

Downregulation of LKB1/AMPK Signaling in Blood Mononuclear Cells Is Associated with the Severity of Guillain–Barre Syndrome

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and PBMCs Isolation
2.2. Retrospective Study of Metformin Influence on GBS
2.3. RT-qPCR Analysis
2.4. Immunoblot Analysis
2.5. Statistical Analysis
3. Results
3.1. Demographic and Clinical Characteristics of Study Subjects
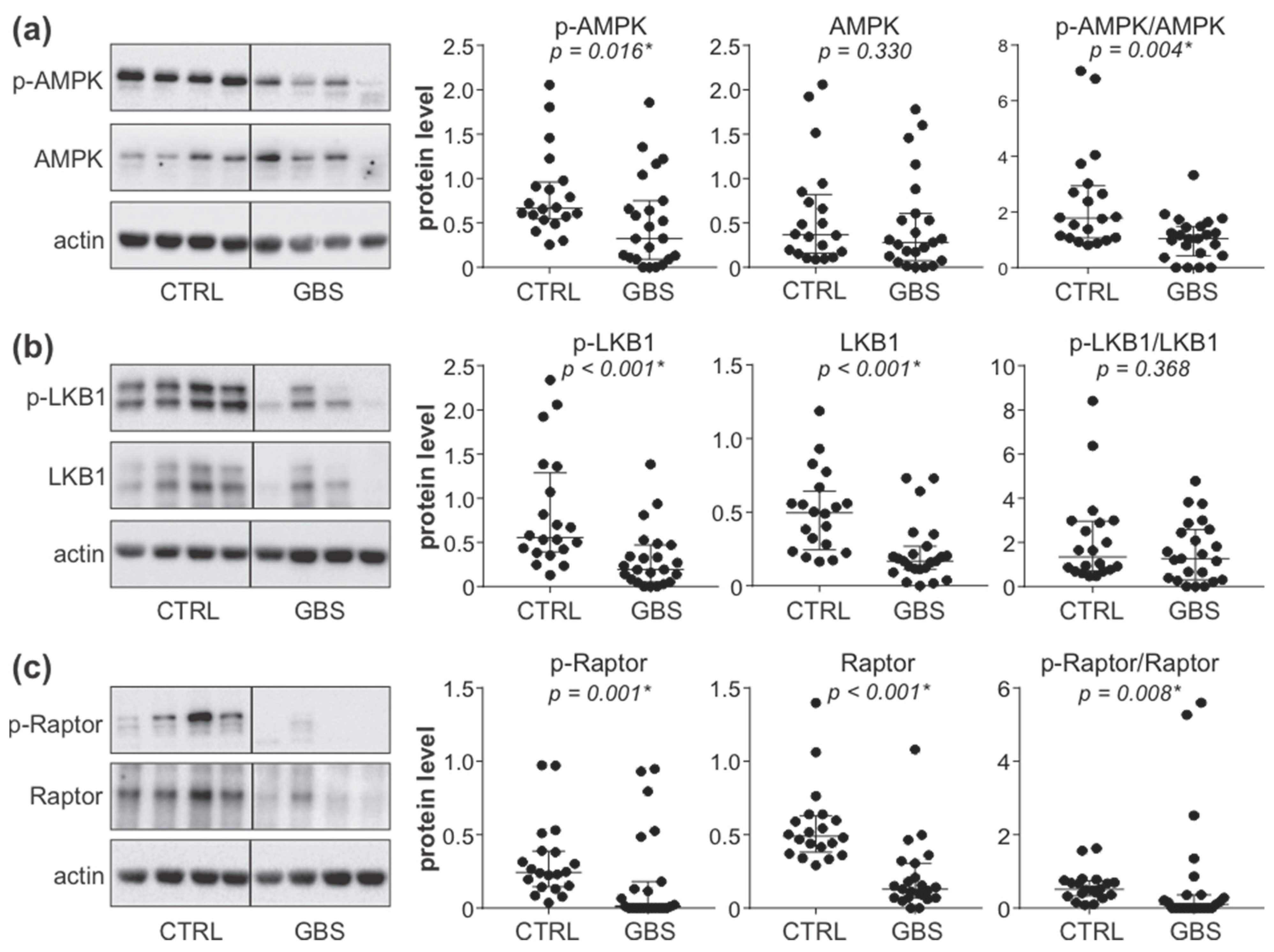
3.2. AMPK Signaling Pathway Is Downregulated in PBMCs of GBS Patients
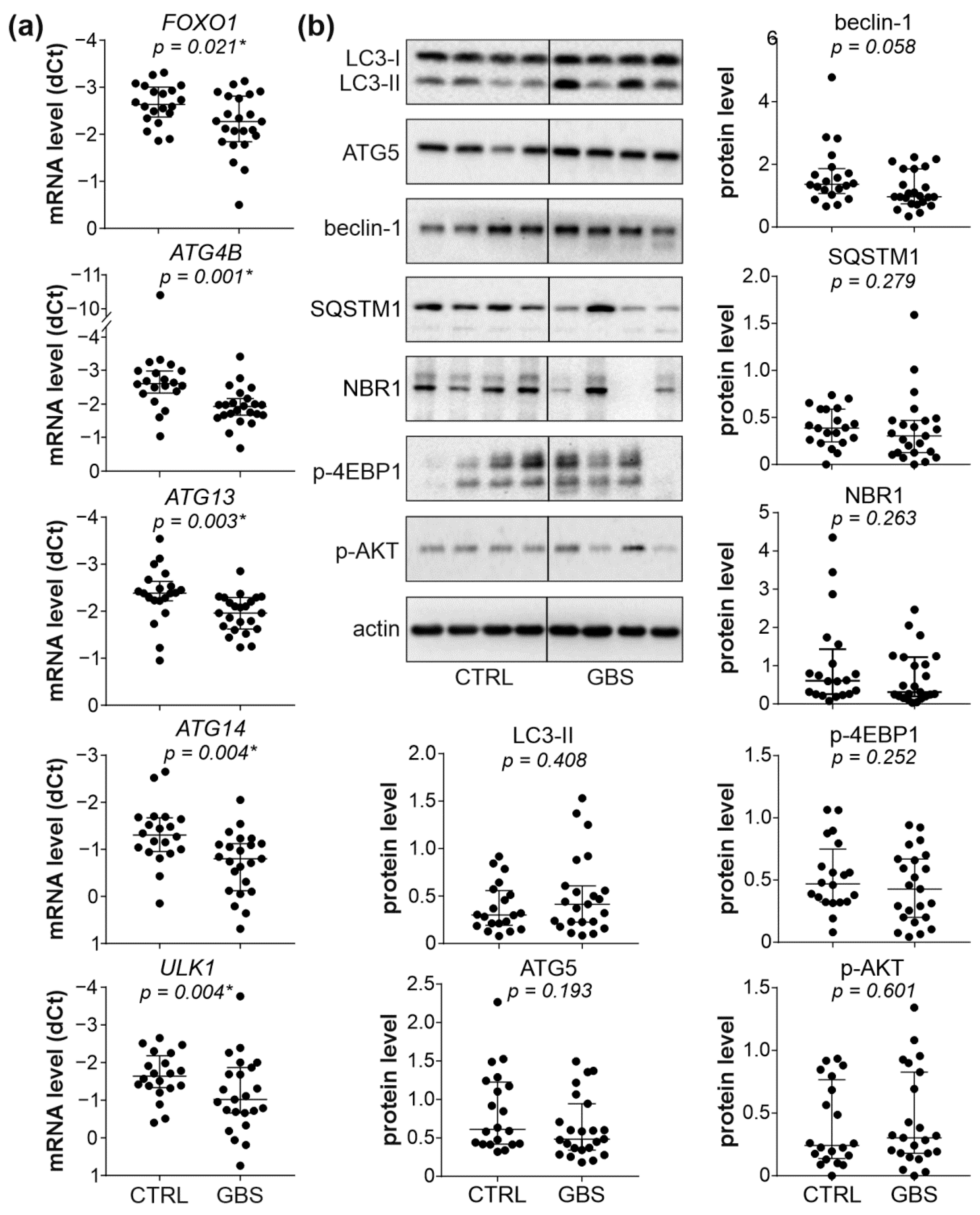
3.3. Autophagy Markers and AKT/mTORC1 Pathway Are Not Affected in PBMCs of GBS Patients
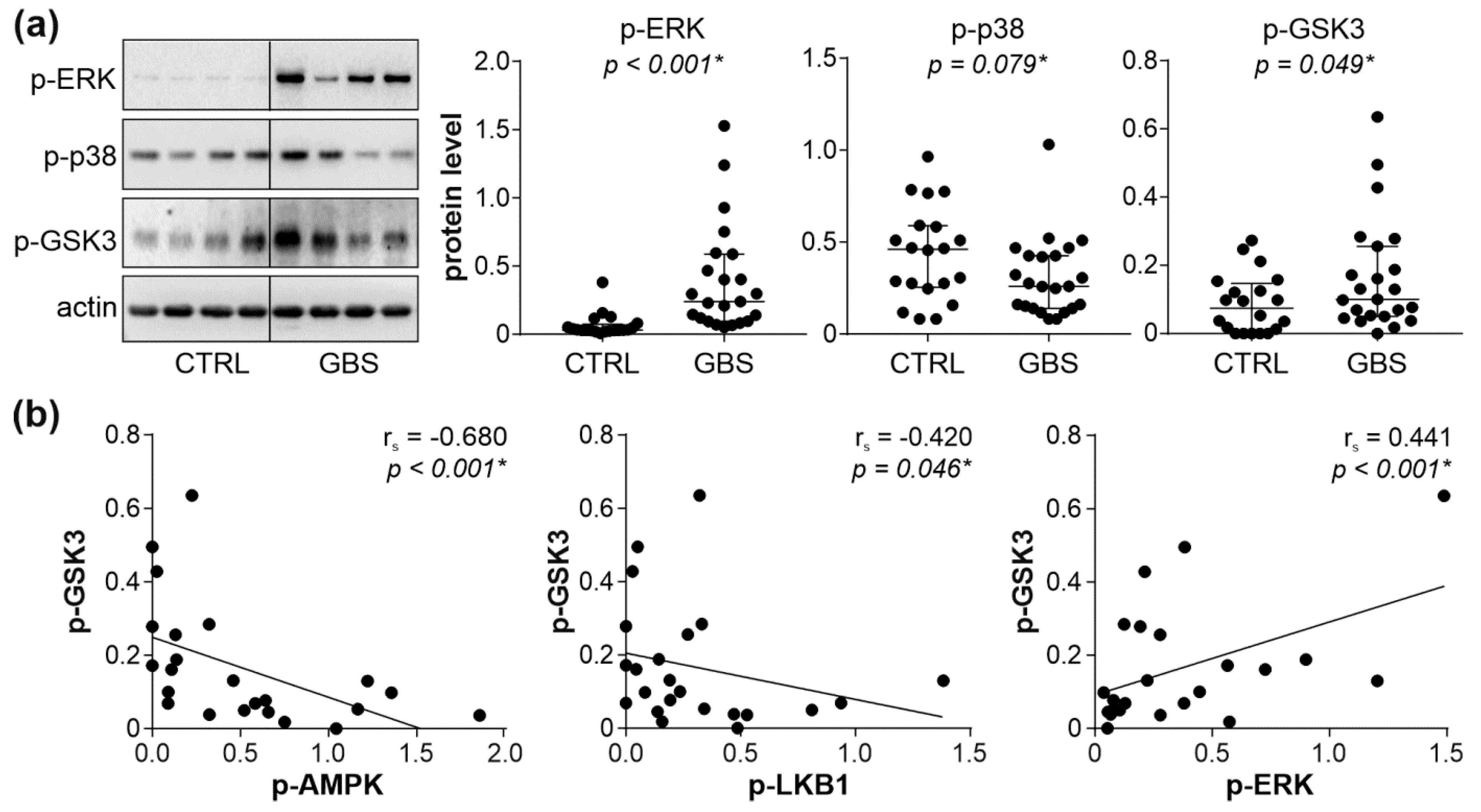
3.4. Interplay between AMPK, ERK, and PKC Signaling Pathways in PBMCs of GBS Patients
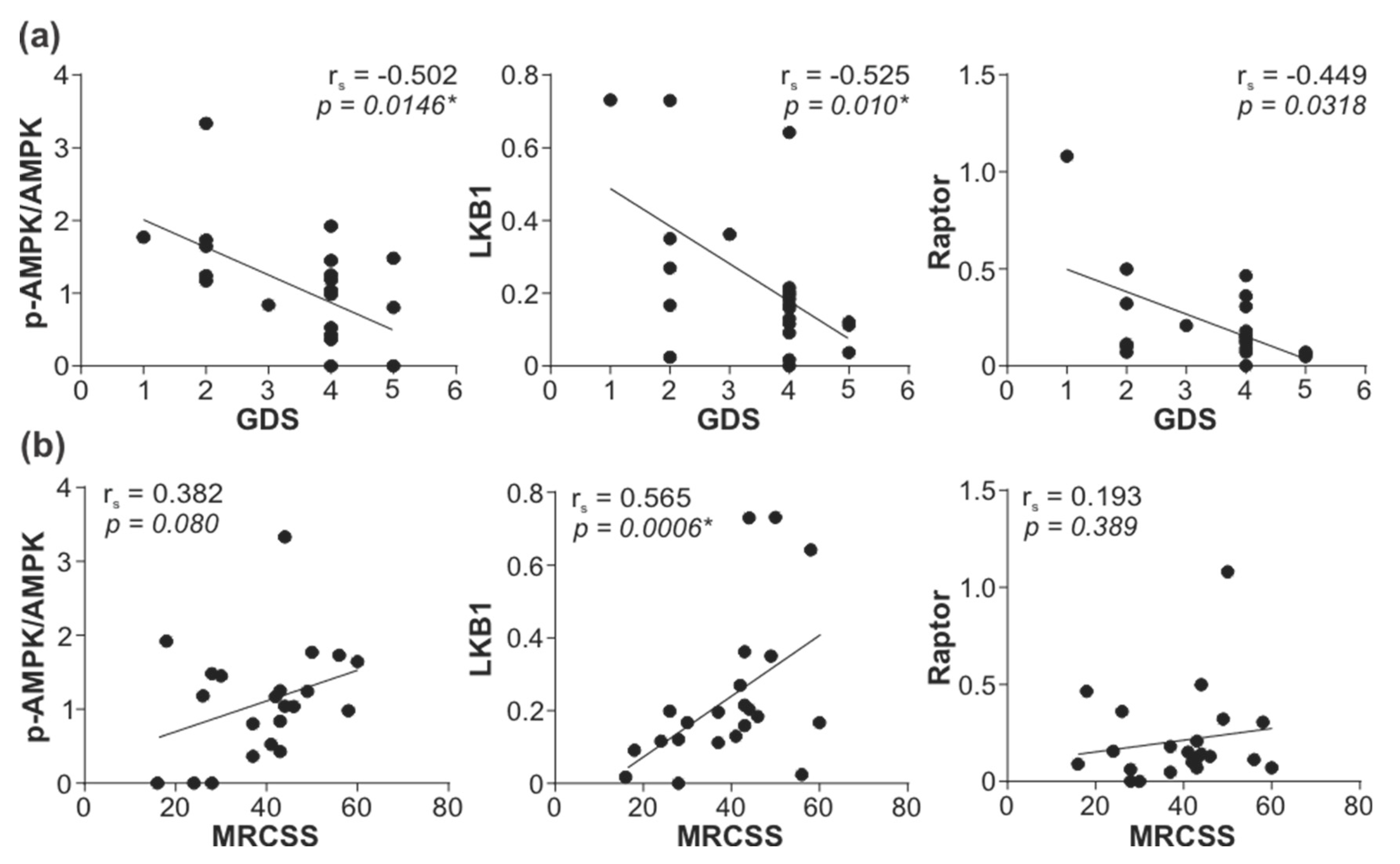
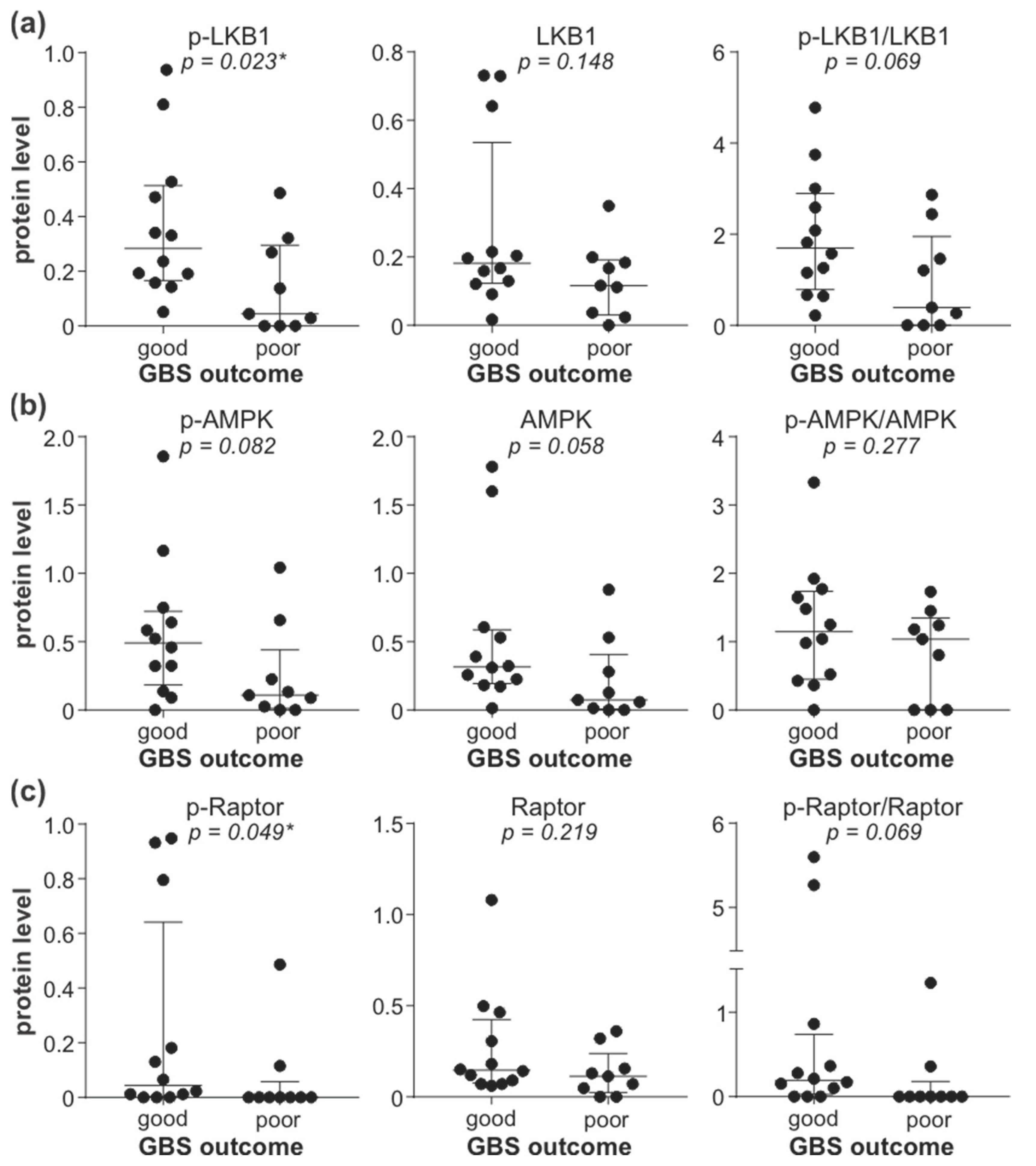
3.5. Downregulation of LKB1/AMPK Pathway Correlates with GBS Severity and Poor Outcome
3.6. Treatment with Metformin Is Associated with Better GBS Outcome in Diabetic Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van den Berg, B.; Walgaard, C.; Drenthen, J.; Fokke, C.; Jacobs, B.C.; van Doorn, P.A. Guillain-Barré syndrome: Pathogenesis, diagnosis, treatment and prognosis. Nat. Rev. Neurol. 2014, 10, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, H.A.; Beydoun, M.A.; Hossain, S.; Zonderman, A.B.; Eid, S.M. Nationwide study of therapeutic plasma exchange vs intravenous immunoglobulin in Guillain-Barré syndrome. Muscle Nerve 2020, 61, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Stojanov, A.; Berisavac, I.; Bozovic, I.; Arsenijevic, M.; Lukic-Rajic, S.; Petrovic, M.; Stojiljkovic-Tamas, O.; Jovin, Z.; Djordjevic, G.; Jovanovic, D.; et al. Incidence and mortality rates of Guillain-Barré syndrome in Serbia. J. Peripher. Nerv. Syst. 2020, 25, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Willison, H.J.; Jacobs, B.C.; van Doorn, P.A. Guillain-Barré syndrome. Lancet 2016, 388, 717–727. [Google Scholar] [CrossRef]
