
Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention

 , and
, and
Abstract
:1. Introduction
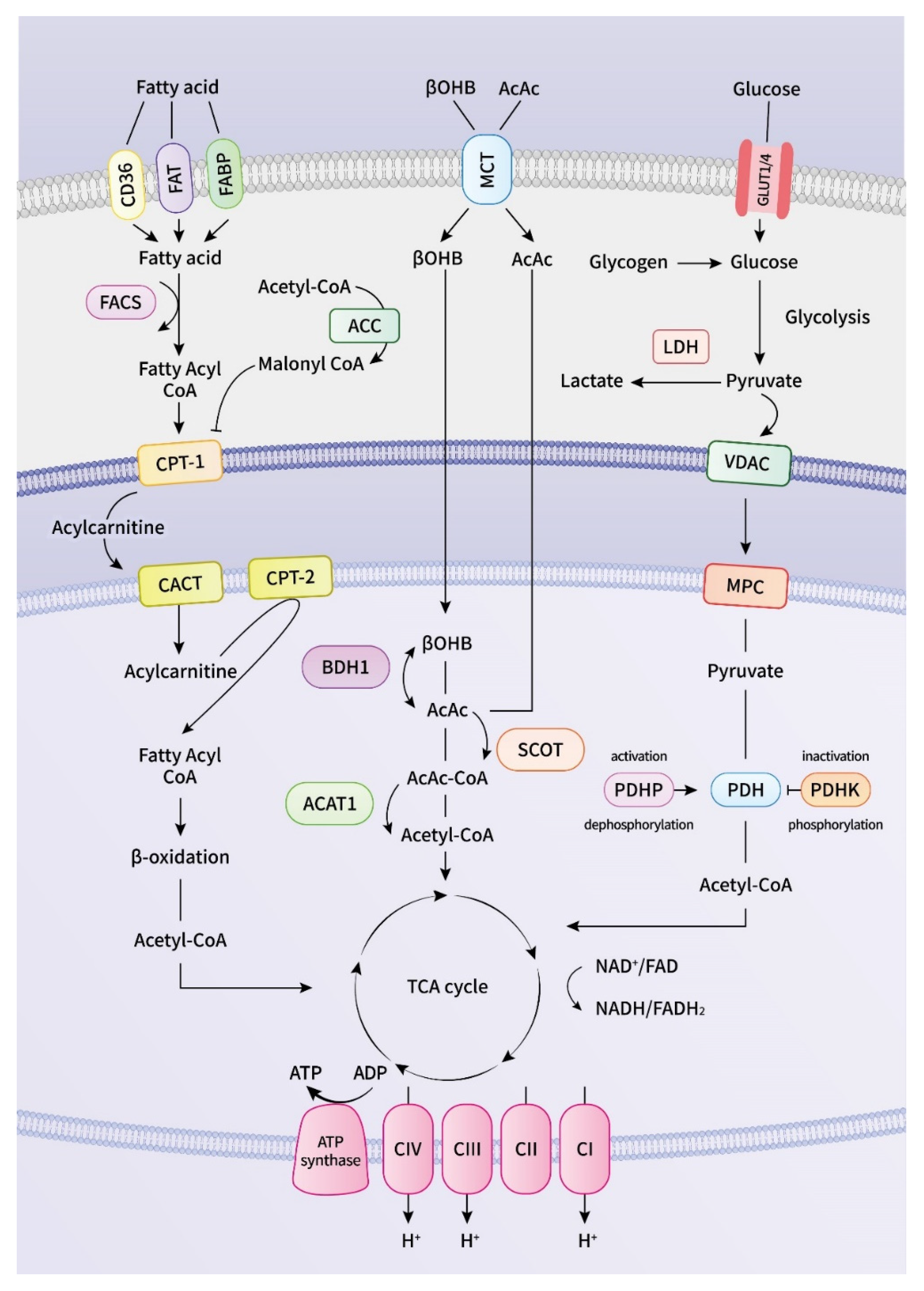
2. Energy Metabolism in the Heart
2.1. Mitochondrial Dysfunction in DM Cardiomyopathy
2.2. Mitochondrial Dysfunction and ATP Deficiency
2.3. Fatty Acid Dysmetabolism in Atrial Arrhythmogenesis
2.4. Glucose Dysmetabolism in Atrial Arrhythmogenesis
2.5. Ketone Body Metabolism and Pathogenesis of Atrial Arrhythmogenesis
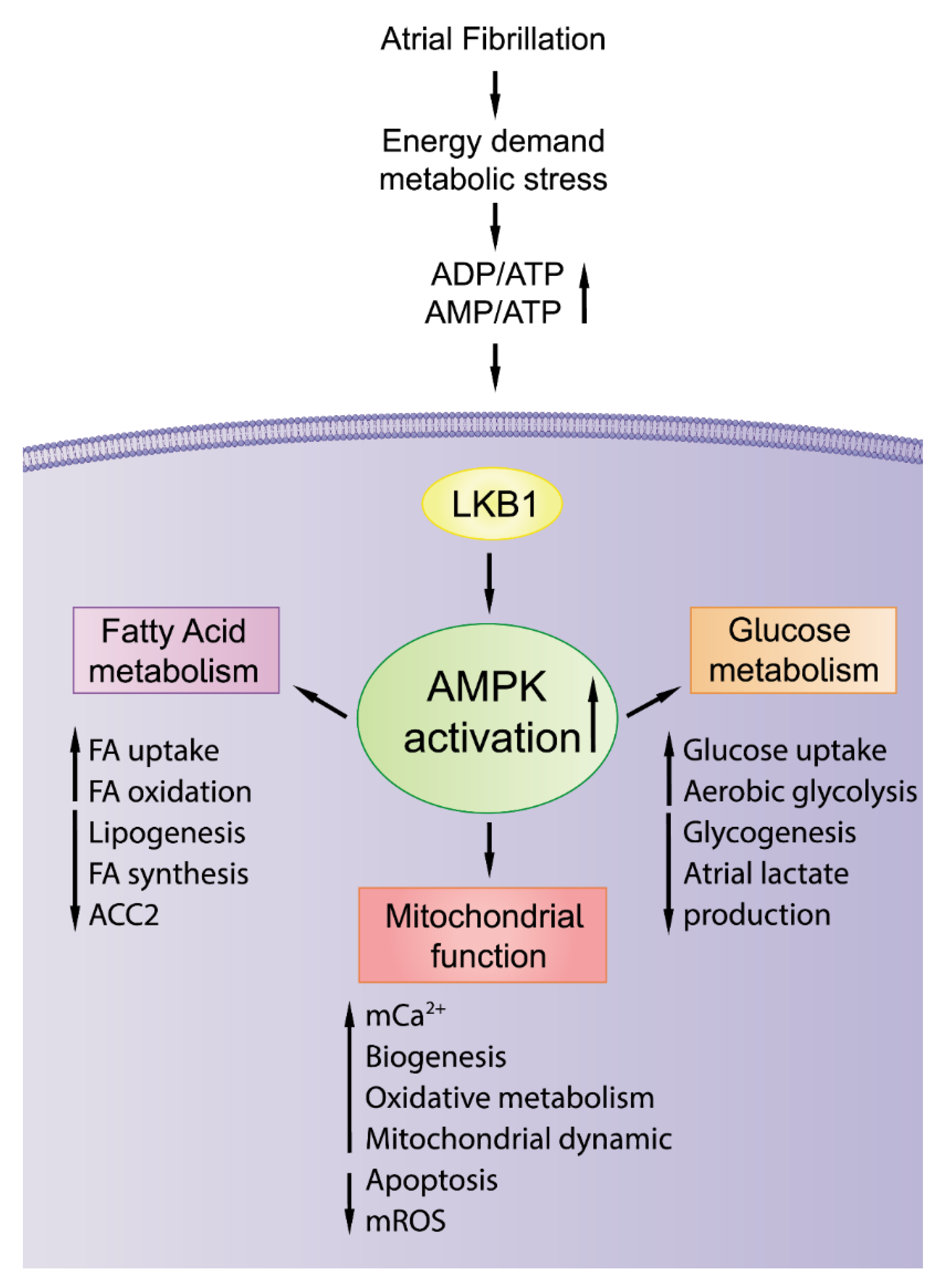
2.6. AMPK Activation with Potential Antiatrial Arrhythmogenesis
2.7. Warburg Effect in Atrial Arrhythmogenesis
3. Targeting Cardiac Metabolism as an Upstream Treatment of AF
3.1. Activation of AMPK: A Potential Therapeutic Strategy
3.2. Ketogenic Diet or Ketone Administration on Risk of Arrhythmogenesis
3.3. Ketone Body Modulation by Using SGLT2 Inhibitor
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Sun, L.; Chen, L.; Liu, S.; Zhong, L.; Cui, M. Comprehensive metabolomic and proteomic analyses reveal candidate biomarkers and related metabolic networks in atrial fibrillation. Metabolomics 2019, 15, 96. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Zhou, S.; Liu, Z.; Li, X.; Liu, Q. Quantitative proteomics of changes in energy metabolism-related proteins in atrial tissue from valvular disease patients with permanent atrial fibrillation. Circ. J. 2014, 78, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Kourliouros, A.; Yin, X.; Didangelos, A.; Hosseini, M.T.; Valencia, O.; Mayr, M.; Jahangiri, M. Substrate modifications precede the development of atrial fibrillation after cardiac surgery: A proteomic study. Ann. Thorac. Surg. 2011, 92, 104–110. [Google Scholar] [CrossRef]
- Mayr, M.; Yusuf, S.; Weir, G.; Chung, Y.L.; Mayr, U.; Yin, X.; Ladroue, C.; Madhu, B.; Roberts, N.; De Souza, A.; et al. Combined metabolomic and proteomic analysis of human atrial fibrillation. J. Am. Coll. Cardiol. 2008, 51, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Sadykhov, N.K.; Kartuesov, A.G.; Borisov, E.E.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Mitochondrial Abnormalities in Diabetic Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 7863. [Google Scholar] [CrossRef]
- Goette, A.; Lendeckel, U. Atrial Cardiomyopathy: Pathophysiology and Clinical Consequences. Cells 2021, 10, 2605. [Google Scholar] [CrossRef] [PubMed]
- Goudis, C.A.; Korantzopoulos, P.; Ntalas, I.V.; Kallergis, E.M.; Liu, T.; Ketikoglou, D.G. Diabetes mellitus and atrial fibrillation: Pathophysiological mechanisms and potential upstream therapies. Int. J. Cardiol. 2015, 184, 617–622. [Google Scholar] [CrossRef]
- Tadic, M.; Cuspidi, C. Type 2 diabetes mellitus and atrial fibrillation: From mechanisms to clinical practice. Arch. Cardiovasc. Dis. 2015, 108, 269–276. [Google Scholar] [CrossRef]
- Lee, T.I.; Chen, Y.C.; Lin, Y.K.; Chung, C.C.; Lu, Y.Y.; Kao, Y.H.; Chen, Y.J. Empagliflozin Attenuates Myocardial Sodium and Calcium Dysregulation and Reverses Cardiac Remodeling in Streptozotocin-Induced Diabetic Rats. Int. J. Mol. Sci. 2019, 20, 1680. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.W.; Lee, T.I.; Lin, Y.K.; Chen, Y.C.; Kao, Y.H.; Chen, Y.J. Effect of antidiabetic drugs on the risk of atrial fibrillation: Mechanistic insights from clinical evidence and translational studies. Cell Mol. Life Sci. 2021, 78, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.; Jaswal, J.S.; Stanley, W.C. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Muller, J.; Bertsch, T.; Volke, J.; Schmid, A.; Klingbeil, R.; Metodiev, Y.; Karaca, B.; Kim, S.H.; Lindner, S.; Schupp, T.; et al. Narrative review of metabolomics in cardiovascular disease. J. Thorac. Dis. 2021, 13, 2532–2550. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Fillmore, N.; Mori, J.; Lopaschuk, G.D. Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 2014, 171, 2080–2090. [Google Scholar] [CrossRef]
