
GSK3 Is a Central Player in Retinal Degenerative Diseases but a Challenging Therapeutic Target

Abstract
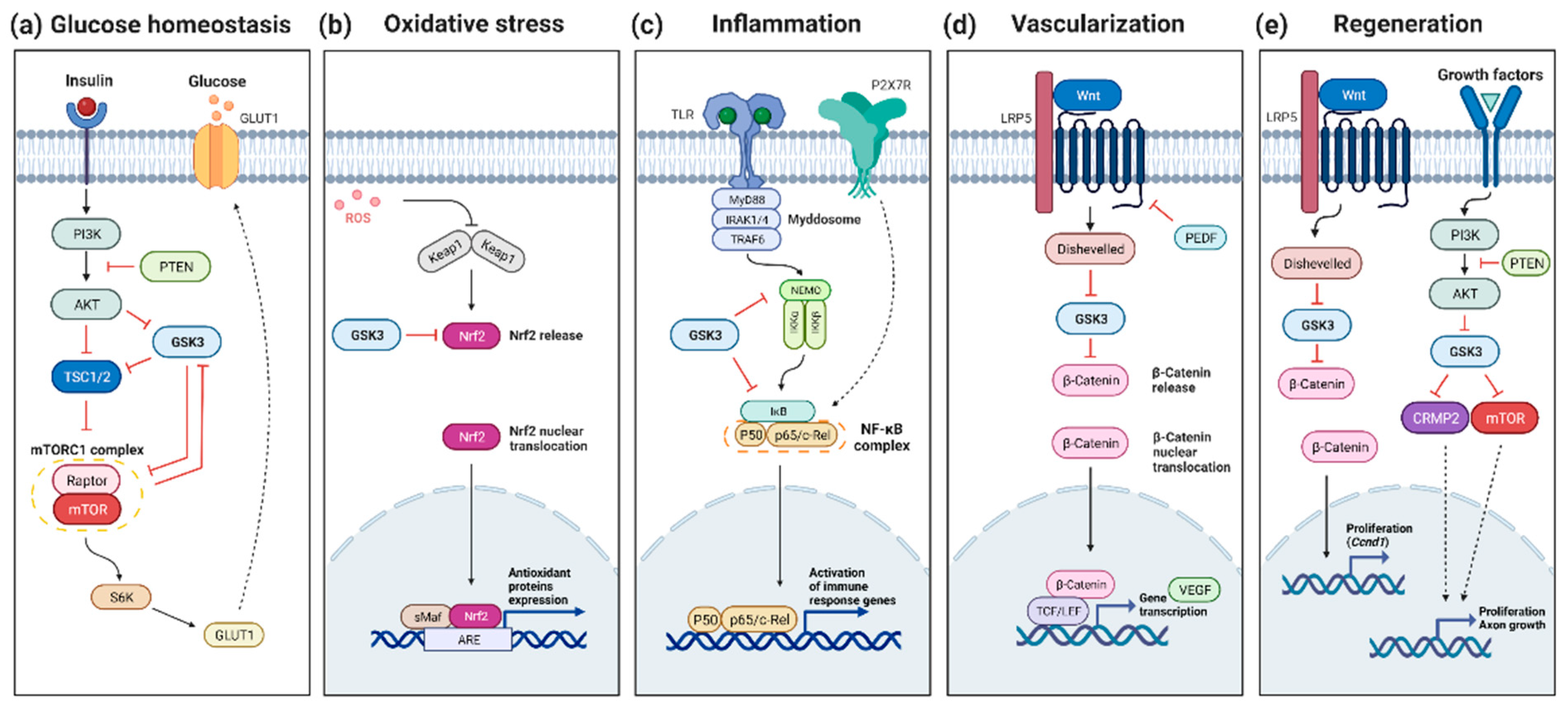
:1. Introduction
2. GSK3 Isoenzymes
3. GSK3 Involvement in Cellular Functions Deregulated during Retinal Degeneration
3.1. GSK3 and Cell Apoptosis
3.2. GSK3 and Retinal Glucose Homeostasis
3.3. GSK3 and Retinal Oxidative Stress
4. GSK3 and Retinal Inflammation
5. GSK3 and Retinal Vascularization
6. GSK3 and Retinal Regeneration
7. Therapeutic Trials Targeting GSK3 in Retinal Degenerative Diseases
8. Precautions and Advantages of Inhibiting GSK3 or GSK3 Targets as a Therapeutic Strategy for Eye Diseases

{kind=link}
| Cell Type | Cell Death Model | GSK3 | Observed Effects | Ref. | |
|---|---|---|---|---|---|
| _ | PRs | RP (rd10) | inactivation | PRs neuroprotection | [96,97] |
| MNU | inhibition (lithium) | [90] | |||
| RGCs | RGCs degeneration | activation | RGCs death | [195] | |
| inhibition | promotes RGCs survival, axon regeneration | [90,195,213] | |||
| DR | activation | mitochondrial oxidative stress increase, RGCs degeneration | [91] | ||
| RGCs degeneration | [211] | ||||
| astrocytes | Ang2 increase, astrocytes apoptosis, BRB disruption | [212] | |||
| RGCs, glial cells, astrocytes | DR | inhibition | RGCs, glial cells and astrocytes neuroprotection | [91,92,211,212] | |
| glucose homeostasis | _ | activation | GS phosphorylation, prevent glucose to glycogen conversion | [56] | |
| CNS | _ | _ | neurodegeneration due to glycogen accumulation | [102] | |
| PRs | DR | _ | hypoglycemia compromise neuronal survival | [103] | |
| RP | _ | mTOR activation preserves cone photoreceptors | [104,108] | ||
| RP | _ | insulin depletion accelerates cone death | [104] | ||
| oxidative stress | PRs | AMD | _ | oxidative stress increase | [115] |
| DR | _ | oxidative stress increase | [116] | ||
| RP | _ | oxidative stress increase | [117,118] | ||
| early DR | activation | NRF2 degradation | [127,128] | ||
| inhibition | NRF2 increased expression, neuroprotection | ||||
| MGCs | high glucose | _ | oxidative stress increase, NRF2 decrease | [126] | |
| RPE | _ | inhibition | NRF2 signaling rescue | [130] | |
| RGCs | inhibition (through PLK2) | cell survival | [225] | ||
| glaucoma | _ | CBD neuroprotection | [226] | ||
| DR | _ | ||||
| inhibition (through CBD) | NRF2 signaling induction | [131,132] | |||
| inflammation | PRs | rd mice | _ | NF-κB activation | [147] |
| light induced | _ | [148] | |||
| RGCs | optic nerve crush | _ | [227] | ||
| microglial cells | _ | inhibition | decrease of LPS-induced inflammation. NF-κB activation, decrease of TNFα secretion | [150] | |
| CNS | AD | Inhibition | P2X7 inhibition via GSK3, neuroprotection | [157,158] | |
| eye | oxidative stress | _ | P2X7 inhibition prevents inflammation and vascularization | [162] | |
| vascularization | PRs | DR | inactivated (through Wnt signaling) | VEGF production, vascularization | [163,164] |
| FEVR | _ | Wnt inhibition is a model of FEVR | [179] | ||
| FEVR | inhibition (inhibitor) | rescue of defective retinal vasculature | [179] | ||
| regeneration | zebrafish retina | retinal damage | inhibition (through Wnt signaling) | Ascl1 expression | [185,186] |
| retinal damage | inhibition (inhibitor) | sufficient for regeneration | [186] | ||
| chick retina | retinal damage | inhibition + FGF2 treatment | MGCs proliferation and dedifferentiation | [187] | |
| PRs | rd mice | inhibition (through Wnt signaling) | necessary for MG-derived progenitor production, proliferation and reprogramming | [189] | |
| RGCs | inhibition (through PTEN inhibition) | RGCs neuroprotection, axon regeneration | [196] | ||
| optic nerve crush | inhibition (through Wnt signaling) | CRMP2 signaling induction, axon regeneration | [198] | ||
| inhibition (inhibitor) | [195] | ||||
| optic nerve crush + inflammation | _ | mTOR signaling increase, optic nerve regeneration, RGCs neuroprotection | [201] | ||
| CNS | _ | inhibition | mTOR signaling induction, axon regeneration | [200] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hur, E.-M.; Zhou, F.-Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Fuster-Matanzo, A.; Llorens-Martín, M.; Sirerol-Piquer, M.S.; García-Verdugo, J.M.; Avila, J.; Hernández, F. Dual Effects of Increased Glycogen Synthase Kinase-3β Activity on Adult Neurogenesis. Hum. Mol. Genet. 2013, 22, 1300–1315. [Google Scholar] [CrossRef] [PubMed]
- Malagon, S.G.G.; Muñoz, A.M.L.; Doro, D.; Bolger, T.G.; Poon, E.; Tucker, E.R.; Adel Al-Lami, H.; Krause, M.; Phiel, C.J.; Chesler, L.; et al. Glycogen Synthase Kinase 3 Controls Migration of the Neural Crest Lineage in Mouse and Xenopus. Nat. Commun. 2018, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Morgan-Smith, M.; Wu, Y.; Zhu, X.; Pringle, J.; Snider, W.D. GSK-3 Signaling in Developing Cortical Neurons Is Essential for Radial Migration and Dendritic Orientation. eLife 2014, 3, e02663. [Google Scholar] [CrossRef]
- Barnat, M.; Benassy, M.N.; Vincensini, L.; Soares, S.; Fassier, C.; Propst, F.; Andrieux, A.; von Boxberg, Y.; Nothias, F. The GSK3-MAP1B Pathway Controls Neurite Branching and Microtubule Dynamics. Mol. Cell. Neurosci. 2016, 72, 9–21. [Google Scholar] [CrossRef]
- Hur, E.-M.; Saijilafu; Lee, B.D.; Kim, S.-J.; Xu, W.-L.; Zhou, F.-Q. GSK3 Controls Axon Growth via CLASP-Mediated Regulation of Growth Cone Microtubules. Genes Dev. 2011, 25, 1968–1981. [Google Scholar] [CrossRef]
- Dill, J.; Wang, H.; Zhou, F.; Li, S. Inactivation of Glycogen Synthase Kinase 3 Promotes Axonal Growth and Recovery in the CNS. J. Neurosci. 2008, 28, 8914–8928. [Google Scholar] [CrossRef]
- Shahab, L.; Plattner, F.; Irvine, E.E.; Cummings, D.M.; Edwards, F.A. Dynamic Range of GSK3α Not GSK3β Is Essential for Bidirectional Synaptic Plasticity at Hippocampal CA3-CA1 Synapses. Hippocampus 2014, 24, 1413–1416. [Google Scholar] [CrossRef]
- Souder, D.C.; Anderson, R.M. An Expanding GSK3 Network: Implications for Aging Research. Geroscience 2019, 41, 369–382. [Google Scholar] [CrossRef]
- Fan, X.; Zhao, Z.; Wang, D.; Xiao, J. Glycogen Synthase Kinase-3 as a Key Regulator of Cognitive Function. Acta Biochim. Biophys. Sin. 2020, 52, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Albeely, A.M.; Ryan, S.D.; Perreault, M.L. Pathogenic Feed-Forward Mechanisms in Alzheimer’s and Parkinson’s Disease Converge on GSK-3. Brain Plast. 2018, 4, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Leroy, K.; Yilmaz, Z.; Brion, J.P. Increased Level of Active GSK-3β in Alzheimer’s Disease and Accumulation in Argyrophilic Grains and in Neurones at Different Stages of Neurofibrillary Degeneration. Neuropathol. Appl. Neurobiol. 2007, 33, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Armentero, M.T.; Sinforiani, E.; Ghezzi, C.; Bazzini, E.; Levandis, G.; Ambrosi, G.; Zangaglia, R.; Pacchetti, C.; Cereda, C.; Cova, E.; et al. Peripheral Expression of Key Regulatory Kinases in Alzheimer’s Disease and Parkinson’s Disease. Neurobiol. Aging 2011, 32, 2142–2151. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Killick, R.; Lovestone, S. The GSK3 Hypothesis of Alzheimer’s Disease. J. Neurochem. 2008, 104, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Mines, M.A.; Beurel, E.; Jope, R.S. Regulation of Cell Survival Mechanisms in Alzheimer’s Disease by Glycogen Synthase Kinase-3. Int. J. Alzheimer’s Dis. 2011, 2011, 861072. [Google Scholar] [CrossRef]