- Ebrahim Soltani, Z.; Rahmani, F.; Rezaei, N. Autoimmunity and cytokines in Guillain-Barré syndrome revisited: Review of pathomechanisms with an eye on therapeutic options. Eur. Cytokine Netw. 2019, 30, 1–14. [Google Scholar] [CrossRef]
- Kusunoki, S.; Willison, H.J.; Jacobs, B.C. Antiglycolipid antibodies in Guillain-Barré and Fisher syndromes: Discovery, current status and future perspective. J. Neurol. Neurosurg. Psychiatry 2021, 92, 311–318. [Google Scholar] [CrossRef]
- Poznanski, S.M.; Barra, N.G.; Ashkar, A.A.; Schertzer, J.D. Immunometabolism of T cells and NK cells: Metabolic control of effector and regulatory function. Inflamm. Res. 2018, 67, 813–828. [Google Scholar] [CrossRef]
- Jellusova, J. Metabolic control of B cell immune responses. Curr. Opin. Immunol. 2020, 63, 21–28. [Google Scholar] [CrossRef]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef]
- Yin, Z.; Pascual, C.; Klionsky, D.J. Autophagy: Machinery and regulation. Microb. Cell 2016, 3, 588–596. [Google Scholar] [CrossRef]
- Feng, Y.; Yao, Z.; Klionsky, D.J. How to control self-digestion: Transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol. 2015, 25, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Rabanal-Ruiz, Y.; Otten, E.G.; Korolchuk, V.I. mTORC1 as the main gateway to autophagy. Essays Biochem. 2017, 61, 565–584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.Y.; Li, Y.; Jiang, W.Q.; Zhou, L.F. MAPK/JNK signalling: A potential autophagy regulation pathway. Biosci. Rep. 2015, 35, e00199. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.H.; Shui, G.; Zhou, J.; Li, J.J.; Bay, B.H.; Wenk, M.R.; Shen, H.M. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin). J. Biol. Chem. 2012, 287, 14364–14376. [Google Scholar] [CrossRef] [PubMed]
- Basu, A. Regulation of autophagy by protein kinase C-ε in breast cancer cells. Int. J. Mol. Sci. 2020, 21, 4247. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem. J. 2008, 412, 179–190. [Google Scholar] [CrossRef]