- Greenwell, A.A.; Gopal, K.; Ussher, J.R. Myocardial Energy Metabolism in Non-ischemic Cardiomyopathy. Front. Physiol. 2020, 11, 570421. [Google Scholar] [CrossRef]
- Jaswal, J.S.; Keung, W.; Wang, W.; Ussher, J.R.; Lopaschuk, G.D. Targeting fatty acid and carbohydrate oxidation--a novel therapeutic intervention in the ischemic and failing heart. Biochim. Biophys. Acta 2011, 1813, 1333–1350. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.S.; Nemeria, N.S.; Furey, W.; Jordan, F. The pyruvate dehydrogenase complexes: Structure-based function and regulation. J. Biol. Chem. 2014, 289, 16615–16623. [Google Scholar] [CrossRef] [PubMed]
- Harada, M.; Melka, J.; Sobue, Y.; Nattel, S. Metabolic Considerations in Atrial Fibrillation- Mechanistic Insights and Therapeutic Opportunities. Circ. J. 2017, 81, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Tumova, J.; Andel, M.; Trnka, J. Excess of free fatty acids as a cause of metabolic dysfunction in skeletal muscle. Physiol. Res. 2016, 65, 193–207. [Google Scholar] [CrossRef]
- Chakraborty, P.; Nattel, S.; Nanthakumar, K. Linking cellular energy state to atrial fibrillation pathogenesis: Potential role of adenosine monophosphate-activated protein kinase. Heart Rhythm. 2020, 17, 1398–1404. [Google Scholar] [CrossRef]
- Cotter, D.G.; Schugar, R.C.; Crawford, P.A. Ketone body metabolism and cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1060–H1076. [Google Scholar] [CrossRef]
- Ho, K.L.; Zhang, L.; Wagg, C.; al Batran, R.; Gopal, K.; Levasseur, J.; Leone, T.; Dyck, J.R.B.; Ussher, J.R.; Muoio, D.M.; et al. Increased ketone body oxidation provides additional energy for the failing heart without improving cardiac efficiency. Cardiovasc. Res. 2019, 115, 1606–1616. [Google Scholar] [CrossRef]
- Schugar, R.C.; Moll, A.R.; d’Avignon, D.A.; Weinheimer, C.J.; Kovacs, A.; Crawford, P.A. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 2014, 3, 754–769. [Google Scholar] [CrossRef]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [Green Version]
- Fukao, T.; Song, X.Q.; Mitchell, G.A.; Yamaguchi, S.; Sukegawa, K.; Orii, T.; Kondo, N. Enzymes of ketone body utilization in human tissues: Protein and messenger RNA levels of succinyl-coenzyme A (CoA):3-ketoacid CoA transferase and mitochondrial and cytosolic acetoacetyl-CoA thiolases. Pediatr. Res. 1997, 42, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Ho, K.L.; Pherwani, S.; Ketema, E.B. Ketone metabolism in the failing heart. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158813. [Google Scholar] [CrossRef] [PubMed]
- Yurista, S.R.; Matsuura, T.R.; Sillje, H.H.W.; Nijholt, K.T.; McDaid, K.S.; Shewale, S.V.; Leone, T.C.; Newman, J.C.; Verdin, E.; van Veldhuisen, D.J.; et al. Ketone Ester Treatment Improves Cardiac Function and Reduces Pathologic Remodeling in Preclinical Models of Heart Failure. Circ. Heart Fail. 2021, 14, e007684. [Google Scholar] [CrossRef]
- Horton, J.L.; Davidson, M.T.; Kurishima, C.; Vega, R.B.; Powers, J.C.; Matsuura, T.R.; Petucci, C.; Lewandowski, E.D.; Crawford, P.A.; Muoio, D.M.; et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 2019, 4, e124079. [Google Scholar] [CrossRef]
- Nagao, M.; Toh, R.; Irino, Y.; Mori, T.; Nakajima, H.; Hara, T.; Honjo, T.; Satomi-Kobayashi, S.; Shinke, T.; Tanaka, H.; et al. beta-Hydroxybutyrate elevation as a compensatory response against oxidative stress in cardiomyocytes. Biochem. Biophys. Res. Commun. 2016, 475, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.C.; Wang, T.T.; Zheng, L.; Hua, F.; Li, J.J. The Role of Mitochondrial Biogenesis Dysfunction in Diabetic Cardiomyopathy. Biomol. Ther. 2022, 30, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Mereweather, L.J.; Aparicio, C.N.M.; Heather, L.C. Positioning Metabolism as a Central Player in the Diabetic Heart. J. Lipid. Atheroscler. 2020, 9, 92–109. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Mao, M.; Zuo, Z. Palmitate Induces Mitochondrial Energy Metabolism Disorder and Cellular Damage via the PPAR Signaling Pathway in Diabetic Cardiomyopathy. Diabetes Metab. Syndr. Obes. 2022, 15, 2287–2299. [Google Scholar] [CrossRef]