- Takahashi, M.; Tomizawa, K.; Kato, R.; Sato, K.; Uchida, T.; Fujita, S.C.; Imahori, K. Localization and Developmental Changes of τ Protein Kinase I/Glycogen Synthase Kinase-3β in Rat Brain. J. Neurochem. 1994, 63, 245–255. [Google Scholar] [CrossRef]
- Toral-Rios, D.; Pichardo-Rojas, P.S.; Alonso-Vanegas, M.; Campos-Peña, V. GSK3β and Tau Protein in Alzheimer’s Disease and Epilepsy. Front. Cell. Neurosci. 2020, 14, 19. [Google Scholar] [CrossRef]
- Phiel, C.J.; Wilson, C.A.; Lee, V.M.Y.; Klein, P.S. GSK-3α Regulates Production of Alzheimer’s Disease Amyloid-β Peptides. Nature 2003, 423, 435–439. [Google Scholar] [CrossRef]
- Golpich, M.; Amini, E.; Hemmati, F.; Ibrahim, N.M.; Rahmani, B.; Mohamed, Z.; Raymond, A.A.; Dargahi, L.; Ghasemi, R.; Ahmadiani, A. Glycogen Synthase Kinase-3 Beta (GSK-3β) Signaling: Implications for Parkinson’s Disease. Pharmacol. Res. 2015, 97, 16–26. [Google Scholar] [CrossRef]
- Credle, J.J.; George, J.L.; Wills, J.; Duka, V.; Shah, K.; Lee, Y.-C.; Rodriguez, O.; Simkins, T.; Winter, M.; Moechars, D.; et al. GSK-3β Dysregulation Contributes to Parkinson’s-like Pathophysiology with Associated Region-Specific Phosphorylation and Accumulation of Tau and α-Synuclein. Cell Death Differ. 2015, 22, 838–851. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Gao, F.; Wang, D.; Li, C.; Fu, Y.; He, W.; Zhang, J. Tau Pathology in Parkinson’s Disease. Front. Neurol. 2018, 9, 809. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Meng, L.; He, M.; Zhang, Z. Tau in the Pathophysiology of Parkinson’s Disease. J. Mol. Neurosci. 2021, 71, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. A-Synuclein Contributes to GSK-3β-catalyzed Tau Phosphorylation in Parkinson’s Disease Models. FASEB J. 2009, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen Synthase Kinase-3 Signaling in Alzheimer’s Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s Disease Multitarget Drug Discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef]
- Wang, W.; Yang, Y.; Ying, C.; Li, W.; Ruan, H.; Zhu, X.; You, Y.; Han, Y.; Chen, R.; Wang, Y.; et al. Inhibition of Glycogen Synthase Kinase-3β Protects Dopaminergic Neurons from MPTP Toxicity. Neuropharmacology 2007, 52, 1678–1684. [Google Scholar] [CrossRef]
- Morales-García, J.A.; Susín, C.; Alonso-Gil, S.; Pérez, D.I.; Palomo, V.; Pérez, C.; Conde, S.; Santos, A.; Gil, C.; Martínez, A.; et al. Glycogen Synthase Kinase-3 Inhibitors as Potent Therapeutic Agents for the Treatment of Parkinson Disease. ACS Chem. Neurosci. 2013, 4, 350–360. [Google Scholar] [CrossRef]
- Jope, R.S.; Roh, M.-S. Glycogen Synthase Kinase-3 (GSK3) in Psychiatric Diseases and Therapeutic Interventions. Curr. Drug Targets 2006, 7, 1421–1434. [Google Scholar] [CrossRef]
- King, M.K.; Pardo, M.; Cheng, Y.; Downey, K.; Jope, R.S.; Beurel, E. Glycogen Synthase Kinase-3 Inhibitors: Rescuers of Cognitive Impairments. Pharmacol. Ther. 2014, 141, 1–12. [Google Scholar] [CrossRef]
- Roca, C.; Campillo, N.E. Glycogen Synthase Kinase 3 (GSK-3) Inhibitors: A Patent Update (2016–2019). Expert Opin. Ther. Pat. 2020, 30, 863–872. [Google Scholar] [CrossRef]
- Palomo, V.; Martinez, A. Glycogen Synthase Kinase 3 (GSK-3) Inhibitors: A Patent Update (2014–2015). Expert Opin. Ther. Pat. 2017, 27, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Alonso, M.; Castro, A.; Pérez, C.; Moreno, F.J. First Non-ATP Competitive Glycogen Synthase Kinase 3 β (GSK-3β) Inhibitors: Thiadiazolidinones (TDZD) as Potential Drugs for the Treatment of Alzheimer’s Disease. J. Med. Chem. 2002, 45, 1292–1299. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.R. When Good Kinases Go Rogue: GSK3, P38 MAPK and CDKs as Therapeutic Targets for Alzheimer’s and Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 5911. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, S.; Chen, J.; Hu, K.; Li, Y.; Zhang, Z.; Su, Z.; Woodgett, J.R.; Li, M.; Huang, Q. GSK-3β Contributes to Parkinsonian Dopaminergic Neuron Death: Evidence From Conditional Knockout Mice and Tideglusib. Front. Mol. Neurosci. 2020, 13, 81. [Google Scholar] [CrossRef]
- Kozlovsky, N.; Belmaker, R.H.; Agam, G. Low GSK-3 Activity in Frontal Cortex of Schizophrenic Patients. Schizophr. Res. 2001, 52, 101–105. [Google Scholar] [CrossRef]
- Li, X.; Friedman, A.B.; Zhu, W.; Wang, L.; Boswell, S.; May, R.S.; Davis, L.L.; Jope, R.S. Lithium Regulates Glycogen Synthase Kinase-3β in Human Peripheral Blood Mononuclear Cells: Implication in the Treatment of Bipolar Disorder. Biol. Psychiatry 2007, 61, 216–222. [Google Scholar] [CrossRef]
- Inkster, B.; Nichols, T.E.; Saemann, P.G.; Auer, D.P.; Holsboer, F.; Muglia, P.; Matthews, P.M. Pathway-Based Approaches to Imaging Genetics Association Studies: Wnt Signaling, GSK3beta Substrates and Major Depression. Neuroimage 2010, 53, 908–917. [Google Scholar] [CrossRef]
- McCallum, R.T.; Perreault, M.L. Glycogen Synthase Kinase-3: A Focal Point for Advancing Pathogenic Inflammation in Depression. Cells 2021, 10, 2270. [Google Scholar] [CrossRef]
- Ruiz, S.M.A.; Eldar-Finkelman, H. Glycogen Synthase Kinase-3 Inhibitors: Preclinical and Clinical Focus on CNS-A Decade Onward. Front. Mol. Neurosci. 2022, 14, 792364. [Google Scholar] [CrossRef]
- Sánchez-Cruz, A.; Martínez, A.; de la Rosa, E.J.; Hernández-Sánchez, C. GSK-3 Inhibitors: From the Brain to the Retina and Back Again. In Advances in Experimental Medicine and Biology; Springer: Singapore, 2019; Volume 1185, pp. 437–441. [Google Scholar]
- Rippin, I.; Eldar-Finkelman, H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells 2021, 10, 262. [Google Scholar] [CrossRef]
- Moore, K.B.; Schneider, M.L.; Vetter, M.L. Posttranslational Mechanisms Control the Timing of BHLH Function and Regulate Retinal Cell Fate. Neuron 2002, 34, 183–195. [Google Scholar] [CrossRef]
- Kisseleff, E.; Vigouroux, R.J.; Hottin, C.; Lourdel, S.; Thomas, L.; Shah, P.; Chédotal, A.; Perron, M.; Swaroop, A.; Roger, J.E. Glycogen Synthase Kinase 3 Regulates the Genesis of Displaced Retinal Ganglion Cells3. eNeuro 2021, 8, ENEURO.0171-21.2021. [Google Scholar] [CrossRef] [PubMed]
- Alon, L.T.; Pietrokovski, S.; Barkan, S.; Avrahami, L.; Kaidanovich-Beilin, O.; Woodgett, J.R.; Barnea, A.; Eldar-Finkelman, H. Selective Loss of Glycogen Synthase Kinase-3α in Birds Reveals Distinct Roles for GSK-3 Isozymes in Tau Phosphorylation. FEBS Lett. 2011, 585, 1158–1162. [Google Scholar] [CrossRef]
