
ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes

 , , , , , , add
Show full author list
, , , , , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation and Culture of hiPSC-CMs in 2D and EHT Formats
2.2. Morphological Analysis of 2D-Cultured hiPSC-CMs
2.3. Production and Purification of Adeno-Associated Virus Vector Particles
2.4. Proteome Analysis
2.5. High-Content Imaging of Autophagy-Lysosomal Pathway in hiPSC-CMs
2.6. Statistics
3. Results
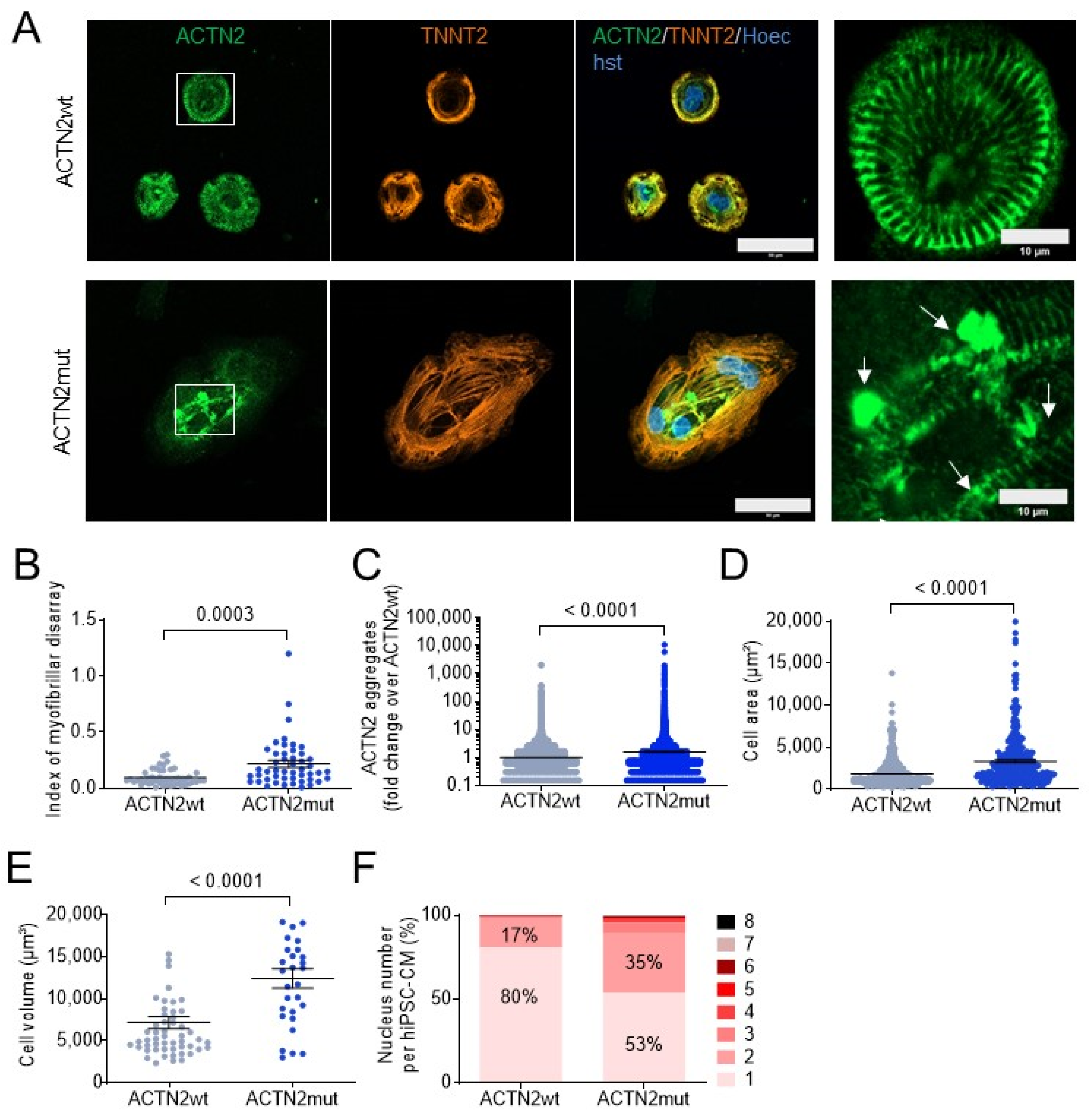
3.1. ACTN2mut 2D-Cultured hiPSC-CMs Display Hypertrophy, Myofibrillar Disarray, Protein Aggregation, and Multinucleation
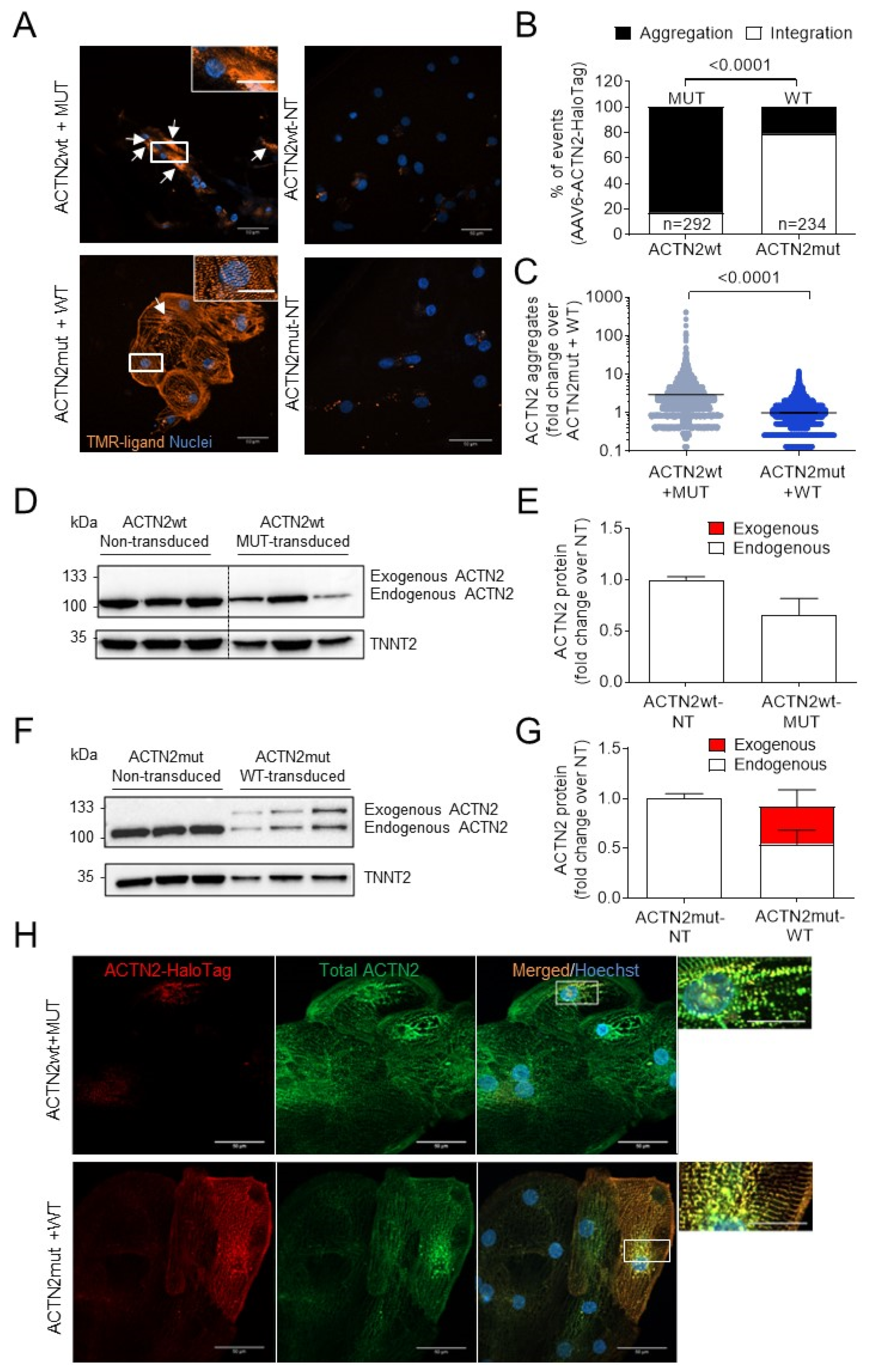
3.2. Exogenous Mutant ACTN2 Causes Aggregate Formation Leading to Sarcomere Disarray
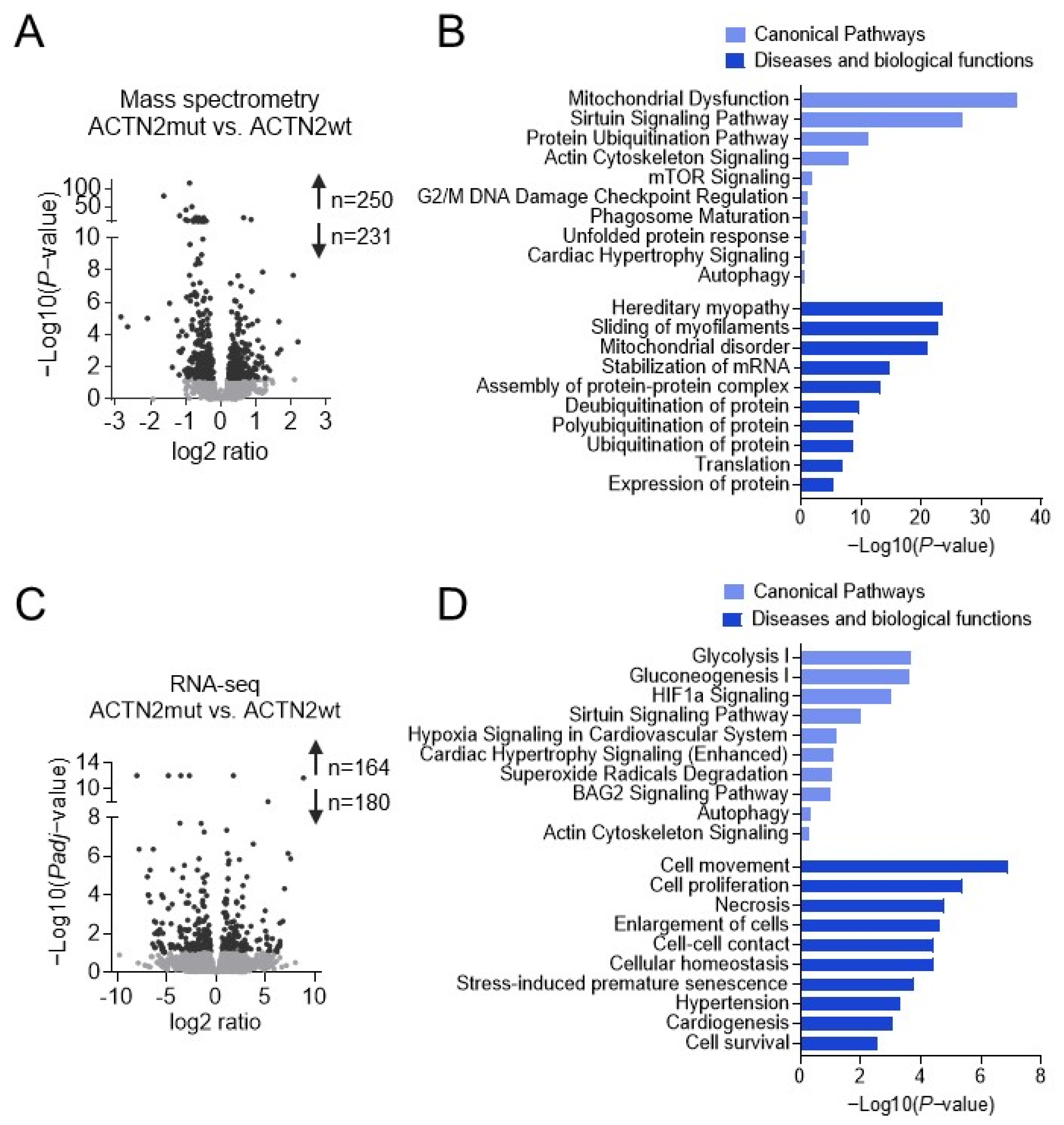
3.3. ACTN2mut hiPSC-CMs Exhibit Alterations of Several Canonical Pathways
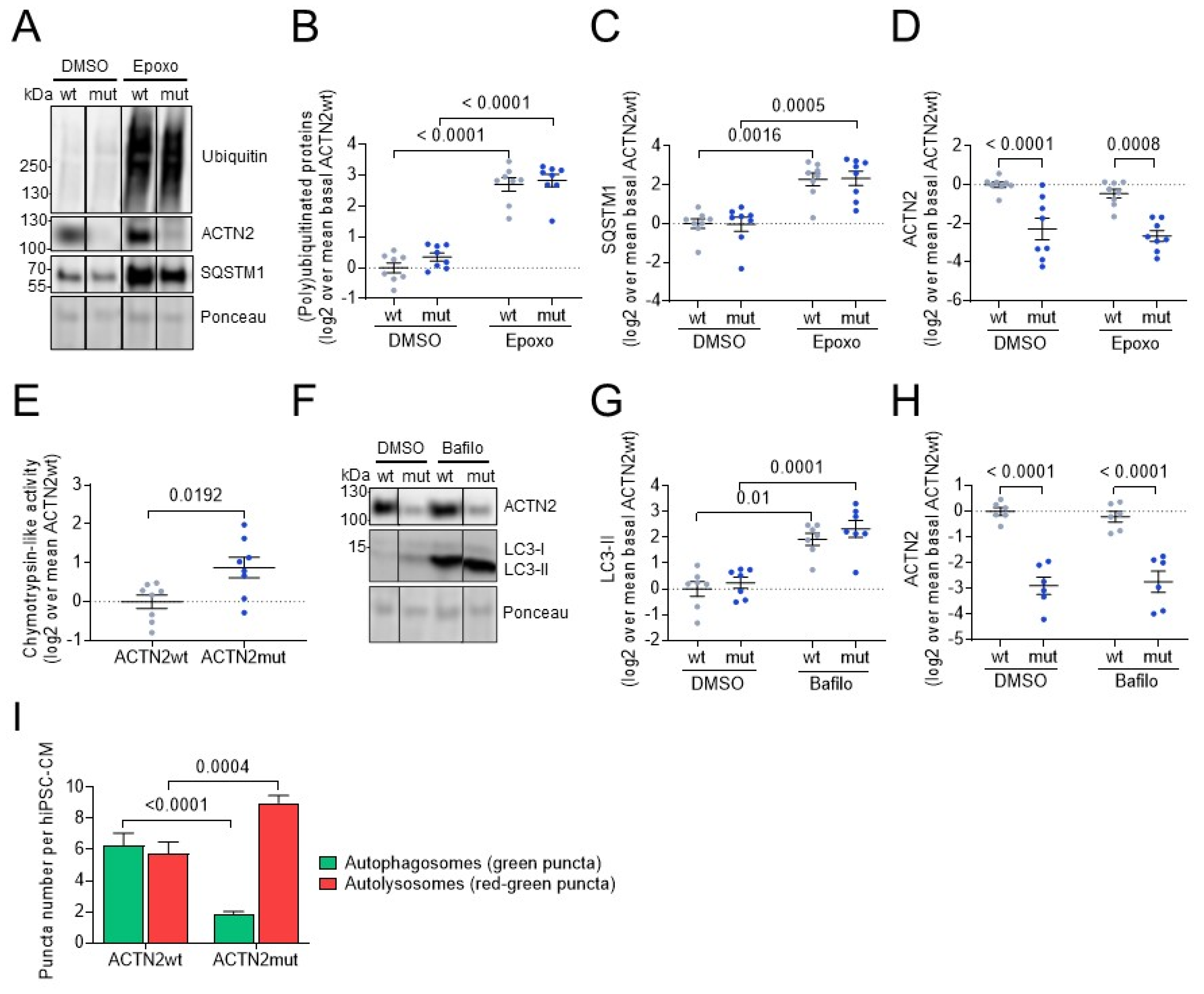
3.4. ACTN2mut hiPSC-CMs Exhibit Higher Activities of the Ubiquitin-Proteasome System and the Autophagy-Lysosomal Pathway
3.5. ACTN2mut hiPSC-CMs Exhibit Force Impairment in Engineered Heart Tissues
4. Discussion
5. Study Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sjoblom, B.; Salmazo, A.; Djinovic-Carugo, K. Alpha-actinin structure and regulation. Cell. Mol. Life Sci. 2008, 65, 2688–2701. [Google Scholar] [CrossRef] [PubMed]
- Gautel, M.; Djinovic-Carugo, K. The sarcomeric cytoskeleton: From molecules to motion. J. Exp. Biol. 2016, 219, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Broderick, M.J.; Winder, S.J. Spectrin, alpha-actinin, and dystrophin. Adv. Protein Chem. 2005, 70, 203–246. [Google Scholar] [PubMed]
- Djinovic-Carugo, K.; Gautel, M.; Ylanne, J.; Young, P. The spectrin repeat: A structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002, 513, 119–123. [Google Scholar] [CrossRef]
- Fan, L.L.; Huang, H.; Jin, J.Y.; Li, J.J.; Chen, Y.Q.; Xiang, R. Whole-Exome Sequencing Identifies a Novel Mutation (p.L320R) of Alpha-Actinin 2 in a Chinese Family with Dilated Cardiomyopathy and Ventricular Tachycardia. Cytogenet. Genome Res. 2019, 157, 148–152. [Google Scholar] [CrossRef]