- Shibutani, S.T.; Saitoh, T.; Nowag, H.; Münz, C.; Yoshimori, T. Autophagy and autophagy-related proteins in the immune system. Nat. Immunol. 2015, 16, 1014–1024. [Google Scholar] [CrossRef]
- Yin, H.; Wu, H.; Chen, Y.; Zhang, J.; Zheng, M.; Chen, G.; Li, L.; Lu, Q. The therapeutic and pathogenic role of autophagy in autoimmune diseases. Front. Immunol. 2018, 9, 1512. [Google Scholar] [CrossRef]
- Brun, S.; Schall, N.; Bonam, S.R.; Bigaut, K.; Mensah-Nyagan, A.G.; de Sèze, J.; Muller, S. An autophagy-targeting peptide to treat chronic inflammatory demyelinating polyneuropathies. J. Autoimmun. 2018, 92, 114–125. [Google Scholar] [CrossRef]
- Fokke, C.; van den Berg, B.; Drenthen, J.; Walgaard, C.; van Doorn, P.A.; Jacobs, B.C. Diagnosis of Guillain-Barré syndrome and validation of Brighton criteria. Brain 2014, 137, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Jauregui, C.E.; Teng, Y. Targeting autophagy as a strategy for drug discovery and therapeutic modulation. Future Med. Chem. 2017, 9, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Berisavac, I.; Arsenijevic, M.; Bozovic, I.; Mladenovic, B.; Kacar, A.; Stojiljkovic Tamas, O.; Petrovic, M.; Stojanovic, M.; Vujovic, B.; Martic, V.; et al. Disability and quality of life in Guillain-Barré syndrome-Longitudinal study. J. Clin. Neurosci. 2020, 78, 185–188. [Google Scholar] [CrossRef]
- Merkies, I.S.; Schmitz, P.I.; van der Meché, F.G.; Samijn, J.P.; van Doorn, P.A. Clinimetric evaluation of a new overall disability scale in immune mediated polyneuropathies. J. Neurol. Neurosurg. Psychiatry 2002, 72, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper–Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Andersen, C.L.; Jensen, J.L.; Ørntoft, T.F. Normalization of real-time quantitative reverse transcription-PCR data: A model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004, 64, 5245–5250. [Google Scholar] [CrossRef]
