; Ishigami, A.; Maruyama, N.; Carp, R.I.; Kim, Y.S.; Choi, E.K. Subcellular localization of peptidylarginine deiminase 2 and citrullinated proteins in brains of scrapie-infected mice: Nuclear localization of PAD2 and membrane fraction-enriched citrullinated proteins. J. Neuropathol. Exp. Neurol. 2011, 70, 116–124. [Google Scholar] [CrossRef]
- Xie, Q.; Wu, Q.; Horbinski, C.M.; Flavahan, W.A.; Yang, K.; Zhou, W.; Dombrowski, S.M.; Huang, Z.; Fang, X.; Shi, Y.; et al. Mitochondrial control by DRP1 in brain tumor initiating cells. Nat. Neurosci. 2015, 18, 501–510. [Google Scholar] [CrossRef]
- Stauffer, W.; Sheng, H.; Lim, H.N. EzColocalization: An ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci. Rep. 2018, 8, 15764. [Google Scholar] [CrossRef]
- Allen, E.A.; Amato, C.; Fortier, T.M.; Velentzas, P.; Wood, W.; Baehrecke, E.H. A conserved myotubularin-related phosphatase regulates autophagy by maintaining autophagic flux. J. Cell Biol. 2020, 219, e201909073. [Google Scholar] [CrossRef]
- Dunn, K.W.; Kamocka, M.M.; McDonald, J.H. A practical guide to evaluating colocalization in biological microscopy. Am. J. Physiol. Cell Physiol. 2011, 300, C723–C742. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B virus disrupts mitochondrial dynamics: Induces fission and mitophagy to attenuate apoptosis. PLoS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Iwashita, H.; Torii, S.; Nagahora, N.; Ishiyama, M.; Shioji, K.; Sasamoto, K.; Shimizu, S.; Okuma, K. Live Cell Imaging of Mitochondrial Autophagy with a Novel Fluorescent Small Molecule. ACS Chem. Biol. 2017, 12, 2546–2551. [Google Scholar] [CrossRef]
- Park, J.W.; Kim, M.J.; Kim, S.E.; Kim, H.J.; Jeon, Y.C.; Shin, H.Y.; Park, S.J.; Jang, M.K.; Kim, D.J.; Park, C.K.; et al. Increased Expression of S100B and RAGE in a Mouse Model of Bile Duct Ligation-induced Liver Fibrosis. J. Korean Med. Sci. 2021, 36, e90. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Sawashita, J.; Kubo, H.; Nishio, S.Y.; Hashimoto, S.; Suzuki, N.; Yoshimura, H.; Tsuruoka, M.; Wang, Y.; Liu, Y.; et al. Ubiquinol-10 supplementation activates mitochondria functions to decelerate senescence in senescence-accelerated mice. Antioxidants Redox Signal. 2014, 20, 2606–2620. [Google Scholar] [CrossRef]
- Lavie, J.; Serrat, R.; Bellance, N.; Courtand, G.; Dupuy, J.W.; Tesson, C.; Coupry, I.; Brice, A.; Lacombe, D.; Durr, A.; et al. Mitochondrial morphology and cellular distribution are altered in SPG31 patients and are linked to DRP1 hyperphosphorylation. Hum. Mol. Genet. 2017, 26, 674–685. [Google Scholar] [CrossRef]
- Ouellet, M.; Guillebaud, G.; Gervais, V.; Lupien St-Pierre, D.; Germain, M. A novel algorithm identifies stress-induced alterations in mitochondrial connectivity and inner membrane structure from confocal images. PLoS Comput. Biol. 2017, 13, e1005612. [Google Scholar] [CrossRef]
- Leonard, A.P.; Cameron, R.B.; Speiser, J.L.; Wolf, B.J.; Peterson, Y.K.; Schnellmann, R.G.; Beeson, C.C.; Rohrer, B. Quantitative analysis of mitochondrial morphology and membrane potential in living cells using high-content imaging, machine learning, and morphological binning. Biochim. Biophys. Acta 2015, 1853, 348–360. [Google Scholar] [CrossRef] [PubMed]
- McClatchey, P.M.; Keller, A.C.; Bouchard, R.; Knaub, L.A.; Reusch, J.E. Fully automated software for quantitative measurements of mitochondrial morphology. Mitochondrion 2016, 26, 58–71. [Google Scholar] [CrossRef]