- Mansor, L.S.; Mehta, K.; Aksentijevic, D.; Carr, C.A.; Lund, T.; Cole, M.A.; le Page, L.; Mda, L.S.F.; Shattock, M.J.; Aasum, E.; et al. Increased oxidative metabolism following hypoxia in the type 2 diabetic heart, despite normal hypoxia signalling and metabolic adaptation. J. Physiol. 2016, 594, 307–320. [Google Scholar] [CrossRef]
- Lou, P.H.; Lucchinetti, E.; Scott, K.Y.; Huang, Y.; Gandhi, M.; Hersberger, M.; Clanachan, A.S.; Lemieux, H.; Zaugg, M. Alterations in fatty acid metabolism and sirtuin signaling characterize early type-2 diabetic hearts of fructose-fed rats. Physiol. Rep. 2017, 5, e13388. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Mori, J.; Alrob, O.A.; Wagg, C.S.; Harris, R.A.; Lopaschuk, G.D.; Oudit, G.Y. ANG II causes insulin resistance and induces cardiac metabolic switch and inefficiency: A critical role of PDK4. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1103–H1113. [Google Scholar] [CrossRef]
- Wang, L.; Cai, Y.; Jian, L.; Cheung, C.W.; Zhang, L.; Xia, Z. Impact of peroxisome proliferator-activated receptor-alpha on diabetic cardiomyopathy. Cardiovasc. Diabetol. 2021, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Parker, A.M.; Tate, M.; Prakoso, D.; Deo, M.; Willis, A.M.; Nash, D.M.; Donner, D.G.; Crawford, S.; Kiriazis, H.; Granata, C.; et al. Characterisation of the Myocardial Mitochondria Structural and Functional Phenotype in a Murine Model of Diabetic Cardiomyopathy. Front. Physiol. 2021, 12, 672252. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Zhu, S.; Wu, J.; Zhang, M.; Xu, Y.; Xu, W.; Cui, J.; Yu, B.; Cao, W.; Liu, J. Exercise enhances cardiac function by improving mitochondrial dysfunction and maintaining energy homoeostasis in the development of diabetic cardiomyopathy. J. Mol. Med. 2020, 98, 245–261. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Kaambre, T.; Han, W.H.; de Goede, P.; et al. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020, 228, e13430. [Google Scholar] [CrossRef]
- Boudina, S.; Sena, S.; O’Neill, B.T.; Tathireddy, P.; Young, M.E.; Abel, E.D. Reduced mitochondrial oxidative capacity and increased mitochondrial uncoupling impair myocardial energetics in obesity. Circulation 2005, 112, 2686–2695. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Kypson, A.P.; Rodriguez, E.; Anderson, C.A.; Lehr, E.J.; Neufer, P.D. Substrate-specific derangements in mitochondrial metabolism and redox balance in the atrium of the type 2 diabetic human heart. J. Am. Coll. Cardiol. 2009, 54, 1891–1898. [Google Scholar] [CrossRef] [PubMed]
- Duicu, O.M.; Lighezan, R.; Sturza, A.; Balica, R.; Vaduva, A.; Feier, H.; Gaspar, M.; Ionac, A.; Noveanu, L.; Borza, C.; et al. Assessment of Mitochondrial Dysfunction and Monoamine Oxidase Contribution to Oxidative Stress in Human Diabetic Hearts. Oxid. Med. Cell. Longev. 2016, 2016, 8470394. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, G.N.; Patten, D.A.; Redpath, C.J.; Harper, M.E. Atrial Fibrillation Is Associated With Impaired Atrial Mitochondrial Energetics and Supercomplex Formation in Adults With Type 2 Diabetes. Can. J. Diabetes 2019, 43, 67–75.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Qiu, W.; Zhou, Y.; Feng, Y.; Chen, J.; Xia, S.; Li, W.; Liao, Y.; Li, X.; Zhou, J.; et al. Clinical characteristics and 1-year outcomes in hospitalized patients with heart failure with preserved ejection fraction: Results from the China Cardiovascular Association Database-Heart Failure Center Registry. Eur. J. Heart Fail. 2022. [Google Scholar] [CrossRef]