- Lindholm, M.E.; Jimenez-Morales, D.; Zhu, H.; Seo, K.; Amar, D.; Zhao, C.; Raja, A.; Madhvani, R.; Abramowitz, S.; Espenel, C.; et al. Mono- and Biallelic Protein-Truncating Variants in Alpha-Actinin 2 Cause Cardiomyopathy Through Distinct Mechanisms. Circ. Genom. Precis. Med. 2021, 14, e003419. [Google Scholar] [CrossRef]
- Mohapatra, B.; Jimenez, S.; Lin, J.H.; Bowles, K.R.; Coveler, K.J.; Marx, J.G.; Chrisco, M.A.; Murphy, R.T.; Lurie, P.R.; Schwartz, R.J.; et al. Mutations in the muscle LIM protein and alpha-actinin-2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol. Genet. Metab. 2003, 80, 207–215. [Google Scholar] [CrossRef]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy. EMBO Mol. Med. 2019, 11, e11115. [Google Scholar] [CrossRef]
- Walsh, R.; Thomson, K.L.; Ware, J.S.; Funke, B.H.; Woodley, J.; McGuire, K.J.; Mazzarotto, F.; Blair, E.; Seller, A.; Taylor, J.C.; et al. Reassessment of Mendelian gene pathogenicity using 7, 855 cardiomyopathy cases and 60, 706 reference samples. Genet. Med. 2017, 19, 192–203. [Google Scholar] [CrossRef]
- Inoue, M.; Noguchi, S.; Sonehara, K.; Nakamura-Shindo, K.; Taniguchi, A.; Kajikawa, H.; Nakamura, H.; Ishikawa, K.; Ogawa, M.; Hayashi, S.; et al. A recurrent homozygous ACTN2 variant associated with core myopathy. Acta Neuropathol. 2021, 142, 785–788. [Google Scholar] [CrossRef]
- Chiu, C.; Bagnall, R.D.; Ingles, J.; Yeates, L.; Kennerson, M.; Donald, J.A.; Jormakka, M.; Lind, J.M.; Semsarian, C. Mutations in alpha-actinin-2 cause hypertrophic cardiomyopathy: A genome-wide analysis. J. Am. Coll. Cardiol. 2010, 55, 1127–1135. [Google Scholar] [CrossRef]
- Sandri, M.; Robbins, J. Proteotoxicity: An underappreciated pathology in cardiac disease. J. Mol. Cell. Cardiol. 2014, 71, 3–10. [Google Scholar] [CrossRef]