- Feise, R.J. Do multiple outcome measures require p-value adjustment? BMC Med. Res. Methodol. 2002, 2, 8. [Google Scholar] [CrossRef]
- Nath, N.; Khan, M.; Rattan, R.; Mangalam, A.; Makkar, R.S.; de Meester, C.; Bertrand, L.; Singh, I.; Chen, Y.; Viollet, B.; et al. Loss of AMPK exacerbates experimental autoimmune encephalomyelitis disease severity. Biochem. Biophys. Res. Commun. 2009, 386, 16–20. [Google Scholar] [CrossRef]
- Singh, J.; Giri, S. Loss of AMP-activated protein kinase in X-linked adrenoleukodystrophy patient-derived fibroblasts and lymphocytes. Biochem. Biophys. Res. Commun. 2014, 445, 126–131. [Google Scholar] [CrossRef]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; van der Windt, G.J.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef]
- Mayer, K.A.; Smole, U.; Zhu, C.; Derdak, S.; Minervina, A.A.; Salnikova, M.; Witzeneder, N.; Christamentl, A.; Boucheron, N.; Waidhofer-Söllner, P.; et al. The energy sensor AMPK orchestrates metabolic and translational adaptation in expanding T helper cells. FASEB J. 2021, 35, e21217. [Google Scholar] [CrossRef]
- Brookens, S.K.; Boothby, M.R. AMPK Metabolism in the B lineage modulates humoral responses. Immunometabolism 2021, 3, e210011. [Google Scholar] [CrossRef] [PubMed]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: Cross talk, shortcuts, and feedbacks. Mol. Cell. Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y. AMPK and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 85–108. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.H.; Chuang, T.J.; Lyu, R.K.; Ro, L.S.; Wu, Y.R.; Chang, H.S.; Huang, C.C.; Kuo, H.C.; Hsu, W.C.; Chu, C.C.; et al. Identification of gene networks and pathways associated with Guillain-Barré syndrome. PLoS ONE 2012, 7, e29506. [Google Scholar] [CrossRef]