- Teramoto, K.; Teng, T.K.; Chandramouli, C.; Tromp, J.; Sakata, Y.; Lam, C.S. Epidemiology and Clinical Features of Heart Failure with Preserved Ejection Fraction. Card. Fail. Rev. 2022, 8, e27. [Google Scholar] [CrossRef] [PubMed]
- van Woerden, G.; Gorter, T.M.; Westenbrink, B.D.; Willems, T.P.; van Veldhuisen, D.J.; Rienstra, M. Epicardial fat in heart failure patients with mid-range and preserved ejection fraction. Eur. J. Heart Fail. 2018, 20, 1559–1566. [Google Scholar] [CrossRef] [PubMed]
- Hahn, V.S.; Knutsdottir, H.; Luo, X.; Bedi, K.; Margulies, K.B.; Haldar, S.M.; Stolina, M.; Yin, J.; Khakoo, A.Y.; Vaishnav, J.; et al. Myocardial Gene Expression Signatures in Human Heart Failure With Preserved Ejection Fraction. Circulation 2021, 143, 120–134. [Google Scholar] [CrossRef]
- Anguita, E.; Chaparro, A.; Candel, F.J.; Ramos-Acosta, C.; Martinez-Micaelo, N.; Amigo, N.; Torrejon, M.J.; Llopis-Garcia, G.; Suarez-Cadenas, M.D.M.; Matesanz, M.; et al. Biomarkers of stable and decompensated phases of heart failure with preserved ejection fraction. Int. J. Cardiol. 2022, 361, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, A.; Perez, G.; Castro, P.F.; Parra, V.; Verdejo, H.E. Mitochondrial function, dynamics and quality control in the pathophysiology of HFpEF. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166208. [Google Scholar] [CrossRef]
- Kumar, A.A.; Kelly, D.P.; Chirinos, J.A. Mitochondrial Dysfunction in Heart Failure With Preserved Ejection Fraction. Circulation 2019, 139, 1435–1450. [Google Scholar] [CrossRef]
- Rizzuto, R.; de Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Kume, O.; Wakisaka, O.; Fukunaga, N.; Teshima, Y.; Hara, M.; Saikawa, T. Novel strategy to prevent atrial fibrosis and fibrillation. Circ. J. 2012, 76, 2318–2326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Cha, Y.M.; Dzeja, P.P.; Shen, W.K.; Jahangir, A.; Hart, C.Y.; Terzic, A.; Redfield, M.M. Failing atrial myocardium: Energetic deficits accompany structural remodeling and electrical instability. Am. J. Physiol. Heart Circ. Physiol. 2003, 284, H1313–H1320. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Lefebvre, P.; Modine, T.; Fayad, G.; Dehondt, H.; Hurt, C.; Coisne, A.; Koussa, M.; Remy-Jouet, I.; et al. Mitochondrial dysfunction as an arrhythmogenic substrate: A translational proof-of-concept study in patients with metabolic syndrome in whom post-operative atrial fibrillation develops. J. Am. Coll. Cardiol. 2013, 62, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Wiersma, M.; van Marion, D.M.S.; Wust, R.C.I.; Houtkooper, R.H.; Zhang, D.; Groot, N.M.S.; Henning, R.H.; Brundel, B. Mitochondrial Dysfunction Underlies Cardiomyocyte Remodeling in Experimental and Clinical Atrial Fibrillation. Cells 2019, 8, 4987. [Google Scholar] [CrossRef]
- Kalifa, J.; Maixent, J.M.; Chalvidan, T.; Dalmasso, C.; Colin, D.; Cozma, D.; Laurent, P.; Deharo, J.C.; Djiane, P.; Cozzone, P.; et al. Energetic metabolism during acute stretch-related atrial fibrillation. Mol. Cell Biochem. 2008, 317, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Barbey, O.; Pierre, S.; Duran, M.J.; Sennoune, S.; Levy, S.; Maixent, J.M. Specific up-regulation of mitochondrial F0F1-ATPase activity after short episodes of atrial fibrillation in sheep. J. Cardiovasc. Electrophysiol. 2000, 11, 432–438. [Google Scholar] [CrossRef]
- Emelyanova, L.; Ashary, Z.; Cosic, M.; Negmadjanov, U.; Ross, G.; Rizvi, F.; Olet, S.; Kress, D.; Sra, J.; Tajik, A.J.; et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H54–H63. [Google Scholar] [CrossRef] [PubMed]
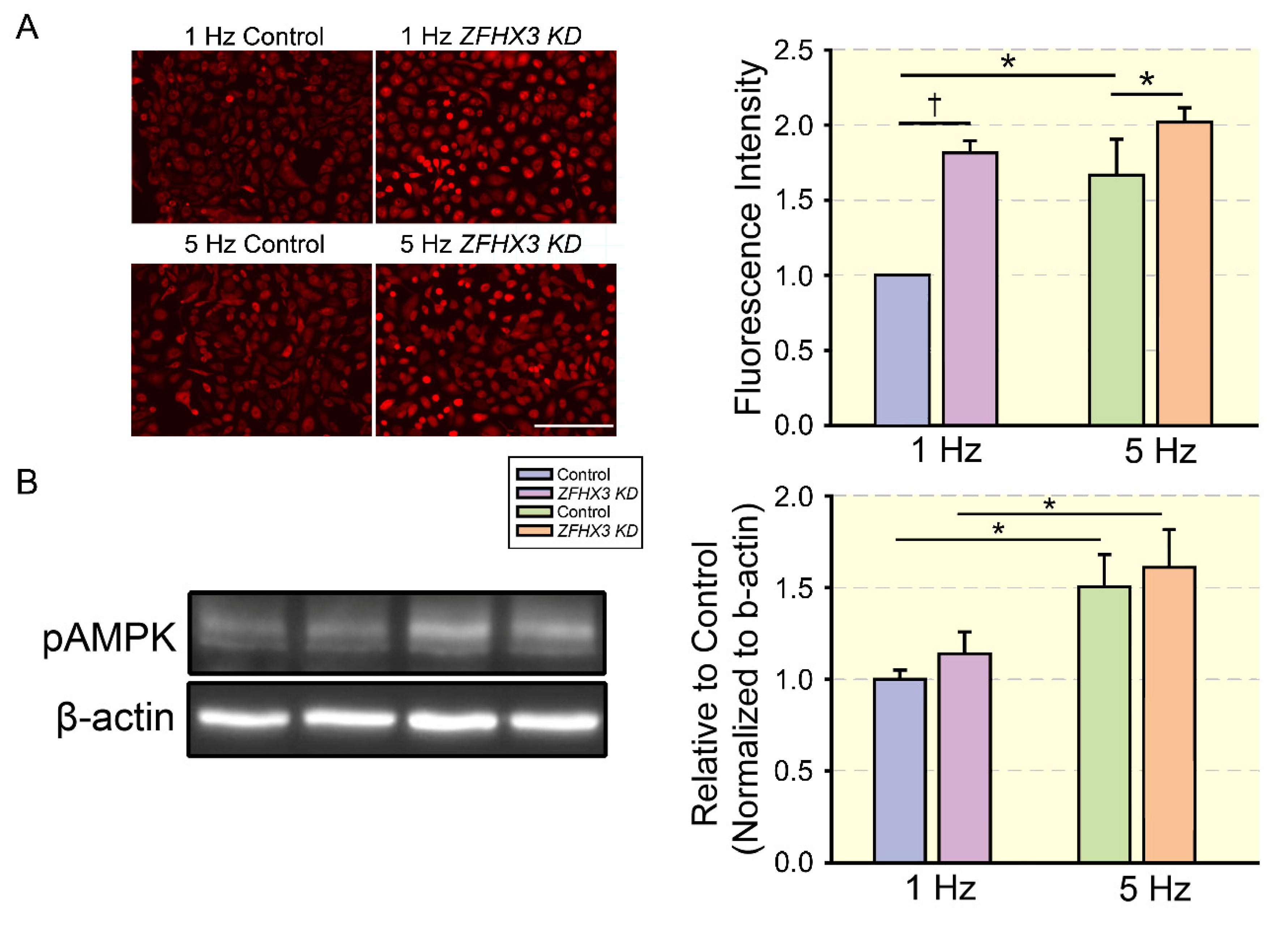