- Tung, H.Y.; Reed, L.J. Purification and Characterization of Protein Phosphatase 1I Activating Kinase from Bovine Brain Cytosolic and Particulate Fractions. J. Biol. Chem. 1989, 264, 2985–2990. [Google Scholar] [CrossRef]
- Woodgett, J.R. Molecular Cloning and Expression of Glycogen Synthase Kinase-3/Factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.-B.; Shaw, P.-C.; Wong, C.-C.; Chi-Cheong Wan, D. Expression of Glycogen Synthase Kinase-3 Isoforms in Mouse Tissues and Their Transcription in the Brain. J. Chem. Neuroanat. 2002, 23, 291–297. [Google Scholar] [CrossRef]
- Lau, K.-F.; Miller, C.C.J.; Anderton, B.H.; Shaw, P.-C. Expression Analysis of Glycogen Synthase Kinase-3 in Human Tissues. J. Pept. Res. 1999, 54, 85–91. [Google Scholar] [CrossRef]
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative Splicing Isoform of Tau Protein Kinase I/Glycogen Synthase Kinase 3β. J. Neurochem. 2002, 81, 1073–1083. [Google Scholar] [CrossRef]
- Soutar, M.P.M.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence That Glycogen Synthase Kinase-3 Isoforms Have Distinct Substrate Preference in the Brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef]
- Dajani, R.; Fraser, E.; Roe, S.M.; Young, N.; Good, V.; Dale, T.C.; Pearl, L.H. Crystal Structure of Glycogen Synthase Kinase 3: Structural Basis for Phosphate-Primed Substrate Specificity and Autoinhibition. Cell 2001, 105, 721–732. [Google Scholar] [CrossRef]
- Ter Haar, E.; Coll, J.T.; Austen, D.A.; Hsiao, H.M.; Swenson, L.; Jain, J. Structure of GSK3β Reveals a Primed Phosphorylation Mechanism. Nat. Struct. Biol. 2001, 8, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Lochhead, P.A.; Kinstrie, R.; Sibbet, G.; Rawjee, T.; Morrice, N.; Cleghon, V. A Chaperone-Dependent GSK3β Transitional Intermediate Mediates Activation-Loop Autophosphorylation. Mol. Cell 2006, 24, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P.; Biondi, R.M. A Common Phosphate Binding Site Explains the Unique Substrate Specificity of GSK3 and Its Inactivation by Phosphorylation. Mol. Cell 2001, 7, 1321–1327. [Google Scholar] [CrossRef]
- Cohen, P.; Frame, S. The Renaissance of GSK3. Nat. Rev. Mol. Cell Biol. 2001, 2, 769–776. [Google Scholar] [CrossRef]
- Parker, P.J.; Caudwell, F.B.; Cohen, P. Glycogen Synthase from Rabbit Skeletal Muscle; Effect of Insulin on the State of Phosphorylation of the Seven Phosphoserine Residues In Vivo. Eur. J. Biochem. 1983, 130, 227–234. [Google Scholar] [CrossRef]
- Embi, N.; Rylatt, D.B.; Cohen, P. Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle. Separation from Cyclic-AMP-Dependent Protein Kinase and Phosphorylase Kinase. Eur. J. Biochem. 1980, 107, 519–527. [Google Scholar] [CrossRef]
- Rylatt, D.B.; Aitken, A.; Bilham, T.; Condon, G.D.; Embi, N.; Cohen, P. Glycogen Synthase from Rabbit Skeletal Muscle. Amino Acid Sequence at the Sites Phosphorylated by Glycogen Synthase Kinase-3, and Extension of the N-Terminal Sequence Containing the Site Phosphorylated by Phosphorylase Kinase. Eur. J. Biochem. 1980, 107, 529–537. [Google Scholar] [CrossRef]
- Sutherland, C. What Are the Bona Fide GSK3 Substrates? Int. J. Alzheimer’s Dis. 2011, 2011, 505607. [Google Scholar] [CrossRef]
- Shinde, M.Y.; Sidoli, S.; Kulej, K.; Mallory, M.J.; Radens, C.M.; Reicherter, A.L.; Myers, R.L.; Barash, Y.; Lynch, K.W.; Garcia, B.A.; et al. Phosphoproteomics Reveals That Glycogen Synthase Kinase-3 Phosphorylates Multiple Splicing Factors and Is Associated with Alternative Splicing. J. Biol. Chem. 2017, 292, 18240–18255. [Google Scholar] [CrossRef]
- Beurel, E.; Grieco, S.F.; Jope, R.S. Glycogen Synthase Kinase-3 (GSK3): Regulation, Actions, and Diseases. Pharmacol. Ther. 2015, 148, 114–131. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.; Woodgett, J.R. Glycogen Synthase Kinase 3: A Kinase for All Pathways? Curr. Top. Dev. Biol. 2017, 123, 277–302. [Google Scholar] [CrossRef] [PubMed]
- Melikian, M.; Eluard, B.; Bertho, G.; Baud, V.; Evrard-Todeschi, N. Model of the Interaction between the NF-ΚB Inhibitory Protein P100 and the E3 Ubiquitin Ligase β-TrCP Based on NMR and Docking Experiments. J. Chem. Inf. Model. 2017, 57, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.; Hayes, J.D.; Sutherland, C. A Partnership with the Proteasome; the Destructive Nature of GSK3. Biochem. Pharmacol. 2018, 147, 77–92. [Google Scholar] [CrossRef] [PubMed]
- MacAulay, K.; Doble, B.W.; Patel, S.; Hansotia, T.; Sinclair, E.M.; Drucker, D.J.; Nagy, A.; Woodgett, J.R. Glycogen Synthase Kinase 3α-Specific Regulation of Murine Hepatic Glycogen Metabolism. Cell Metab. 2007, 6, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; Luo, J.; Rubie, E.A.; Tsao, M.-S.; Jin, O.; Woodgett, J.R. Requirement for Glycogen Synthase Kinase-3β in Cell Survival and NF-ΚB Activation. Nature 2000, 406, 86–90. [Google Scholar] [CrossRef]
- Xu, C.; Kim, N.-G.; Gumbiner, B.M. Regulation of Protein Stability by GSK3 Mediated Phosphorylation. Cell Cycle 2009, 8, 4032–4039. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt Signaling Pathway in Development and Disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Komiya, Y.; Habas, R. Wnt Signal Transduction Pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef]
- Veleri, S.; Lazar, C.H.; Chang, B.; Sieving, P.A.; Banin, E.; Swaroop, A. Biology and Therapy of Inherited Retinal Degenerative Disease: Insights from Mouse Models. Dis. Models Mech. 2015, 8, 109–129. [Google Scholar] [CrossRef]
- Swaroop, A.; Kim, D.; Forrest, D. Transcriptional Regulation of Photoreceptor Development and Homeostasis in the Mammalian Retina. Nat. Rev. Neurosci. 2010, 11, 563–576. [Google Scholar] [CrossRef] [Green Version]
- Bringmann, A.; Pannicke, T.; Grosche, J.; Francke, M.; Wiedemann, P.; Skatchkov, S.; Osborne, N.; Reichenbach, A. Müller Cells in the Healthy and Diseased Retina. Prog. Retin. Eye Res. 2006, 25, 397–424. [Google Scholar] [CrossRef] [PubMed]
- Masland, R.H. The Fundamental Plan of the Retina. Nat. Neurosci. 2001, 4, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Wässle, H. Parallel Processing in the Mammalian Retina. Nat. Rev. Neurosci. 2004, 5, 747–757. [Google Scholar] [CrossRef] [PubMed]
- Harris, A. Wirostko Age-Related Macular Degeneration and the Aging Eye. Clin. Interv. Aging 2008, 3, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Fariss, R.N.; Stambolian, D.; Abecasis, G.R.; Curcio, C.A.; Swaroop, A. Age-Related Macular Degeneration: Genetics and Biology Coming Together. Annu. Rev. Genom. Hum. Genet 2014, 15, 151–171. [Google Scholar] [CrossRef]
- O’Neal, T.B.; Luther, E.E. Retinitis Pigmentosa; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Tang, J.; Kern, T.S. Inflammation in Diabetic Retinopathy. Prog. Retin. Eye Res. 2011, 30, 343–358. [Google Scholar] [CrossRef]