- Basu, H.; Pekkurnaz, G.; Falk, J.; Wei, W.; Chin, M.; Steen, J.; Schwarz, T.L. FHL2 anchors mitochondria to actin and adapts mitochondrial dynamics to glucose supply. J. Cell Biol. 2021, 220, e201912077. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Castillo, K.; Armisen, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Binyamin, O.; Frid, K.; Saada, A.; Gabizon, R. Mitochondrial dysfunction in preclinical genetic prion disease: A target for preventive treatment? Neurobiol. Dis. 2019, 124, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.I.; Ju, W.K.; Choi, E.K.; Kim, J.; Lea, H.Z.; Carp, R.I.; Wisniewski, H.M.; Kim, Y.S. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. 1998, 96, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, M.J.; Mostafa, M.N.; Park, J.H.; Choi, H.S.; Kim, Y.S.; Choi, E.K. RhoA/ROCK Regulates Prion Pathogenesis by Controlling Connexin 43 Activity. Int J. Mol. Sci. 2020, 21, 1255. [Google Scholar] [CrossRef]
- Brand, C.S.; Tan, V.P.; Brown, J.H.; Miyamoto, S. RhoA regulates Drp1 mediated mitochondrial fission through ROCK to protect cardiomyocytes. Cell Signal. 2018, 50, 48–57. [Google Scholar] [CrossRef]
- Gao, L.P.; Xiao, K.; Wu, Y.Z.; Chen, D.D.; Yang, X.H.; Shi, Q.; Dong, X.P. Enhanced Mitophagy Activity in Prion-Infected Cultured Cells and Prion-Infected Experimental Mice via a Pink1/Parkin-Dependent Mitophagy Pathway. ACS Chem. Neurosci. 2020, 11, 814–829. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Yang, L. Mitochondrial Dysfunction and Oxidative Stress in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 617588. [Google Scholar] [CrossRef]
- Area-Gomez, E.; Guardia-Laguarta, C.; Schon, E.A.; Przedborski, S. Mitochondria, OxPhos, and neurodegeneration: Cells are not just running out of gas. J. Clin. Investig. 2019, 129, 34–45. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders-A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Bourgognon, J.M.; Spiers, J.G.; Scheiblich, H.; Antonov, A.; Bradley, S.J.; Tobin, A.B.; Steinert, J.R. Alterations in neuronal metabolism contribute to the pathogenesis of prion disease. Cell Death Differ. 2018, 25, 1408–1425. [Google Scholar] [CrossRef]
- Arnould, H.; Baudouin, V.; Baudry, A.; Ribeiro, L.W.; Ardila-Osorio, H.; Pietri, M.; Caradeuc, C.; Soultawi, C.; Williams, D.; Alvarez, M.; et al. Loss of prion protein control of glucose metabolism promotes neurodegeneration in model of prion diseases. PLoS Pathog. 2021, 17, e1009991. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, E.; Musich, P.R.; Lin, F. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci. Ther. 2019, 25, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Tatsuta, T.; Langer, T. Quality control of mitochondria: Protection against neurodegeneration and ageing. EMBO J. 2008, 27, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef]
- Martinez-Cue, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Li, X.; Zhang, W.; Cao, Q.; Wang, Z.; Zhao, M.; Xu, L.; Zhuang, Q. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov. 2020, 6, 80. [Google Scholar] [CrossRef]
- Pajares, M.; Jimenez-Moreno, N.; Dias, I.H.K.; Debelec, B.; Vucetic, M.; Fladmark, K.E.; Basaga, H.; Ribaric, S.; Milisav, I.; Cuadrado, A. Redox control of protein degradation. Redox Biol. 2015, 6, 409–420. [Google Scholar] [CrossRef]
- Mattei, V.; Matarrese, P.; Garofalo, T.; Tinari, A.; Gambardella, L.; Ciarlo, L.; Manganelli, V.; Tasciotti, V.; Misasi, R.; Malorni, W.; et al. Recruitment of cellular prion protein to mitochondrial raft-like microdomains contributes to apoptosis execution. Mol. Biol. Cell 2011, 22, 4842–4853. [Google Scholar] [CrossRef]
- Li, C.; Wang, D.; Wu, W.; Yang, W.; Ali Shah, S.Z.; Zhao, Y.; Duan, Y.; Wang, L.; Zhou, X.; Zhao, D.; et al. DLP1-dependent mitochondrial fragmentation and redistribution mediate prion-associated mitochondrial dysfunction and neuronal death. Aging Cell 2018, 17, e12693. [Google Scholar] [CrossRef] [PubMed]