- Zech, A.T.L.; Singh, S.R.; Schlossarek, S.; Carrier, L. Autophagy in cardiomyopathies. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118432. [Google Scholar] [CrossRef]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Prondzynski, M.; Kramer, E.; Laufer, S.D.; Shibamiya, A.; Pless, O.; Flenner, F.; Muller, O.J.; Munch, J.; Redwood, C.; Hansen, A.; et al. Evaluation of MYBPC3 trans-Splicing and Gene Replacement as Therapeutic Options in Human iPSC-Derived Cardiomyocytes. Mol. Ther. Nucleic Acids 2017, 7, 475–486. [Google Scholar] [CrossRef]
- Saleem, U.; Mannhardt, I.; Braren, I.; Denning, C.; Eschenhagen, T.; Hansen, A. Force and Calcium Transients Analysis in Human Engineered Heart Tissues Reveals Positive Force-Frequency Relation at Physiological Frequency. Stem Cell Rep. 2020, 14, 312–324. [Google Scholar] [CrossRef]
- Palma Medina, L.M.; Becker, A.K.; Michalik, S.; Yedavally, H.; Raineri, E.J.M.; Hildebrandt, P.; Gesell Salazar, M.; Surmann, K.; Pfortner, H.; Mekonnen, S.A.; et al. Metabolic Cross-talk Between Human Bronchial Epithelial Cells and Internalized Staphylococcus aureus as a Driver for Infection. Mol. Cell Proteom. 2019, 18, 892–908. [Google Scholar] [CrossRef]
- Seidl, M.D.; Stein, J.; Hamer, S.; Pluteanu, F.; Scholz, B.; Wardelmann, E.; Huge, A.; Witten, A.; Stoll, M.; Hammer, E.; et al. Characterization of the Genetic Program Linked to the Development of Atrial Fibrillation in CREM-IbDeltaC-X Mice. Circ. Arrhythm. Electrophysiol. 2017, 10, e005075. [Google Scholar] [CrossRef]
- Prondzynski, M.; Lemoine, M.D.; Zech, A.T.; Horvath, A.; Di Mauro, V.; Koivumaki, J.T.; Kresin, N.; Busch, J.; Krause, T.; Kramer, E.; et al. Disease modeling of a mutation in alpha-actinin 2 guides clinical therapy in hypertrophic cardiomyopathy—Addendum. EMBO Mol. Med. 2022, 14, e16423. [Google Scholar] [CrossRef]
- Derks, W.; Bergmann, O. Polyploidy in Cardiomyocytes: Roadblock to Heart Regeneration? Circ. Res. 2020, 126, 552–565. [Google Scholar] [CrossRef]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.R.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.C.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef] [PubMed]
- Ladha, F.A.; Thakar, K.; Pettinato, A.M.; Legere, N.; Cohn, R.; Romano, R.; Meredith, E.; Chen, Y.S.; Hinson, J.T. Identifying cardiac actinin interactomes reveals sarcomere crosstalk with RNA-binding proteins. bioRxiv 2020, 2020, 994004. [Google Scholar]