- Lkhagva, B.; Lin, Y.K.; Chen, Y.C.; Cheng, W.L.; Higa, S.; Kao, Y.H.; Chen, Y.J. ZFHX3 knockdown dysregulates mitochondrial adaptations to tachypacing in atrial myocytes through enhanced oxidative stress and calcium overload. Acta Physiol. 2021, 231, e13604. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Zhao, J.; Zhang, M.; Liu, G.; Wang, X.; Liu, Y.; Yang, N.; Liu, Y.; Zhao, G.; Sun, J.; et al. beta3-Adrenoceptor Impairs Mitochondrial Biogenesis and Energy Metabolism During Rapid Atrial Pacing-Induced Atrial Fibrillation. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.F.; Chen, T.; Johnson, S.C.; Szeto, H.; Rabinovitch, P.S. Cardiac aging: From molecular mechanisms to significance in human health and disease. Antioxid Redox Signal. 2012, 16, 1492–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Li, X.; Zhang, L.; Zhu, M.; Gao, L. A high-fat diet impairs mitochondrial biogenesis, mitochondrial dynamics, and the respiratory chain complex in rat myocardial tissues. J. Cell Biochem. 2018, 119, 9602. [Google Scholar] [CrossRef] [PubMed]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ. Res. 2001, 88, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovasc. Res. 2009, 81, 449–456. [Google Scholar] [CrossRef]
- Saadeh, K.; Fazmin, I.T. Mitochondrial Dysfunction Increases Arrhythmic Triggers and Substrates; Potential Anti-arrhythmic Pharmacological Targets. Front. Cardiovasc. Med. 2021, 8, 646932. [Google Scholar] [CrossRef]
- Faivre, J.F.; Findlay, I. Action potential duration and activation of ATP-sensitive potassium current in isolated guinea-pig ventricular myocytes. Biochim. Biophys. Acta. 1990, 1029, 167–172. [Google Scholar] [CrossRef]
- Muszynski, P.; Bonda, T.A. Mitochondrial Dysfunction in Atrial Fibrillation-Mechanisms and Pharmacological Interventions. J. Clin. Med. 2021, 10, 2385. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Bai, F.; Liu, N.; Ouyang, F.; Liu, Q. The Warburg effect: A new insight into atrial fibrillation. Clin. Chim. Acta 2019, 499, 4–12. [Google Scholar] [CrossRef]
- Sutanto, H.; Lyon, A.; Lumens, J.; Schotten, U.; Dobrev, D.; Heijman, J. Cardiomyocyte calcium handling in health and disease: Insights from in vitro and in silico studies. Prog. Biophys. Mol. Biol. 2020, 157, 54–75. [Google Scholar] [CrossRef]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef]
- Finkel, T.; Menazza, S.; Holmstrom, K.M.; Parks, R.J.; Liu, J.; Sun, J.; Liu, J.; Pan, X.; Murphy, E. The ins and outs of mitochondrial calcium. Circ. Res. 2015, 116, 1810–1819. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.; Matsuoka, S. Cytoplasmic Na+-dependent modulation of mitochondrial Ca2+ via electrogenic mitochondrial Na+-Ca2+ exchange. J. Physiol. 2008, 586, 1683–1697. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Enhancing mitochondrial Ca2+ uptake in myocytes from failing hearts restores energy supply and demand matching. Circ. Res. 2008, 103, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Paucek, P. Mitochondrial potassium transport: The K(+) cycle. Biochim. Biophys. Acta. 2003, 1606, 23–41. [Google Scholar] [CrossRef]
- Kaasik, A.; Safiulina, D.; Zharkovsky, A.; Veksler, V. Regulation of mitochondrial matrix volume. Am. J. Physiol. Cell Physiol. 2007, 292, C157–C163. [Google Scholar] [CrossRef] [PubMed]
- Djousse, L.; Benkeser, D.; Arnold, A.; Kizer, J.R.; Zieman, S.J.; Lemaitre, R.N.; Tracy, R.P.; Gottdiener, J.S.; Mozaffarian, D.; Siscovick, D.S.; et al. Plasma free fatty acids and risk of heart failure: The Cardiovascular Health Study. Circ. Heart Fail. 2013, 6, 964–969. [Google Scholar] [CrossRef]
- Khawaja, O.; Bartz, T.M.; Ix, J.H.; Heckbert, S.R.; Kizer, J.R.; Zieman, S.J.; Mukamal, K.J.; Tracy, R.P.; Siscovick, D.S.; Djousse, L. Plasma free fatty acids and risk of atrial fibrillation (from the Cardiovascular Health Study). Am. J. Cardiol. 2012, 110, 212–216. [Google Scholar] [CrossRef]
- Choi, J.Y.; Jung, J.M.; Kwon, D.Y.; Park, M.H.; Kim, J.H.; Oh, K.; Koh, S.B.; Seo, W.K. Free fatty acid as an outcome predictor of atrial fibrillation-associated stroke. Ann. Neurol. 2016, 79, 317–325. [Google Scholar] [CrossRef]