- O’Donovan, C.N.; Tobin, D.; Cotter, T.G. Prion protein fragment PrP-(106–126) induces apoptosis via mitochondrial disruption in human neuronal SH-SY5Y cells. J. Biol Chem 2001, 276, 43516–43523. [Google Scholar] [CrossRef] [PubMed]
- Bialik, S.; Dasari, S.K.; Kimchi, A. Autophagy-dependent cell death-where, how and why a cell eats itself to death. J. Cell Sci. 2018, 131, jcs215152. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, D.C. Mitochondrial Dynamics in Regulating the Unique Phenotypes of Cancer and Stem Cells. Cell Metab. 2017, 26, 39–48. [Google Scholar] [CrossRef]
- Han, S.; Zhang, M.; Jeong, Y.Y.; Margolis, D.J.; Cai, Q. The role of mitophagy in the regulation of mitochondrial energetic status in neurons. Autophagy 2021, 17, 4182–4201. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Kroemer, G.; Kepp, O. Mitophagy: An Emerging Role in Aging and Age-Associated Diseases. Front. Cell Dev. Biol. 2020, 8, 200. [Google Scholar] [CrossRef]
- Forrester, S.J.; Kikuchi, D.S.; Hernandes, M.S.; Xu, Q.; Griendling, K.K. Reactive Oxygen Species in Metabolic and Inflammatory Signaling. Circ. Res. 2018, 122, 877–902. [Google Scholar] [CrossRef]
- Gray, L.R.; Tompkins, S.C.; Taylor, E.B. Regulation of pyruvate metabolism and human disease. Cell Mol. Life Sci. 2014, 71, 2577–2604. [Google Scholar] [CrossRef] [Green Version]
- Albanese, M.; Zagaglia, S.; Landi, D.; Boffa, L.; Nicoletti, C.G.; Marciani, M.G.; Mandolesi, G.; Marfia, G.A.; Buttari, F.; Mori, F.; et al. Cerebrospinal fluid lactate is associated with multiple sclerosis disease progression. J. Neuroinflammation 2016, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subject | Diagnosis | Sex | Age | Brain Weight (g) | PrPSc Type | PMD (Hours) | Applications |
|---|---|---|---|---|---|---|---|
| Control 1 | Non-CJD | M | 83 | 1220 | ND | 12 | WB |
| Control 2 | Non-CJD | M | 86 | 1100 | ND | 2.5 | WB |
| Control 3 | Non-CJD | M | 71 | 1225 | ND | 7 | WB |
| # Control 4 | Non-CJD | F | 70 | 1300 | ND | 12 | ICC |
| CJD 1 | Sporadic CJD | M | 77 | 1600 | 1 | 2.5 | WB |
| CJD 2 | Sporadic CJD | M | 66 | 1380 | 1 | 13 | WB, ICC |
| CJD 3 | Sporadic CJD | F | 65 | 1170 | 1 | 24 | WB |
| CJD 4 | Sporadic CJD | M | 49 | 1150 | 1 and 2 | 120 | WB |
| CJD 5 | Sporadic CJD | M | 50 | 1550 | 1 | 12 | WB |
| CJD 6 | Sporadic CJD | M | 58 | 1040 | 1 | 12 | WB |
| CJD 7 | Sporadic CJD | M | 58 | 1250 | 1 | 12 | ICC |
| CJD 8 | Sporadic CJD | M | 66 | 1510 | 1 | 24 | ICC |
| Gene | Sequence (Forward/Reverse) | Product Length (bp) |
|---|---|---|
| Drp1 | 5′-CGTGACAAATGAAATGGTGC-3′ 5′-CATTAGCCCACAG-GCATCAG-3′ | 216 |
| Actin | 5′-GACCTCTATGCCAACACAGT-3′ 5′-AGTACTTGCGC-TCAGGAGGA-3′ | 139 |
| mtDNA (MT-ND2, NADH dehydrogenase) | 5′-CCTATCACCCTTGCCATCAT-3′ 5′-GAGGCT GTTGCTTGTGTGAC-3‘ | 193 |
| nucDNA (Pecam 1) | 5′-ATGGAAAGCCTGCCATCATG-3′ 5′-TCCT TGTTGTTCAGCATCAC-3′ | 235 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.-J.; Kim, H.-J.; Jang, B.; Kim, H.-J.; Mostafa, M.N.; Park, S.-J.; Kim, Y.-S.; Choi, E.-K. Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration. Cells 2022, 11, 2744. https://doi.org/10.3390/cells11172744
Kim M-J, Kim H-J, Jang B, Kim H-J, Mostafa MN, Park S-J, Kim Y-S, Choi E-K. Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration. Cells. 2022; 11(17):2744. https://doi.org/10.3390/cells11172744
Chicago/Turabian StyleKim, Mo-Jong, Hee-Jun Kim, Byungki Jang, Hyun-Ji Kim, Mohd Najib Mostafa, Seok-Joo Park, Yong-Sun Kim, and Eun-Kyoung Choi. 2022. "Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration" Cells 11, no. 17: 2744. https://doi.org/10.3390/cells11172744
APA StyleKim, M.-J., Kim, H.-J., Jang, B., Kim, H.-J., Mostafa, M. N., Park, S.-J., Kim, Y.-S., & Choi, E.-K. (2022). Impairment of Neuronal Mitochondrial Quality Control in Prion-Induced Neurodegeneration. Cells, 11(17), 2744. https://doi.org/10.3390/cells11172744