- Wan, C.; Borgeson, B.; Phanse, S.; Tu, F.; Drew, K.; Clark, G.; Xiong, X.; Kagan, O.; Kwan, J.; Bezginov, A.; et al. Panorama of ancient metazoan macromolecular complexes. Nature 2015, 525, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Linnemann, A.; van der Ven, P.F.; Vakeel, P.; Albinus, B.; Simonis, D.; Bendas, G.; Schenk, J.A.; Micheel, B.; Kley, R.A.; Furst, D.O. The sarcomeric Z-disc component myopodin is a multiadapter protein that interacts with filamin and alpha-actinin. Eur. J. Cell Biol. 2010, 89, 681–692. [Google Scholar] [CrossRef]
- Friedrich, F.W.; Wilding, B.R.; Reischmann, S.; Crocini, C.; Lang, P.; Charron, P.; Muller, O.J.; McGrath, M.J.; Vollert, I.; Hansen, A.; et al. Evidence for FHL1 as a novel disease gene for isolated hypertrophic cardiomyopathy. Hum. Mol. Genet. 2012, 21, 3237–3254. [Google Scholar] [CrossRef]
- Friedrich, F.W.; Reischmann, S.; Schwalm, A.; Unger, A.; Ramanujam, D.; Munch, J.; Muller, O.J.; Hengstenberg, C.; Galve, E.; Charron, P.; et al. FHL2 expression and variants in hypertrophic cardiomyopathy. Basic Res. Cardiol. 2014, 109, 451. [Google Scholar] [CrossRef]
- Hinson, J.T.; Chopra, A.; Nafissi, N.; Polacheck, W.J.; Benson, C.C.; Swist, S.; Gorham, J.; Yang, L.; Schafer, S.; Sheng, C.C.; et al. HEART DISEASE. Titin mutations in iPS cells define sarcomere insufficiency as a cause of dilated cardiomyopathy. Science 2015, 349, 982–986. [Google Scholar] [CrossRef]
- Cuello, F.; Knaust, A.E.; Saleem, U.; Loos, M.; Raabe, J.; Mosqueira, D.; Laufer, S.; Schweizer, M.; van der Kraak, P.; Flenner, F.; et al. Impairment of the ER/mitochondria compartment in human cardiomyocytes with PLN p.Arg14del mutation. EMBO Mol. Med. 2021, 13, e13074. [Google Scholar] [CrossRef]
- Feyen, D.A.M.; Perea-Gil, I.; Maas, R.G.C.; Harakalova, M.; Gavidia, A.A.; Arthur Ataam, J.; Wu, T.H.; Vink, A.; Pei, J.; Vadgama, N.; et al. Unfolded Protein Response as a Compensatory Mechanism and Potential Therapeutic Target in PLN R14del Cardiomyopathy. Circulation 2021, 144, 382–392. [Google Scholar] [CrossRef]