- Shingu, Y.; Takada, S.; Yokota, T.; Shirakawa, R.; Yamada, A.; Ooka, T.; Katoh, H.; Kubota, S.; Matsui, Y. Correlation between increased atrial expression of genes related to fatty acid metabolism and autophagy in patients with chronic atrial fibrillation. PLoS ONE 2020, 15, e0224713. [Google Scholar] [CrossRef]
- Rennison, J.H.; Li, L.; Lin, C.R.; Lovano, B.S.; Castel, L.; Wass, S.Y.; Cantlay, C.C.; McHale, M.; Gillinov, A.M.; Mehra, R.; et al. Atrial fibrillation rhythm is associated with marked changes in metabolic and myofibrillar protein expression in left atrial appendage. Pflugers Arch. 2021, 473, 461–475. [Google Scholar] [CrossRef]
- Liu, Y.; Geng, J.; Liu, Y.; Li, Y.; Shen, J.; Xiao, X.; Sheng, L.; Yang, B.; Cheng, C.; Li, W. beta3-adrenoceptor mediates metabolic protein remodeling in a rabbit model of tachypacing-induced atrial fibrillation. Cell Physiol. Biochem. 2013, 32, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Z.; Hou, T.T.; Yuan, Y.; Hang, P.Z.; Zhao, J.J.; Sun, L.; Zhao, G.Q.; Zhao, J.; Dong, J.M.; Wang, X.B.; et al. Fenofibrate inhibits atrial metabolic remodelling in atrial fibrillation through PPAR-alpha/sirtuin 1/PGC-1alpha pathway. Br. J. Pharmacol. 2016, 173, 1095–1109. [Google Scholar] [CrossRef] [PubMed]
- Shingu, Y.; Yokota, T.; Takada, S.; Niwano, H.; Ooka, T.; Katoh, H.; Tachibana, T.; Kubota, S.; Matsui, Y. Decreased gene expression of fatty acid binding protein 3 in the atrium of patients with new onset of atrial fibrillation in cardiac perioperative phase. J. Cardiol. 2018, 71, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Fernandez, C.; Melander, O.; Ottosson, F. Altered Acylcarnitine Metabolism Is Associated With an Increased Risk of Atrial Fibrillation. J. Am. Heart Assoc. 2020, 9, e016737. [Google Scholar] [CrossRef]
- Harskamp, R.E.; Granger, T.M.; Clare, R.M.; White, K.R.; Lopes, R.D.; Pieper, K.S.; Granger, C.B.; Newgard, C.B.; Shah, S.H.; Newby, L.K. Peripheral blood metabolite profiles associated with new onset atrial fibrillation. Am. Heart J. 2019, 211, 54–59. [Google Scholar] [CrossRef]
- Strand, E.; Pedersen, E.R.; Svingen, G.F.; Olsen, T.; Bjorndal, B.; Karlsson, T.; Dierkes, J.; Njolstad, P.R.; Mellgren, G.; Tell, G.S.; et al. Serum Acylcarnitines and Risk of Cardiovascular Death and Acute Myocardial Infarction in Patients With Stable Angina Pectoris. J. Am. Heart Assoc. 2017, 6, e003620. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.E.; Evans, A.M. Carnitine and acylcarnitines: Pharmacokinetic, pharmacological and clinical aspects. Clin. Pharmacokinet. 2012, 51, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Lenski, M.; Schleider, G.; Kohlhaas, M.; Adrian, L.; Adam, O.; Tian, Q.; Kaestner, L.; Lipp, P.; Lehrke, M.; Maack, C.; et al. Arrhythmia causes lipid accumulation and reduced glucose uptake. Basic Res Cardiol. 2015, 110, 40. [Google Scholar] [CrossRef] [PubMed]
- Miles, J.M.; Wooldridge, D.; Grellner, W.J.; Windsor, S.; Isley, W.L.; Klein, S.; Harris, W.S. Nocturnal and postprandial free fatty acid kinetics in normal and type 2 diabetic subjects: Effects of insulin sensitization therapy. Diabetes 2003, 52, 675–681. [Google Scholar] [CrossRef]
- Gowen, B.H.; Reyes, M.V.; Joseph, L.C.; Morrow, J.P. Mechanisms of Chronic Metabolic Stress in Arrhythmias. Antioxidants 2020, 9, 1012. [Google Scholar] [CrossRef]
- Lambertucci, R.H.; Hirabara, S.M.; Ldos, R.S.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef]
- Nakamura, S.; Takamura, T.; Matsuzawa-Nagata, N.; Takayama, H.; Misu, H.; Noda, H.; Nabemoto, S.; Kurita, S.; Ota, T.; Ando, H.; et al. Palmitate induces insulin resistance in H4IIEC3 hepatocytes through reactive oxygen species produced by mitochondria. J. Biol. Chem. 2009, 284, 14809–14818. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Nam, S.M.; Kim, J.H.; Das, R.; Choi, S.K.; Nguyen, T.T.; Quan, X.; Choi, S.J.; Chung, C.H.; Lee, E.Y.; et al. Palmitate induces ER calcium depletion and apoptosis in mouse podocytes subsequent to mitochondrial oxidative stress. Cell Death Dis. 2015, 6, e1976. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Ladriere, L.; Igoillo-Esteve, M.; Moura, R.F.; Cunha, D.A. Causes and cures for endoplasmic reticulum stress in lipotoxic beta-cell dysfunction. Diabetes Obes. Metab. 2010, 12 (Suppl. 2), 76–82. [Google Scholar] [CrossRef]