- Maurer, U.; Preiss, F.; Brauns-Schubert, P.; Schlicher, L.; Charvet, C. GSK-3—At the Crossroads of Cell Death and Survival. J. Cell Sci. 2014, 127, 1369–1378. [Google Scholar] [CrossRef]
- Cross, D.A.E.; Culbert, A.A.; Chalmers, K.A.; Facci, L.; Skaper, S.D.; Reith, A.D. Selective Small-Molecule Inhibitors of Glycogen Synthase Kinase-3 Activity Protect Primary Neurones from Death. J. Neurochem. 2001, 77, 94–102. [Google Scholar] [CrossRef]
- Beurel, E.; Jope, R.S. The Paradoxical Pro- and Anti-Apoptotic Actions of GSK3 in the Intrinsic and Extrinsic Apoptosis Signaling Pathways. Prog. Neurobiol. 2006, 79, 173–189. [Google Scholar] [CrossRef]
- Jacobs, K.M.; Bhave, S.R.; Ferraro, D.J.; Jaboin, J.J.; Hallahan, D.E.; Thotala, D. GSK-3: A Bifunctional Role in Cell Death Pathways. Int. J. Cell Biol. 2012, 2012, 930710. [Google Scholar] [CrossRef] [Green Version]
- Hongisto, V.; Smeds, N.; Brecht, S.; Herdegen, T.; Courtney, M.J.; Coffey, E.T. Lithium Blocks the C-Jun Stress Response and Protects Neurons via Its Action on Glycogen Synthase Kinase 3. Mol. Cell Biol. 2003, 23, 6027–6036. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.; Allouche, M.; Esposti, D.D.; Fanelli, E.; Boursier, C.; Henry, C.; Chopineau, J.; Calamita, G.; Kroemer, G.; Lemoine, A.; et al. Glycogen Synthase Kinase 3-Mediated Voltage-Dependent Anion Channel Phosphorylation Controls Outer Mitochondrial Membrane Permeability during Lipid Accumulation. Hepatology 2013, 57, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Linseman, D.A. Glycogen Synthase Kinase-3 Phosphorylates Bax and Promotes Its Mitochondrial Localization during Neuronal Apoptosis. J. Neurosci. 2004, 24, 9993–10002. [Google Scholar] [CrossRef]
- Gomez-Sintes, R.; Hernandez, F.; Lucas, J.J.; Avila, J. GSK-3 Mouse Models to Study Neuronal Apoptosis and Neurodegeneration. Front. Mol. Neurosci. 2011, 4, 45. [Google Scholar] [CrossRef] [PubMed]
- Crowder, R.J.; Freeman, R.S. Phosphatidylinositol 3-Kinase and Akt Protein Kinase Are Necessary and Sufficient for the Survival of Nerve Growth Factor-Dependent Sympathetic Neurons. J. Neurosci. 1998, 18, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Pap, M.; Cooper, G.M. Role of Glycogen Synthase Kinase-3 in the Phosphatidylinositol 3-Kinase/Akt Cell Survival Pathway. J. Biol. Chem. 1998, 273, 19929–19932. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Liu, S.L.; Hu, D.P.; Xing, Y.Q.; Shen, Y. N-Methyl-N-Nitrosourea-Induced Retinal Degeneration in Mice. Exp. Eye Res. 2014, 121, 102–113. [Google Scholar] [CrossRef]
- Wang, B.; Hu, C.; Yang, X.; Du, F.; Feng, Y.; Li, H.; Zhu, C.; Yu, X. Inhibition of GSK-3β Activation Protects SD Rat Retina Against N-Methyl-N-Nitrosourea-Induced Degeneration by Modulating the Wnt/β-Catenin Signaling Pathway. J. Mol. Neurosci. 2017, 63, 233–242. [Google Scholar] [CrossRef]
- Shu, X.-S.; Zhu, H.; Huang, X.; Yang, Y.; Wang, D.; Zhang, Y.; Zhang, W.; Ying, Y. Loss of β-Catenin via Activated GSK3β Causes Diabetic Retinal Neurodegeneration by Instigating a Vicious Cycle of Oxidative Stress-Driven Mitochondrial Impairment. Aging 2020, 12, 13437–13462. [Google Scholar] [CrossRef]
- Li, Z.; Ma, L.; Chen, X.; Li, Y.; Li, S.; Zhang, J.; Lu, L. Glycogen Synthase Kinase-3: A Key Kinase in Retinal Neuron Apoptosis in Early Diabetic Retinopathy. Chin. Med. J. 2014, 127, 3464–3470. [Google Scholar] [CrossRef]
- Li, C.L.; Sathyamurthy, A.; Oldenborg, A.; Tank, D.; Ramanan, N. SRF Phosphorylation by Glycogen Synthase Kinase-3 Promotes Axon Growth in Hippocampal Neurons. J. Neurosci. 2014, 34, 4027–4042. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.H.; Zhang, S.H.; Gao, F.J.; Lei, Y.; Chen, X.Y.; Gao, F.; Zhang, S.J.; Sun, X.H. RNAi Screening Identifies GSK3β as a Regulator of DRP1 and the Neuroprotection of Lithium Chloride against Elevated Pressure Involved in Downregulation of DRP1. Neurosci. Lett. 2013, 554, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Gargini, C.; Terzibasi, E.; Mazzoni, F.; Strettoi, E. Retinal Organization in the Retinal Degeneration 10 (Rd10) Mutant Mouse: A Morphological and ERG Study. J. Comp. Neurol. 2007, 500, 222–238. [Google Scholar] [CrossRef]
- Marchena, M.; Villarejo-Zori, B.; Zaldivar-Diez, J.; Palomo, V.; Gil, C.; Hernández-Sánchez, C.; Martínez, A.; de la Rosa, E.J. Small Molecules Targeting Glycogen Synthase Kinase 3 as Potential Drug Candidates for the Treatment of Retinitis Pigmentosa. J. Enzym. Inhib. Med. Chem. 2017, 32, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cruz, A.; Villarejo-Zori, B.; Marchena, M.; Zaldivar-Díez, J.; Palomo, V.; Gil, C.; Lizasoain, I.; de la Villa, P.; Martínez, A.; de la Rosa, E.J.; et al. Modulation of GSK-3 Provides Cellular and Functional Neuroprotection in the Rd10 Mouse Model of Retinitis Pigmentosa. Mol. Neurodegener. 2018, 13, 19. [Google Scholar] [CrossRef] [PubMed]
- PérezLeón, J.A.; Osorio-Paz, I.; Francois, L.; Salceda, R. Immunohistochemical Localization of Glycogen Synthase and GSK3β: Control of Glycogen Content in Retina. Neurochem. Res. 2013, 38, 1063–1069. [Google Scholar] [CrossRef]
- Rungger-Brändle, E.; Kolb, H.; Niemeyer, G. Histochemical Demonstration of Glycogen in Neurons of the Cat Retina. Investig. Ophthalmol. Vis. Sci. 1996, 37, 702–715. [Google Scholar]
- Poitry-Yamate, C.L.; Tsacopoulos, M. Glucose Metabolism in Freshly Isolated Müller Glial Cells from a Mammalian Retina. J. Comp. Neurol. 1992, 320, 257–266. [Google Scholar] [CrossRef]
- Coffe, V.; Carbajal, R.C.; Salceda, R. Glycogen Metabolism in the Rat Retina. J. Neurochem. 2004, 88, 885–890. [Google Scholar] [CrossRef]
- Duran, J.; Tevy, M.F.; Garcia-Rocha, M.; Calbó, J.; Milán, M.; Guinovart, J.J. Deleterious Effects of Neuronal Accumulation of Glycogen in Flies and Mice. EMBO Mol. Med. 2012, 4, 719–729. [Google Scholar] [CrossRef]
- Osorio-Paz, I.; Sánchez-Chávez, G.; Salceda, R. Control of Glycogen Content in Retina: Allosteric Regulation of Glycogen Synthase. PLoS ONE 2012, 7, e30822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Punzo, C.; Kornacker, K.; Cepko, C.L. Stimulation of the Insulin/MTOR Pathway Delays Cone Death in a Mouse Model of Retinitis Pigmentosa. Nat. Neurosci. 2009, 12, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, H.; Wang, W.; Piri, N.; Dean, D. Metabolic Rescue of Cone Photoreceptors in Retinitis Pigmentosa. Taiwan J. Ophthalmol. 2021, 11, 331–335. [Google Scholar] [CrossRef] [PubMed]
- Aït-Ali, N.; Fridlich, R.; Millet-Puel, G.; Clérin, E.; Delalande, F.; Jaillard, C.; Blond, F.; Perrocheau, L.; Reichman, S.; Byrne, L.C.; et al. Rod-Derived Cone Viability Factor Promotes Cone Survival by Stimulating Aerobic Glycolysis. Cell 2015, 161, 817–832. [Google Scholar] [CrossRef]
- Düvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of MTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef]