- Davis, J.; Davis, L.C.; Correll, R.N.; Makarewich, C.A.; Schwanekamp, J.A.; Moussavi-Harami, F.; Wang, D.; York, A.J.; Wu, H.; Houser, S.R.; et al. A Tension-Based Model Distinguishes Hypertrophic versus Dilated Cardiomyopathy. Cell 2016, 165, 1147–1159. [Google Scholar] [CrossRef]
- Haywood, N.J.; Wolny, M.; Rogers, B.; Trinh, C.H.; Shuping, Y.; Edwards, T.A.; Peckham, M. Hypertrophic cardiomyopathy mutations in the calponin-homology domain of ACTN2 affect actin binding and cardiomyocyte Z-disc incorporation. Biochem. J. 2016, 473, 2485–2493. [Google Scholar] [CrossRef] [Green Version]
- Dorsch, L.M.; Schuldt, M.; dos Remedios, C.G.; Schinkel, A.F.L.; de Jong, P.L.; Michels, M.; Kuster, D.W.D.; Brundel, B.; van der Velden, J. Protein Quality Control Activation and Microtubule Remodeling in Hypertrophic Cardiomyopathy. Cells 2019, 8, 741. [Google Scholar] [CrossRef]
- Spaich, S.; Will, R.D.; Just, S.; Kuhn, C.; Frank, D.; Berger, I.M.; Wiemann, S.; Korn, B.; Koegl, M.; Backs, J.; et al. F-box and leucine-rich repeat protein 22 is a cardiac-enriched F-box protein that regulates sarcomeric protein turnover and is essential for maintenance of contractile function in vivo. Circ. Res. 2012, 111, 1504–1516. [Google Scholar] [CrossRef]
- Lewis, Y.E.; Moskovitz, A.; Mutlak, M.; Heineke, J.; Caspi, L.H.; Kehat, I. Localization of transcripts, translation, and degradation for spatiotemporal sarcomere maintenance. J. Mol. Cell. Cardiol. 2018, 116, 16–28. [Google Scholar] [CrossRef]
- Agarwal, R.; Paulo, J.A.; Toepfer, C.N.; Ewoldt, J.K.; Sundaram, S.; Chopra, A.; Zhang, Q.; Gorham, J.; DePalma, S.R.; Chen, C.S.; et al. Filamin C Cardiomyopathy Variants Cause Protein and Lysosome Accumulation. Circ. Res. 2021, 129, 751–766. [Google Scholar] [CrossRef]
- Valdes-Mas, R.; Gutierrez-Fernandez, A.; Gomez, J.; Coto, E.; Astudillo, A.; Puente, D.A.; Reguero, J.R.; Alvarez, V.; Moris, C.; Leon, D.; et al. Mutations in filamin C cause a new form of familial hypertrophic cardiomyopathy. Nat. Commun. 2014, 5, 5326. [Google Scholar] [CrossRef]