- Hu, H.J.; Zhang, C.; Tang, Z.H.; Qu, S.L.; Jiang, Z.S. Regulating the Warburg effect on metabolic stress and myocardial fibrosis remodeling and atrial intracardiac waveform activity induced by atrial fibrillation. Biochem. Biophys. Res. Commun. 2019, 516, 653–660. [Google Scholar] [CrossRef]
- Bedi, K.C., Jr.; Snyder, N.W.; Brandimarto, J.; Aziz, M.; Mesaros, C.; Worth, A.J.; Wang, L.L.; Javaheri, A.; Blair, I.A.; Margulies, K.B.; et al. Evidence for Intramyocardial Disruption of Lipid Metabolism and Increased Myocardial Ketone Utilization in Advanced Human Heart Failure. Circulation 2016, 133, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Ho, K.L.; Karwi, Q.G.; Wagg, C.; Zhang, L.; Vo, K.; Altamimi, T.; Uddin, G.M.; Ussher, J.R.; Lopaschuk, G.D. Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc. Res. 2021, 117, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Klos, M.; Morgenstern, S.; Hicks, K.; Suresh, S.; Devaney, E.J. The effects of the ketone body beta-hydroxybutyrate on isolated rat ventricular myocyte excitation-contraction coupling. Arch. Biochem. Biophys. 2019, 662, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Zhang, C.; Xie, M. Beta-Hydroxybutyrate, Friend or Foe for Stressed Hearts. Front. Aging 2021, 2, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Daskalopoulos, E.P.; Dufeys, C.; Beauloye, C.; Bertrand, L.; Horman, S. AMPK in Cardiovascular Diseases. Exp. Suppl. 2016, 107, 179–201. [Google Scholar]
- Harada, M.; Nattel, S.N.; Nattel, S. AMP-activated protein kinase: Potential role in cardiac electrophysiology and arrhythmias. Circ. Arrhythm. Electrophysiol. 2012, 5, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zou, M.H. AMPK, Mitochondrial Function, and Cardiovascular Disease. Int. J. Mol. Sci. 2020, 21, 4987. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, Y.; Sato, K.; Pimentel, D.R.; Sam, F.; Shaw, R.J.; Dyck, J.R.; Walsh, K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 2009, 284, 35839–35849. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Battaglia, E.; Young, R.; Suzuki, G. LKB1 knockout mouse develops spontaneous atrial fibrillation and provides mechanistic insights into human disease process. J. Am. Heart Assoc. 2015, 4, e001733. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Dixit, G.; Li, Z. Activation of AMP-Activated Protein Kinases Prevents Atrial Fibrillation. J. Cardiovasc. Transl. Res. 2021, 14, 492–502. [Google Scholar] [CrossRef]
- Harada, M.; Tadevosyan, A.; Qi, X.; Xiao, J.; Liu, T.; Voigt, N.; Karck, M.; Kamler, M.; Kodama, I.; Murohara, T.; et al. Atrial Fibrillation Activates AMP-Dependent Protein Kinase and its Regulation of Cellular Calcium Handling: Potential Role in Metabolic Adaptation and Prevention of Progression. J. Am. Coll. Cardiol. 2015, 66, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Liu, Y.; Tu, T.; Li, B.; Xiao, Y.; Ma, Y.; Qin, F.; Xie, J.; Zhou, S.; Liu, Q. Metformin regulates lipid metabolism in a canine model of atrial fibrillation through AMPK/PPAR-alpha/VLCAD pathway. Lipids Health Dis. 2019, 18, 109. [Google Scholar] [CrossRef] [PubMed]