- Venkatesh, A.; Ma, S.; Le, Y.Z.; Hall, M.N.; Rüegg, M.A.; Punzo, C. Activated MTORC1 Promotes Long-Term Cone Survival in Retinitis Pigmentosa Mice. J. Clin. Investig. 2015, 125, 1446–1458. [Google Scholar] [CrossRef]
- Hermida, M.A.; Kumar, J.D.; Leslie, N.R. GSK3 and Its Interactions with the PI3K/AKT/MTOR Signalling Network. Adv. Biol. Regul. 2017, 65, 5–15. [Google Scholar] [CrossRef]
- Evangelisti, C.; Chiarini, F.; Paganelli, F.; Marmiroli, S.; Martelli, A.M. Crosstalks of GSK3 Signaling with the MTOR Network and Effects on Targeted Therapy of Cancer. Biochim. Biophys. Acta BBA Mol. Cell Res. 2020, 1867, 118635. [Google Scholar] [CrossRef]
- Stretton, C.; Hoffmann, T.M.; Munson, M.J.; Prescott, A.; Taylor, P.M.; Ganley, I.G.; Hundal, H.S. GSK3-Mediated Raptor Phosphorylation Supports Amino-Acid-Dependent MTORC1-Directed Signalling. Biochem. J. 2015, 470, 207–221. [Google Scholar] [CrossRef]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Bratic, A.; Larsson, N.-G. The Role of Mitochondria in Aging. J. Clin. Investig. 2013, 123, 951–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eells, J.T. Mitochondrial Dysfunction in the Aging Retina. Biology 2019, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D. Mechanisms of Mitochondrial Dysfunction and Their Impact on Age-Related Macular Degeneration. Prog. Retin. Eye Res. 2020, 79, 100858. [Google Scholar] [CrossRef] [PubMed]
- Kang, Q.; Yang, C. Oxidative Stress and Diabetic Retinopathy: Molecular Mechanisms, Pathogenetic Role and Therapeutic Implications. Redox Biol. 2020, 37, 101799. [Google Scholar] [CrossRef]
- Murakami, Y.; Nakabeppu, Y.; Sonoda, K.-H. Oxidative Stress and Microglial Response in Retinitis Pigmentosa. Int. J. Mol. Sci. 2020, 21, 7170. [Google Scholar] [CrossRef]
- Gallenga, C.E.; Lonardi, M.; Pacetti, S.; Violanti, S.S.; Tassinari, P.; Di Virgilio, F.; Tognon, M.; Perri, P. Molecular Mechanisms Related to Oxidative Stress in Retinitis Pigmentosa. Antioxidants 2021, 10, 848. [Google Scholar] [CrossRef]
- Dammak, A.; Huete-Toral, F.; Carpena-Torres, C.; Martin-Gil, A.; Pastrana, C.; Carracedo, G. From Oxidative Stress to Inflammation in the Posterior Ocular Diseases: Diagnosis and Treatment. Pharmaceutics 2021, 13, 1376. [Google Scholar] [CrossRef]
- Ryu, J.; Gulamhusein, H.; Oh, J.K.; Chang, J.H.; Chen, J.; Tsang, S.H. Nutrigenetic Reprogramming of Oxidative Stress. Taiwan J. Ophthalmol. 2021, 11, 207–215. [Google Scholar] [CrossRef]
- Nakagami, Y. Nrf2 is an Attractive Therapeutic Target for Retinal Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 7469326. [Google Scholar] [CrossRef]
- Xu, Z.; Wei, Y.; Gong, J.; Cho, H.; Park, J.K.; Sung, E.-R.; Huang, H.; Wu, L.; Eberhart, C.; Handa, J.T.; et al. NRF2 Plays a Protective Role in Diabetic Retinopathy in Mice. Diabetologia 2014, 57, 204–213. [Google Scholar] [CrossRef]
- Albert-Garay, J.S.; Riesgo-Escovar, J.R.; Sánchez-Chávez, G.; Salceda, R. Retinal Nrf2 Expression in Normal and Early Streptozotocin-Diabetic Rats. Neurochem. Int. 2021, 145, 105007. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmström, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 Regulates ROS Production by Mitochondria and NADPH Oxidase. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 794–801. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Lv, Y.-F.; Zhao, J.-L.; You, Q.-D.; Jiang, Z.-Y. Regulation of Nrf2 by Phosphorylation: Consequences for Biological Function and Therapeutic Implications. Free Radic. Biol. Med. 2021, 168, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Albert-Garay, J.S.; Riesgo-Escovar, J.R.; Salceda, R. High Glucose Concentrations Induce Oxidative Stress by Inhibiting Nrf2 Expression in Rat Müller Retinal Cells In Vitro. Sci. Rep. 2022, 12, 1261. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The Stress Response Protein REDD1 Promotes Diabetes-Induced Oxidative Stress in the Retina by Keap1-Independent Nrf2 Degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef]
- Miller, W.P.; Toro, A.L.; Barber, A.J.; Dennis, M.D. REDD1 Activates a ROS-Generating Feedback Loop in the Retina of Diabetic Mice. Investig. Opthalmol. Vis. Sci. 2019, 60, 2369–2379. [Google Scholar] [CrossRef]
- Sachdeva, M.M.; Cano, M.; Handa, J.T. Nrf2 Signaling Is Impaired in the Aging RPE given an Oxidative Insult. Exp. Eye Res. 2013, 119, 111–114. [Google Scholar] [CrossRef]
- Ebrahimi, K.B.; Cano, M.; Rhee, J.; Datta, S.; Wang, L.; Handa, J.T. Oxidative Stress Induces an Interactive Decline in Wnt and Nrf2 Signaling in Degenerating Retinal Pigment Epithelium. Antioxid. Redox Signal. 2018, 29, 389–407. [Google Scholar] [CrossRef]
- Ozaita, A.; Puighermanal, E.; Maldonado, R. Regulation of PI3K/Akt/GSK-3 Pathway by Cannabinoids in the Brain. J. Neurochem. 2007, 102, 1105–1114. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y.; Vallée, J.N. Cannabidiol and the Canonical Wnt/Β-catenin Pathway in Glaucoma. Int. J. Mol. Sci. 2021, 22, 3798. [Google Scholar] [CrossRef]
- Castillo-Quan, J.I.; Li, L.; Kinghorn, K.J.; Ivanov, D.K.; Tain, L.S.; Slack, C.; Kerr, F.; Nespital, T.; Thornton, J.; Hardy, J.; et al. Lithium Promotes Longevity through GSK3/NRF2-Dependent Hormesis. Cell Rep. 2016, 15, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, I.; Michalska, P.; Tenti, G.; Cores, Á.; Buendia, I.; Rojo, A.I.; Georgakopoulos, N.D.; Hernández-Guijo, J.M.; Teresa Ramos, M.; Wells, G.; et al. Discovery of the First Dual GSK3β Inhibitor/Nrf2 Inducer. A New Multitarget Therapeutic Strategy for Alzheimer’s Disease. Sci. Rep. 2017, 7, 45701. [Google Scholar] [CrossRef] [PubMed]
- Xiang, J.; Cao, K.; Dong, Y.T.; Xu, Y.; Li, Y.; Song, H.; Zeng, X.X.; Ran, L.Y.; Hong, W.; Guan, Z.Z. Lithium Chloride Reduced the Level of Oxidative Stress in Brains and Serums of APP/PS1 Double Transgenic Mice via the Regulation of GSK3β/Nrf2/HO-1 Pathway. Int. J. Neurosci. 2020, 130, 564–573. [Google Scholar] [CrossRef]
- Concetta, R.M.; Martino, D.; Pruccoli, L.; Bisi, A.; Gobbi, S.; Rampa, A.; Martinez, A.; Concepción Pérezpérez, C.; Martinez-Gonzalez, L.; Paglione, M.; et al. Novel Curcumin-Diethyl Fumarate Hybrid as a Dualistic GSK-3β Inhibitor/Nrf2 Inducer for the Treatment of Parkinson’s Disease. ACS Chem. Neurosci. 2020, 11, 2728–2740. [Google Scholar] [CrossRef]
- Cuadrado, A.; Kügler, S.; Lastres-Becker, I. Pharmacological Targeting of GSK-3 and NRF2 Provides Neuroprotection in a Preclinical Model of Tauopathy. Redox Biol. 2018, 14, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Buschini, E.; Piras, A.; Nuzzi, R.; Vercelli, A. Age Related Macular Degeneration and Drusen: Neuroinflammation in the Retina. Prog. Neurobiol. 2011, 95, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A Central Role for Inflammation in the Pathogenesis of Diabetic Retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Yoshida, N.; Ikeda, Y.; Notomi, S.; Ishikawa, K.; Murakami, Y.; Hisatomi, T.; Enaida, H.; Ishibashi, T. Clinical Evidence of Sustained Chronic Inflammatory Reaction in Retinitis Pigmentosa. Ophthalmology 2013, 120, 100–105. [Google Scholar] [CrossRef]