- Saberi, B.; Ybanez, M.D.; Johnson, H.S.; Gaarde, W.A.; Han, D.; Kaplowitz, N. Protein kinase C (PKC) participates in acetaminophen hepatotoxicity through c-jun-N-terminal kinase (JNK)-dependent and -independent signaling pathways. Hepatology 2014, 59, 1543–1554. [Google Scholar] [CrossRef]
- Heathcote, H.R.; Mancini, S.J.; Strembitska, A.; Jamal, K.; Reihill, J.A.; Palmer, T.M.; Gould, G.W.; Salt, I.P. Protein kinase C phosphorylates AMP-activated protein kinase α1 Ser487. Biochem. J. 2016, 473, 4681–4697. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Denison, F.C.; Heath, R.B.; Carling, D.; Saggerson, D. 5’-AMP-activated protein kinase is inactivated by adrenergic signalling in adult cardiac myocytes. Biosci. Rep. 2012, 32, 197–213. [Google Scholar] [CrossRef]
- Saberi, B.; Shinohara, M.; Ybanez, M.D.; Hanawa, N.; Gaarde, W.A.; Kaplowitz, N.; Han, D. Regulation of H2O2-induced necrosis by PKC and AMP-activated kinase signaling in primary cultured hepatocytes. Am. J. Physiol. Cell Physiol. 2008, 295, 50–63. [Google Scholar] [CrossRef]
- Mandic, M.; Misirkic Marjanovic, M.; Vucicevic, L.; Jovanovic, M.; Bosnjak, M.; Perovic, V.; Ristic, B.; Ciric, D.; Harhaji-Trajkovic, L.; Trajkovic, V. MAP kinase-dependent autophagy controls phorbol myristate acetate-induced macrophage differentiation of HL-60 leukemia cells. Life Sci. 2022, 297, 120481. [Google Scholar] [CrossRef]
- Brändlin, I.; Hübner, S.; Eiseler, T.; Martinez-Moya, M.; Horschinek, A.; Hausser, A.; Link, G.; Rupp, S.; Storz, P.; Pfizenmaier, K.; et al. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J. Biol. Chem. 2002, 277, 6490–6496. [Google Scholar] [CrossRef] [Green Version]
- Puente, L.G.; He, J.S.; Ostergaard, H.L. A novel PKC regulates ERK activation and degranulation of cytotoxic T lymphocytes: Plasticity in PKC regulation of ERK. Eur. J. Immunol. 2006, 36, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Kang, C.; Philp, R.J.; Li, B. PKC delta phosphorylates p52ShcA at Ser29 to regulate ERK activation in response to H2O2. Cell. Signal. 2007, 19, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef]