- Eijgenraam, T.R.; Boogerd, C.J.; Stege, N.M.; Oliveira Nunes Teixeira, V.; Dokter, M.M.; Schmidt, L.E.; Yin, X.; Theofilatos, K.; Mayr, M.; van der Meer, P.; et al. Protein Aggregation Is an Early Manifestation of Phospholamban p.(Arg14del)-Related Cardiomyopathy: Development of PLN-R14del-Related Cardiomyopathy. Circ. Heart Fail. 2021, 4, e008532. [Google Scholar] [CrossRef]
- Arvanitis, M.; Tampakakis, E.; Zhang, Y.; Wang, W.; Auton, A.; Me Research, T.; Dutta, D.; Glavaris, S.; Keramati, A.; Chatterjee, N.; et al. Genome-wide association and multi-omic analyses reveal ACTN2 as a gene linked to heart failure. Nat. Commun. 2020, 11, 1122. [Google Scholar] [CrossRef]
- Eschenhagen, T.; Carrier, L. Cardiomyopathy phenotypes in human-induced pluripotent stem cell-derived cardiomyocytes-a systematic review. Pflug. Arch. 2019, 471, 755–768. [Google Scholar] [CrossRef]
- Brandao, K.O.; Tabel, V.A.; Atsma, D.E.; Mummery, C.L.; Davis, R.P. Human pluripotent stem cell models of cardiac disease: From mechanisms to therapies. Dis. Model. Mech. 2017, 10, 1039–1059. [Google Scholar] [CrossRef]
- Puri, M.C.; Nagy, A. Concise review: Embryonic stem cells versus induced pluripotent stem cells: The game is on. Stem Cells 2012, 30, 10–14. [Google Scholar] [CrossRef]
- Shibamiya, A.; Schulze, E.; Krauss, D.; Augustin, C.; Reinsch, M.; Schulze, M.L.; Steuck, S.; Mearini, G.; Mannhardt, I.; Schulze, T.; et al. Cell Banking of hiPSCs: A Practical Guide to Cryopreservation and Quality Control in Basic Research. Curr. Protoc. Stem Cell Biol. 2020, 55, e127. [Google Scholar] [CrossRef]
- Breckwoldt, K.; Letuffe-Breniere, D.; Mannhardt, I.; Schulze, T.; Ulmer, B.; Werner, T.; Benzin, A.; Klampe, B.; Reinsch, M.C.; Laufer, S.; et al. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat. Protoc. 2017, 12, 1177–1197. [Google Scholar] [CrossRef]
- Skryabin, B.V.; Kummerfeld, D.-M.; Gubar, L.; Seeger, B.; Kaiser, H.; Stegemann, A.; Roth, J.; Meuth, S.G.; Pavenstädt, H.; Sherwood, J.; et al. Pervasive head-to-tail insertions of DNA templates mask desired CRISPR-Cas9–mediated genome editing events. Sci. Adv. 2020, 6, eaax2941. [Google Scholar] [CrossRef]