- Whitcup, S.M.; Nussenblatt, R.B.; Lightman, S.L.; Hollander, D.A. Inflammation in Retinal Disease. Int. J. Inflamm. 2013, 2013, 724648. [Google Scholar] [CrossRef]
- Wang, H.; Brown, J.; Martin, M. Glycogen Synthase Kinase 3: A Point of Convergence for the Host Inflammatory Response. Cytokine 2011, 53, 130–140. [Google Scholar] [CrossRef]
- Jope, R.S.; Yuskaitis, C.J.; Beurel, E. Glycogen Synthase Kinase-3 (GSK3): Inflammation, Diseases, and Therapeutics. Neurochem. Res. 2007, 32, 577–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmeister, L.; Diekmann, M.; Brand, K.; Huber, R. GSK3: A Kinase Balancing Promotion and Resolution of Inflammation. Cells 2020, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Rehani, K.; Jope, R.S.; Michalek, S.M. Toll-like Receptor–Mediated Cytokine Production Is Differentially Regulated by Glycogen Synthase Kinase 3. Nat. Immunol. 2005, 6, 777–784. [Google Scholar] [CrossRef]
- Zeng, H.-Y.; Tso, M.O.M.; Lai, S.; Lai, H. Activation of Nuclear Factor-ΚB during Retinal Degeneration in Rd Mice. Mol. Vis. 2008, 14, 1075–1080. [Google Scholar]
- Wu, T.; Chen, Y.; Chiang, S.K.S.; Tso, M.O.M. NF-ΚB Activation in Light-Induced Retinal Degeneration in a Mouse Model. Investig. Ophthalmol. Vis. Sci. 2002, 43, 2834–2840. [Google Scholar]
- Schwabe, R.F.; Brenner, D.A. Role of Glycogen Synthase Kinase-3 in TNF-Alpha-Induced NF-ΚB Activation and Apoptosis in Hepatocytes. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 283, G204-11. [Google Scholar] [CrossRef]
- Wang, M.-J.; Huang, H.-Y.; Chen, W.-F.; Chang, H.-F.; Kuo, J.-S. Glycogen Synthase Kinase-3β Inactivation Inhibits Tumor Necrosis Factor-α Production in Microglia by Modulating Nuclear Factor ΚB and MLK3/JNK Signaling Cascades. J. Neuroinflamm. 2010, 7, 99. [Google Scholar] [CrossRef]
- Busino, L.; Millman, S.E.; Scotto, L.; Kyratsous, C.A.; Basrur, V.; O’Connor, O.; Hoffmann, A.; Elenitoba-Johnson, K.S.; Pagano, M. Fbxw7α- and GSK3-Mediated Degradation of P100 Is a Pro-Survival Mechanism in Multiple Myeloma. Nat. Cell Biol. 2012, 14, 375–385. [Google Scholar] [CrossRef]
- Deng, J.; Miller, S.A.; Wang, H.-Y.; Xia, W.; Wen, Y.; Zhou, B.P.; Li, Y.; Lin, S.-Y.; Hung, M.-C. β-Catenin Interacts with and Inhibits NF-ΚB in Human Colon and Breast Cancer. Cancer Cell 2002, 2, 323–334. [Google Scholar] [CrossRef]
- Demarchi, F.; Bertoli, C.; Sandy, P.; Schneider, C. Glycogen Synthase Kinase-3β Regulates NF-ΚB1/P105 Stability. J. Biol. Chem. 2003, 278, 39583–39590. [Google Scholar] [CrossRef] [PubMed]
- Graham, J.R.; Tullai, J.W.; Cooper, G.M. GSK-3 Represses Growth Factor-Inducible Genes by Inhibiting NF-ΚB in Quiescent Cells. J. Biol. Chem. 2010, 285, 4472–4480. [Google Scholar] [CrossRef] [PubMed]
- Buss, H.; Dörrie, A.; Schmitz, M.L.; Frank, R.; Livingstone, M.; Resch, K.; Kracht, M. Phosphorylation of Serine 468 by GSK-3β Negatively Regulates Basal P65 NF-ΚB Activity. J. Biol. Chem. 2004, 279, 49571–49574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrejew, R.; Oliveira-Giacomelli, Á.; Ribeiro, D.E.; Glaser, T.; Arnaud-Sampaio, V.F.; Lameu, C.; Ulrich, H. The P2X7 Receptor: Central Hub of Brain Diseases. Front. Mol. Neurosci. 2020, 13, 124. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Gomez-Villafuertes, R.; León-Otegui, M.; Hontecillas-Prieto, L.; del Puerto, A.; Trejo, J.L.; Lucas, J.J.; Garrido, J.J.; Gualix, J.; Miras-Portugal, M.T.; et al. In Vivo P2X7 Inhibition Reduces Amyloid Plaques in Alzheimer’s Disease through GSK3β and Secretases. Neurobiol. Aging 2012, 33, 1816–1828. [Google Scholar] [CrossRef]
- Di Lauro, C.; Bianchi, C.; Sebastián-Serrano, Á.; Soria-Tobar, L.; Alvarez-Castelao, B.; Nicke, A.; Díaz-Hernández, M. P2X7 Receptor Blockade Reduces Tau Induced Toxicity, Therapeutic Implications in Tauopathies. Prog. Neurobiol. 2021, 208, 102173. [Google Scholar] [CrossRef]
- Nadal-Nicolás, F.M.; Galindo-Romero, C.; Valiente-Soriano, F.J.; Barberà-Cremades, M.; DeTorre-Minguela, C.; Salinas-Navarro, M.; Pelegrín, P.; Agudo-Barriuso, M. Involvement of P2X7 Receptor in Neuronal Degeneration Triggered by Traumatic Injury. Sci. Rep. 2016, 6, 38499. [Google Scholar] [CrossRef]
- Sugiyama, T. Role of P2X7 Receptors in Neuronal Death in the Retina. Neural Regen. Res. 2014, 9, 579–581. [Google Scholar] [CrossRef]
- De Lara, M.J.P.; Avilés-Trigueros, M.; Guzmán-Aránguez, A.; Valiente-Soriano, F.J.; de la Villa, P.; Vidal-Sanz, M.; Pintor, J. Potential Role of P2X7 Receptor in Neurodegenerative Processes in a Murine Model of Glaucoma. Brain Res. Bull. 2019, 150, 61–74. [Google Scholar] [CrossRef]
- Yang, M.; Qiu, R.; Wang, W.; Liu, J.; Jin, X.; Li, Y.; Li, L.; Lei, B. P2X7 Receptor Antagonist Attenuates Retinal Inflammation and Neovascularization Induced by Oxidized Low-Density Lipoprotein. Oxid. Med. Cell. Longev. 2021, 2021, 5520644. [Google Scholar] [CrossRef]
- Yang, J.; Caldwell, R.B.; Behzadian, M.A. Blockade of VEGF-Induced GSK/β-Catenin Signaling, UPAR Expression and Increased Permeability by Dominant Negative P38α. Exp. Eye Res 2012, 100, 101–108. [Google Scholar] [CrossRef] [PubMed]
- El-Remessy, A.B.; Franklin, T.; Ghaley, N.; Yang, J.; Brands, M.W.; Caldwell, R.B.; Behzadian, M.A. Diabetes-Induced Superoxide Anion and Breakdown of the Blood-Retinal Barrier: Role of the VEGF/UPAR Pathway. PLoS ONE 2013, 8, e71868. [Google Scholar] [CrossRef]
- Lanzetta, P. Anti-VEGF Therapies for Age-Related Macular Degeneration: A Powerful Tactical Gear or a Blunt Weapon? The Choice Is Ours. Graefe’s Arch. Clin. Exp. Ophthalmol. 2021, 259, 3561–3567. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Lee, D.; Tsubota, K.; Negishi, K.; Kurihara, T. Updates on the Current Treatments for Diabetic Retinopathy and Possibility of Future Oral Therapy. J. Clin. Med. 2021, 10, 4666. [Google Scholar] [CrossRef]
- Osaadon, P.; Fagan, X.J.; Lifshitz, T.; Levy, J. A Review of Anti-VEGF Agents for Proliferative Diabetic Retinopathy. Eye 2014, 28, 510–520. [Google Scholar] [CrossRef]
- Khanna, S.; Komati, R.; Eichenbaum, D.A.; Hariprasad, I.; Ciulla, T.A.; Hariprasad, S.M. Current and Upcoming Anti-VEGF Therapies and Dosing Strategies for the Treatment of Neovascular AMD: A Comparative Review. BMJ Open Ophthalmol. 2019, 4, e000398. [Google Scholar] [CrossRef]
- Kovach, J.L.; Schwartz, S.G.; Flynn, H.W.; Scott, I.U. Anti-VEGF Treatment Strategies for Wet AMD. J. Ophthalmol. 2012, 2012, 786870. [Google Scholar] [CrossRef]
- Maguire, M.G.; Martin, D.F.; Ying, G.; Jaffe, G.J.; Daniel, E.; Grunwald, J.E.; Toth, C.A.; Ferris, F.L.; Fine, S.L. Five-Year Outcomes with Anti–Vascular Endothelial Growth Factor Treatment of Neovascular Age-Related Macular Degeneration. Ophthalmology 2016, 123, 1751–1761. [Google Scholar] [CrossRef]