- Bensalem, J.; Hattersley, K.J.; Hein, L.K.; Teong, X.T.; Carosi, J.M.; Hassiotis, S.; Grose, R.H.; Fourrier, C.; Heilbronn, L.K.; Sargeant, T.J. Measurement of autophagic flux in humans: An optimized method for blood samples. Autophagy 2021, 17, 3238–3255. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- van Koningsveld, R.; Steyerberg, E.W.; Hughes, R.A.; Swan, A.V.; van Doorn, P.A.; Jacobs, B.C. A clinical prognostic scoring system for Guillain-Barré syndrome. Lancet Neurol. 2007, 6, 589–594. [Google Scholar] [CrossRef]
- Nath, N.; Khan, M.; Paintlia, M.K.; Singh, I.; Hoda, M.N.; Giri, S. Metformin attenuated the autoimmune disease of the central nervous system in animal models of multiple sclerosis. J. Immunol. 2009, 182, 8005–8014. [Google Scholar] [CrossRef]
- Sun, Y.; Tian, T.; Gao, J.; Liu, X.; Hou, H.; Cao, R.; Li, B.; Quan, M.; Guo, L. Metformin ameliorates the development of experimental autoimmune encephalomyelitis by regulating T helper 17 and regulatory T cells in mice. J. Neuroimmunol. 2016, 292, 58–67. [Google Scholar] [CrossRef]
- Peric, S.; Bozovic, I.; Bjelica, B.; Berisavac, I.; Stojiljkovic, O.; Basta, I.; Beslac-Bumbasirevic, L.; Rakocevic-Stojanovic, V.; Lavrnic, D.; Stevic, Z. Diabetes mellitus may affect short-term outcome of Guillain-Barré syndrome. J. Peripher. Nerv. Syst. 2017, 22, 127–130. [Google Scholar] [CrossRef]
- Bae, J.S.; Kim, Y.J.; Kim, J.K. Diabetes mellitus exacerbates the clinical and electrophysiological features of Guillain-Barré syndrome. Eur. J. Neurol. 2016, 23, 439–446. [Google Scholar] [CrossRef]
- Gong, Q.; Liu, S.; Xiao, Z.; Fu, X.; Lu, Z. Elevated blood and cerebrospinal fluid glucose levels affect the severity and short-term prognosis of Guillain-Barré syndrome. Neurol. Res. 2022, 44, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Polydefkis, M.; Hauer, P.; Sheth, S.; Sirdofsky, M.; Griffin, J.W.; McArthur, J.C. The time course of epidermal nerve fibre regeneration: Studies in normal controls and people with diabetes, with and without neuropathy. Brain 2004, 127, 1606–1615. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, G.J.; O’Donnell, R.; Hauer, P.; Cimino, N.P.; McArthur, J.C.; Polydefkis, M. Impaired neurovascular repair in subjects with diabetes following experimental intracutaneous axotomy. Brain 2011, 134, 1853–1863. [Google Scholar] [CrossRef] [PubMed]
- Pietropaolo, M.; Barinas-Mitchell, E.; Kuller, L.H. The heterogeneity of diabetes: Unraveling a dispute: Is systemic inflammation related to islet autoimmunity? Diabetes 2007, 56, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A. Second line therapy: Type 2 diabetic subjects failing on metformin GLP-1/DPP-IV inhibitors versus sulphonylurea/insulin: For GLP-1/DPP-IV inhibitors. Diabetes Metab. Res. Rev. 2012, 28 (Suppl. 2), 21–25. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Characteristics | Values |
|---|---|
| GDS | |
| –at admission (n = 23) | 4 (2–4) a |
| –at 6 months (n = 21) b | 1 (0.5–3) a |
| MRCSS at admission (n = 22) c | 42.5 (28–47) a |
| Previous infection | 14 (60.8%) |
| –gastrointestinal | 7 (30.4%) |
| –respiratory | 7 (30.4%) |
| IVIG therapy | 23 (100%) |
| Deaths | 3 (13%) |
| Clinical Characteristics | Metformin Therapy | p Value | |
|---|---|---|---|
| yes (n =32) | no (n = 17) | ||
| sex (male/female) | 17/15 | 14/3 | 0.063 b |
| age (years) | 65 (57–68) a | 68 (61–72.5) a | 0.066 c |
| fasting glycemia (mmol/L) | 8.5 (6.0–12.2) a | 9.3 (7.1–12.1) a | 0.690 c |
| Previous infection | 13 (40.6%) | 7 (41.2%) | 1.000 b |
| –gastrointestinal | 5 (15.6%) | 5 (29.4%) | 0.285 b |
| –respiratory | 8 (25%) | 2 (11.8%) | 0.459 b |
| IVIG and/or PE therapy | 20 (62.5%) | 11 (64.7%) | 1.000 b |
| GBS variant | |||
| –AIDP | 11 (34.4%) | 8 (47.1%) | 0.539 b |
| –AMAN or AMSAN | 10 (31.2%) | 3 (17.6%) | 0.498 b |
| –undefined | 11 (34.4%) | 6 (35.3%) | 1.000 b |