- Mosqueira, D.; Lis-Slimak, K.; Denning, C. High-Throughput Phenotyping Toolkit for Characterizing Cellular Models of Hypertrophic Cardiomyopathy In Vitro. Methods Protoc. 2019, 2, 83. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Strimmer, K. fdrtool: A versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 2008, 24, 1461–1462. [Google Scholar] [CrossRef]
- Wenzel, K.; Kramer, E.; Geertz, B.; Carrier, L.; Felix, S.B.; Konemann, S.; Schlossarek, S. A Transgenic Mouse Model of Eccentric Left Ventricular Hypertrophy With Preserved Ejection Fraction Exhibits Alterations in the Autophagy-Lysosomal Pathway. Front. Physiol. 2021, 12, 614878. [Google Scholar] [CrossRef]
- Singh, S.R.; Meyer-Jens, M.; Alizoti, E.; Bacon, W.C.; Davis, G.; Osinska, H.; Gulick, J.; Reischmann-Düsener, S.; Orthey, E.; McLendon, P.M.; et al. A high-throughput screening identifies ZNF418 as a novel regulator of the ubiquitin-proteasome system and autophagy-lysosomal pathway. Autophagy 2020, 17, 3124–3139. [Google Scholar] [CrossRef] [PubMed]
- Vignier, N.; Schlossarek, S.; Fraysse, B.; Mearini, G.; Kramer, E.; Pointu, H.; Mougenot, N.; Guiard, J.; Reimer, R.; Hohenberg, H.; et al. Nonsense-Mediated mRNA Decay and UbiquitinProteasome System Regulate Cardiac Myosin-Binding Protein C Mutant Levels in Cardiomyopathic Mice. Circ. Res. 2009, 105, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Schlossarek, S.; Englmann, D.R.; Sultan, K.R.; Sauer, M.; Eschenhagen, T.; Carrier, L. Defective proteolytic systems in Mybpc3-targeted mice with cardiac hypertrophy. Basic Res. Cardiol. 2011, 107, 235. [Google Scholar] [CrossRef]
- Suomi, T.; Elo, L.L. Enhanced differential expression statistics for data-independent acquisition proteomics. Sci. Rep. 2017, 7, 5869. [Google Scholar] [CrossRef] [PubMed]