- Motevasseli, T.; Mohammadi, S.; Abdi, F.; Freeman, W.R. Side Effects of Brolucizumab. J. Ophthalmic Vis. Res. 2021, 16, 670–675. [Google Scholar] [CrossRef]
- Masckauchán, T.N.H.; Kitajewski, J. Wnt/Frizzled Signaling in the Vasculature: New Angiogenic Factors in Sight. Physiology 2006, 21, 181–188. [Google Scholar] [CrossRef]
- Goodwin, A.; D’Amore, P. Wnt Signaling in the Vasculature. Angiogenesis 2002, 5, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Drenser, K.A. Wnt Signaling Pathway in Retinal Vascularization. Eye Brain 2016, 8, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Yemanyi, F.; Bora, K.; Blomfield, A.K.; Wang, Z.; Chen, J. Wnt Signaling in Inner Blood–Retinal Barrier Maintenance. Int. J. Mol. Sci. 2021, 22, 11877. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Stahl, A.; Krah, N.M.; Seaward, M.R.; Dennison, R.J.; Sapieha, P.; Hua, J.; Hatton, C.J.; Juan, A.M.; Aderman, C.M.; et al. Wnt Signaling Mediates Pathological Vascular Growth in Proliferative Retinopathy. Circulation 2011, 124, 1871–1881. [Google Scholar] [CrossRef]
- Huang, W.; Li, Q.; Amiry-Moghaddam, M.; Hokama, M.; Sardi, S.H.; Nagao, M.; Warman, M.L.; Olsen, B.R. Critical Endothelial Regulation by LRP5 during Retinal Vascular Development. PLoS ONE 2016, 11, e0152833. [Google Scholar] [CrossRef]
- Gilmour, D.F. Familial Exudative Vitreoretinopathy and Related Retinopathies. Eye 2015, 29, 1–14. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, C.-H.; Sun, Y.; Gong, Y.; Favazza, T.L.; Morss, P.C.; Saba, N.J.; Fredrick, T.W.; He, X.; Akula, J.D.; et al. Pharmacologic Activation of Wnt Signaling by Lithium Normalizes Retinal Vasculature in a Murine Model of Familial Exudative Vitreoretinopathy. Am. J. Pathol. 2016, 186, 2588–2600. [Google Scholar] [CrossRef]
- Hamon, A.; Roger, J.E.; Yang, X.J.; Perron, M. Müller Glial Cell-Dependent Regeneration of the Neural Retina: An Overview across Vertebrate Model Systems. Dev. Dyn. 2016, 245, 727–738. [Google Scholar] [CrossRef]
- García-García, D.; Locker, M.; Perron, M. Update on Müller Glia Regenerative Potential for Retinal Repair. Curr. Opin. Genet. Dev. 2020, 64, 52–59. [Google Scholar] [CrossRef]
- Goldman, D. Müller Glial Cell Reprogramming and Retina Regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef]
- Salman, A.; McClements, M.; MacLaren, R. Insights on the Regeneration Potential of Müller Glia in the Mammalian Retina. Cells 2021, 10, 1957. [Google Scholar] [CrossRef] [PubMed]
- Wilken, M.S.; Reh, T.A. Retinal Regeneration in Birds and Mice. Curr. Opin. Genet. Dev. 2016, 40, 57–64. [Google Scholar] [CrossRef]
- Ramachandran, R.; Fausett, B.V.; Goldman, D. Ascl1a Regulates Müller Glia Dedifferentiation and Retina Regeneration via a Lin-28-Dependent, Let-7 MiRNA Signaling Pathway HHS Public Access. Nat. Cell Biol. 2010, 12, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramachandran, R.; Zhao, X.-F.; Goldman, D. Ascl1a/Dkk/β-Catenin Signaling Pathway Is Necessary and Glycogen Synthase Kinase-3β Inhibition Is Sufficient for Zebrafish Retina Regeneration. Proc. Natl. Acad. Sci. USA 2011, 108, 15858–15863. [Google Scholar] [CrossRef]
- Gallina, D.; Palazzo, I.; Steffenson, L.; Todd, L.; Fischer, A.J. Wnt/Βcatenin-Signaling and the Formation of Müller Glia-Derived Progenitors in the Chick Retina. Dev. Neurobiol. 2016, 76, 983–1002. [Google Scholar] [CrossRef]
- Ooto, S.; Akagi, T.; Kageyama, R.; Akita, J.; Mandai, M.; Honda, Y.; Takahashi, M. Potential for Neural Regeneration after Neurotoxic Injury in the Adult Mammalian Retina. Proc. Natl. Acad. Sci. USA 2004, 101, 13654–13659. [Google Scholar] [CrossRef] [PubMed]
- Osakada, F.; Ooto, S.; Akagi, T.; Mandai, M.; Akaike, A.; Takahashi, M. Wnt Signaling Promotes Regeneration in the Retina of Adult Mammals. J. Neurosci. 2007, 27, 4210–4219. [Google Scholar] [CrossRef]
- Suga, A.; Sadamoto, K.; Fujii, M.; Mandai, M.; Takahashi, M. Proliferation Potential of Müller Glia after Retinal Damage Varies between Mouse Strains. PLoS ONE 2014, 9, e94556. [Google Scholar] [CrossRef]
- Zhou, F.-Q.; Zhou, J.; Dedhar, S.; Wu, Y.-H.; Snider, W.D. NGF-Induced Axon Growth Is Mediated by Localized Inactivation of GSK-3β and Functions of the Microtubule Plus End Binding Protein APC. Neuron 2004, 42, 897–912. [Google Scholar] [CrossRef]
- Alabed, Y.Z.; Pool, M.; Tone, S.O.; Sutherland, C.; Fournier, A.E. GSK3β regulates Myelin-Dependent Axon Outgrowth Inhibition through CRMP4. J. Neurosci. 2010, 30, 5635–5643. [Google Scholar] [CrossRef]
- Saijilafu; Hur, E.-M.; Jiao, Z.; Liu, C.-M.; Xu, W.-L.; Zhou, F.-Q. PI3K-GSK3 Signaling Regulates Mammalian Axon Regeneration by Inducing the Expression of Smad1. Nat. Commun. 2013, 4, 2690. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F. Coordinating Gene Expression and Axon Assembly to Control Axon Growth: Potential Role of GSK3 Signaling. Front. Mol. Neurosci. 2012, 5, 3. [Google Scholar] [CrossRef]
- Leibinger, M.; Andreadaki, A.; Golla, R.; Levin, E.; Hilla, A.M.; Diekmann, H.; Fischer, D. Boosting CNS Axon Regeneration by Harnessing Antagonistic Effects of GSK3 Activity. Proc. Natl. Acad. Sci. USA 2017, 114, E5454–E5463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibinger, M.; Hilla, A.M.; Andreadaki, A.; Fischer, D. GSK3-CRMP2 Signaling Mediates Axonal Regeneration Induced by Pten Knockout. Commun. Biol. 2019, 2, 318. [Google Scholar] [CrossRef]
- Ahmed, Z. The GSK3β Pathway in Optic Nerve Regeneration. Arch. Clin. Exp. Ophthalmol. 2020, 2, 10–16. [Google Scholar]
- Patel, A.K.; Park, K.K.; Hackam, A.S. Wnt Signaling Promotes Axonal Regeneration Following Optic Nerve Injury in the Mouse. Neuroscience 2017, 343, 372–383. [Google Scholar] [CrossRef]
- Inoki, K.; Ouyang, H.; Zhu, T.; Lindvall, C.; Wang, Y.; Zhang, X.; Yang, Q.; Bennett, C.; Harada, Y.; Stankunas, K.; et al. TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. Cell 2006, 126, 955–968. [Google Scholar] [CrossRef]
- Miao, L.; Yang, L.; Huang, H.; Liang, F.; Ling, C.; Hu, Y. MTORC1 is Necessary but MTORC2 and GSK3β Are Inhibitory for AKT3-Induced Axon Regeneration in the Central Nervous System. eLife 2016, 5, e14908. [Google Scholar] [CrossRef]
- Leibinger, M.; Andreadaki, A.; Fischer, D. Role of MTOR in Neuroprotection and Axon Regeneration after Inflammatory Stimulation. Neurobiol. Dis. 2012, 46, 314–324. [Google Scholar] [CrossRef]
- Wenzel, A.; Grimm, C.; Samardzija, M.; Reme´ã, C.E.R. Molecular Mechanisms of Light-Induced Photoreceptor Apoptosis and Neuroprotection for Retinal Degeneration. Prog. Retin. Eye Res. 2005, 24, 275–306. [Google Scholar] [CrossRef]