| Mild GBS d at nadir | 9 (28.1%) | 0 (0%) | 0.049 *b |
| Mild GBS d at discharge | 21 (65.6%) | 4 (23.5%) | 0.007 *b |
| Variable | B | S.E. | Wald | df | p | OR | 95% CI |
|---|---|---|---|---|---|---|---|
| Age | −0.033 | 0.046 | 0.531 | 1 | 0.466 | 0.967 | 0.885–1.058 |
| Prior GIT infection | −2.600 | 1.154 | 5.078 | 1 | 0.024 * | 0.074 | 0.008–0.713 |
| Metformin therapy | 1.729 | 0.747 | 5.367 | 1 | 0.021 * | 5.637 | 1.305–24.352 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paunovic, V.; Peric, S.; Vukovic, I.; Stamenkovic, M.; Milosevic, E.; Stevanovic, D.; Mandic, M.; Basta, I.; Berisavac, I.; Arsenijevic, M.; et al. Downregulation of LKB1/AMPK Signaling in Blood Mononuclear Cells Is Associated with the Severity of Guillain–Barre Syndrome. Cells 2022, 11, 2897. https://doi.org/10.3390/cells11182897
Paunovic V, Peric S, Vukovic I, Stamenkovic M, Milosevic E, Stevanovic D, Mandic M, Basta I, Berisavac I, Arsenijevic M, et al. Downregulation of LKB1/AMPK Signaling in Blood Mononuclear Cells Is Associated with the Severity of Guillain–Barre Syndrome. Cells. 2022; 11(18):2897. https://doi.org/10.3390/cells11182897
Chicago/Turabian StylePaunovic, Verica, Stojan Peric, Irena Vukovic, Marina Stamenkovic, Emina Milosevic, Danijela Stevanovic, Milos Mandic, Ivana Basta, Ivana Berisavac, Mirjana Arsenijevic, and et al. 2022. "Downregulation of LKB1/AMPK Signaling in Blood Mononuclear Cells Is Associated with the Severity of Guillain–Barre Syndrome" Cells 11, no. 18: 2897. https://doi.org/10.3390/cells11182897
APA StylePaunovic, V., Peric, S., Vukovic, I., Stamenkovic, M., Milosevic, E., Stevanovic, D., Mandic, M., Basta, I., Berisavac, I., Arsenijevic, M., Bozovic, I., Nikolic, M., Stevic, Z., & Trajkovic, V. (2022). Downregulation of LKB1/AMPK Signaling in Blood Mononuclear Cells Is Associated with the Severity of Guillain–Barre Syndrome. Cells, 11(18), 2897. https://doi.org/10.3390/cells11182897