- Phipson, B.; Lee, S.; Majewski, I.J.; Alexander, W.S.; Smyth, G.K. Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann. Appl. Stat. 2016, 10, 946–963. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Zhou, C.; Zhong, W.; Zhou, J.; Sheng, F.; Fang, Z.; Wei, Y.; Chen, Y.; Deng, X.; Xia, B.; Lin, J. Monitoring autophagic flux by an improved tandem fluorescenttagged LC3 (mTagRFP-mWasabi-LC3) reveals that high-dose rapamycin impairs autophagic flux in cancer cells. Autophagy 2012, 8, 1215–1226. [Google Scholar] [CrossRef]
- Wasilko, D.J.; Lee, S.E.; Stutzman-Engwall, K.J.; Reitz, B.A.; Emmons, T.L.; Mathis, K.J.; Bienkowski, M.J.; Tomasselli, A.G.; Fischer, H.D. The titerless infected-cells preservation and scale-up (TIPS) method for large-scale production of NO-sensitive human soluble guanylate cyclase (sGC) from insect cells infected with recombinant baculovirus. Protein Expr. Purif. 2009, 65, 122–132. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zech, A.T.L.; Prondzynski, M.; Singh, S.R.; Pietsch, N.; Orthey, E.; Alizoti, E.; Busch, J.; Madsen, A.; Behrens, C.S.; Meyer-Jens, M.; et al. ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes. Cells 2022, 11, 2745. https://doi.org/10.3390/cells11172745
Zech ATL, Prondzynski M, Singh SR, Pietsch N, Orthey E, Alizoti E, Busch J, Madsen A, Behrens CS, Meyer-Jens M, et al. ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes. Cells. 2022; 11(17):2745. https://doi.org/10.3390/cells11172745
Chicago/Turabian StyleZech, Antonia T. L., Maksymilian Prondzynski, Sonia R. Singh, Niels Pietsch, Ellen Orthey, Erda Alizoti, Josefine Busch, Alexandra Madsen, Charlotta S. Behrens, Moritz Meyer-Jens, and et al. 2022. "ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes" Cells 11, no. 17: 2745. https://doi.org/10.3390/cells11172745
APA StyleZech, A. T. L., Prondzynski, M., Singh, S. R., Pietsch, N., Orthey, E., Alizoti, E., Busch, J., Madsen, A., Behrens, C. S., Meyer-Jens, M., Mearini, G., Lemoine, M. D., Krämer, E., Mosqueira, D., Virdi, S., Indenbirken, D., Depke, M., Salazar, M. G., Völker, U., ... Carrier, L. (2022). ACTN2 Mutant Causes Proteopathy in Human iPSC-Derived Cardiomyocytes. Cells, 11(17), 2745. https://doi.org/10.3390/cells11172745