- Fudalej, E.; Justyniarska, M.; Kasarełło, K.; Dziedziak, J.; Szaflik, J.P.; Cudnoch-Jędrzejewska, A. Neuroprotective Factors of the Retina and Their Role in Promoting Survival of Retinal Ganglion Cells: A Review. Ophthalmic Res. 2021, 64, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Trifunovic, D.; Sahaboglu, A.; Kaur, J.; Mencl, S.; Zrenner, E.; Ueffing, M.; Arango-Gonzalez, B.; Paquet-Durand, F. Neuroprotective Strategies for the Treatment of Inherited Photoreceptor Degeneration. Curr. Mol. Med. 2012, 12, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Iraha, S.; Hirami, Y.; Ota, S.; Sunagawa, G.A.; Mandai, M.; Tanihara, H.; Takahashi, M.; Kurimoto, Y. Efficacy of Valproic Acid for Retinitis Pigmentosa Patients: A Pilot Study. Clin. Ophthalmol. 2016, 10, 1375–1384. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.R.; Correia, A.O.; dos Santos, G.C.A.; Parente, L.L.T.; de Siqueira, K.P.; Lima, D.G.S.; Moura, J.A.; da Silva Ribeiro, A.E.; Costa, R.O.; Lucetti, D.L.; et al. Neuroprotective Effects of Valproic Acid on Brain Ischemia Are Related to Its HDAC and GSK3 Inhibitions. Pharmacol. Biochem. Behav. 2018, 167, 17–28. [Google Scholar] [CrossRef] [PubMed]
- De Sarno, P.; Li, X.; Jope, R.S. Regulation of Akt and Glycogen Synthase Kinase-3β Phosphorylation by Sodium Valproate and Lithium. Neuropharmacology 2002, 43, 1158–1164. [Google Scholar] [CrossRef]
- Chen, G.; Huang, L.-D.; Jiang, Y.-M.; Manji, H.K. The Mood-Stabilizing Agent Valproate Inhibits the Activity of Glycogen Synthase Kinase-3. J. Neurochem. 2008, 72, 1327–1330. [Google Scholar] [CrossRef]
- Birch, D.G.; Bernstein, P.S.; Iannacone, A.; Pennesi, M.E.; Lam, B.L.; Heckenlively, J.; Csaky, K.; Hartnett, M.E.; Winthrop, K.L.; Jayasundera, T.; et al. Effect of Oral Valproic Acid vs. Placebo for Vision Loss in Patients with Autosomal Dominant Retinitis Pigmentosa. JAMA Ophthalmol. 2018, 136, 849–856. [Google Scholar] [CrossRef]
- Vent-Schmidt, R.Y.J.; Wen, R.H.; Zong, Z.; Chiu, C.N.; Tam, B.M.; May, C.G.; Moritz, O.L. Opposing Effects of Valproic Acid Treatment Mediated by Histone Deacetylase Inhibitor Activity in Four Transgenic X. laevis Models of Retinitis Pigmentosa. J. Neurosci. 2017, 37, 1039–1054. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, W.; Zhao, Y.; Shu, X.; Wang, W.; Wang, D.; Yang, Y.; He, Z.; Wang, X.; Ying, Y. GSK3β-Mediated Tau Hyperphosphorylation Triggers Diabetic Retinal Neurodegeneration by Disrupting Synaptic and Mitochondrial Functions. Mol. Neurodegener. 2018, 13, 62. [Google Scholar] [CrossRef]
- Yun, J.-H.; Park, S.W.; Kim, J.H.; Park, Y.-J.; Cho, C.-H.; Kim, J.H. Angiopoietin 2 Induces Astrocyte Apoptosis via Avβ5-Integrin Signaling in Diabetic Retinopathy. Cell Death Dis. 2016, 7, e2101. [Google Scholar] [CrossRef]
- Ahmed, Z.; Morgan-Warren, P.J.; Berry, M.; Scott, R.A.H.; Logan, A. Effects of SiRNA-Mediated Knockdown of GSK3β on Retinal Ganglion Cell Survival and Neurite/Axon Growth. Cells 2019, 8, 956. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Peng, Z.; Seven, E.S.; Leblanc, R.M. Crossing the Blood-Brain Barrier with Nanoparticles. J. Control. Release 2018, 270, 290–303. [Google Scholar] [CrossRef] [PubMed]
- Matos, A.L.; Bruno, D.F.; Ambrósio, A.F.; Santos, P.F. The Benefits of Flavonoids in Diabetic Retinopathy. Nutrients 2020, 12, 3169. [Google Scholar] [CrossRef] [PubMed]
- Maher, P.; Hanneken, A. Flavonoids Protect Retinal Ganglion Cells from Oxidative Stress–Induced Death. Investig. Opthalmol. Vis. Sci. 2005, 46, 4796–4803. [Google Scholar] [CrossRef]
- Campbell, W.A.; Blum, S.; Reske, A.; Hoang, T.; Blackshaw, S.; Fischer, A.J. Cannabinoid Signaling Promotes the Reprogramming of Muller Glia into Proliferating Progenitor Cells. Glia 2021, 69, 2503–2521. [Google Scholar] [CrossRef]
- Fujita, K.; Nishiguchi, K.M.; Shiga, Y.; Nakazawa, T. Spatially and Temporally Regulated NRF2 Gene Therapy Using Mcp-1 Promoter in Retinal Ganglion Cell Injury. Mol. Ther. Methods Clin. Dev. 2017, 5, 130–141. [Google Scholar] [CrossRef]
- Chen, W.-J.; Wu, C.; Xu, Z.; Kuse, Y.; Hara, H.; Duh, E.J. Nrf2 Protects Photoreceptor Cells from Photo-Oxidative Stress Induced by Blue Light. Exp. Eye Res. 2017, 154, 151–158. [Google Scholar] [CrossRef]
- Chiha, W.; Bartlett, C.A.; Petratos, S.; Fitzgerald, M.; Harvey, A.R. Intravitreal Application of AAV-BDNF or Mutant AAV-CRMP2 Protects Retinal Ganglion Cells and Stabilizes Axons and Myelin after Partial Optic Nerve Injury. Exp. Neurol. 2020, 326, 113167. [Google Scholar] [CrossRef]
- Yao, A.; van Wijngaarden, P. Metabolic Pathways in Context: MTOR Signalling in the Retina and Optic Nerve—A Review. Clin. Exp. Ophthalmol. 2020, 48, 1072–1084. [Google Scholar] [CrossRef]
- Park, T.K.; Lee, S.H.; Choi, J.S.; Nah, S.K.; Kim, H.J.; Park, H.Y.; Lee, H.; Lee, S.H.S.; Park, K. Adeno-Associated Viral Vector-Mediated MTOR Inhibition by Short Hairpin RNA Suppresses Laser-Induced Choroidal Neovascularization. Mol. Ther. Nucleic Acids 2017, 8, 26–35. [Google Scholar] [CrossRef]
- Lee, S.H.S.; Lee, J.Y.; Choi, J.-S.; Kim, H.J.; Kim, J.; Cha, S.; Lee, K.J.; Woo, H.-N.; Park, K.; Lee, H. MTOR Inhibition as a Novel Gene Therapeutic Strategy for Diabetic Retinopathy. PLoS ONE 2022, 17, e0269951. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Seo, W.l.; Woo, H.N.; Kim, H.J.; Lee, S.H.S.; Kim, J.; Choi, J.S.; Park, K.; Lee, J.Y.; Lee, B.J.; et al. AAV Expressing an MTOR-Inhibiting SiRNA Exhibits Therapeutic Potential in Retinal Vascular Disorders by Preserving Endothelial Integrity. FEBS Open Bio 2022, 12, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Wang, J.; He, N.; Feng, H. PLK2 Protects Retinal Ganglion Cells from Oxidative Stress by Potentiating Nrf2 Signaling via GSK-3β. J. Biochem. Mol. Toxicol. 2021, 35, e22815. [Google Scholar] [CrossRef]
- Rapino, C.; Tortolani, D.; Scipioni, L.; Maccarrone, M. Neuroprotection by (Endo)Cannabinoids in Glaucoma and Retinal Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Haenold, R.; Weih, F.; Herrmann, K.-H.; Schmidt, K.-F.; Krempler, K.; Engelmann, C.; Nave, K.-A.; Reichenbach, J.R.; Löwel, S.; Witte, O.W.; et al. NF-ΚB Determines Axonal Re- and Degeneration by Cell-Specific Balance of RelA and P50 Subunits in the Adult CNS. J. Cell Sci. 2014, 127, 3052–3065. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hottin, C.; Perron, M.; Roger, J.E. GSK3 Is a Central Player in Retinal Degenerative Diseases but a Challenging Therapeutic Target. Cells 2022, 11, 2898. https://doi.org/10.3390/cells11182898
Hottin C, Perron M, Roger JE. GSK3 Is a Central Player in Retinal Degenerative Diseases but a Challenging Therapeutic Target. Cells. 2022; 11(18):2898. https://doi.org/10.3390/cells11182898
Chicago/Turabian StyleHottin, Catherine, Muriel Perron, and Jérôme E. Roger. 2022. "GSK3 Is a Central Player in Retinal Degenerative Diseases but a Challenging Therapeutic Target" Cells 11, no. 18: 2898. https://doi.org/10.3390/cells11182898
APA StyleHottin, C., Perron, M., & Roger, J. E. (2022). GSK3 Is a Central Player in Retinal Degenerative Diseases but a Challenging Therapeutic Target. Cells, 11(18), 2898. https://doi.org/10.3390/cells11182898