- Bensinger, S.J.; Christofk, H.R. New aspects of the Warburg effect in cancer cell biology. Semin. Cell Dev. Biol. 2012, 23, 352–361. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Heiden, M.G.V. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xu, X.; Si, L.; Xue, L.; Zhang, S.; Qin, J.; Wu, Y.; Shao, Y.; Chen, Y.; Wang, X. Intracellular lactate signaling cascade in atrial remodeling of mitral valvular patients with atrial fibrillation. J. Cardiothorac. Surg. 2013, 8, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jie, Q.Q.; Li, G.; Duan, J.B.; Li, X.B.; Yang, W.; Chu, Y.P.; Yu, S.D.; Liu, X.Y.; Wang, C.Y.; Liu, F.F.; et al. Remodeling of myocardial energy and metabolic homeostasis in a sheep model of persistent atrial fibrillation. Biochem. Biophys. Res. Commun. 2019, 517, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.F.; Chen, Y.J.; Lin, Y.J.; Chen, S.A. Inflammation and the pathogenesis of atrial fibrillation. Nat. Rev. Cardiol. 2015, 12, 230–243. [Google Scholar] [CrossRef] [PubMed]
- Horng, T. Calcium signaling and mitochondrial destabilization in the triggering of the NLRP3 inflammasome. Trends Immunol. 2014, 35, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Kunzel, S.; et al. Atrial Myocyte NLRP3/CaMKII Nexus Forms a Substrate for Postoperative Atrial Fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.; Brickey, W.J.; Ting, J.P. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Guo, M.; Zeng, W.; Zeng, M.; Zhang, X.; Zhang, Y.; Zhang, W.; Jiang, X.; Yu, B. Interactions between NLRP3 inflammasome and glycolysis in macrophages: New insights into chronic inflammation pathogenesis. Immun. Inflamm. Dis. 2022, 10, e581. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Nakahira, K.; Chung, K.P.; DeNicola, G.M.; Koo, M.J.; Pabon, M.A.; Rooney, K.T.; Yoon, J.H.; Ryter, S.W.; Stout-Delgado, H.; et al. NOX4-dependent fatty acid oxidation promotes NLRP3 inflammasome activation in macrophages. Nat. Med. 2016, 22, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.H.; Wu, L.S.; Chiou, M.J.; Liu, J.R.; Yu, K.H.; Kuo, C.F.; Wen, M.S.; Chen, W.J.; Yeh, Y.H.; See, L.C. Association of metformin with lower atrial fibrillation risk among patients with type 2 diabetes mellitus: A population-based dynamic cohort and in vitro studies. Cardiovasc. Diabetol. 2014, 13, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.H. Metformin Use Is Associated With a Lower Incidence of Hospitalization for Atrial Fibrillation in Patients With Type 2 Diabetes Mellitus. Front. Med. 2020, 7, 592901. [Google Scholar] [CrossRef] [PubMed]
- Bueno, N.B.; de Melo, I.S.; de Oliveira, S.L.; Ataide, T.d. Very-low-carbohydrate ketogenic diet v. low-fat diet for long-term weight loss: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2013, 110, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, M.S.; Dong, T.A.; Ragazzo, G.; Dhindsa, D.S.; Mehta, A.; Sandesara, P.B.; Freeman, A.M.; Taub, P.; Sperling, L.S. From Fad to Fact: Evaluating the Impact of Emerging Diets on the Prevention of Cardiovascular Disease. Am. J. Med. 2020, 133, 1126–1134. [Google Scholar] [CrossRef]
- Al-Zaid, N.S.; Dashti, H.M.; Mathew, T.C.; Juggi, J.S. Low carbohydrate ketogenic diet enhances cardiac tolerance to global ischaemia. Acta Cardiol. 2007, 62, 381–389. [Google Scholar] [CrossRef]
- Krebs, P.; Fan, W.; Chen, Y.H.; Tobita, K.; Downes, M.R.; Wood, M.R.; Sun, L.; Li, X.; Xia, Y.; Ding, N.; et al. Lethal mitochondrial cardiomyopathy in a hypomorphic Med30 mouse mutant is ameliorated by ketogenic diet. Proc. Natl. Acad. Sci. USA 2011, 108, 19678–19682. [Google Scholar] [CrossRef]
- Jarrett, S.G.; Milder, J.B.; Liang, L.P.; Patel, M. The ketogenic diet increases mitochondrial glutathione levels. J. Neurochem. 2008, 106, 1044–1051. [Google Scholar] [CrossRef]
- Milder, J.B.; Liang, L.P.; Patel, M. Acute oxidative stress and systemic Nrf2 activation by the ketogenic diet. Neurobiol. Dis. 2010, 40, 238–244. [Google Scholar] [CrossRef]
- Bough, K.J.; Wetherington, J.; Hassel, B.; Pare, J.F.; Gawryluk, J.W.; Greene, J.G.; Shaw, R.; Smith, Y.; Geiger, J.D.; Dingledine, R.J. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Ann. Neurol. 2006, 60, 223–235. [Google Scholar] [CrossRef]
- Elamin, M.; Ruskin, D.N.; Masino, S.A.; Sacchetti, P. Ketogenic Diet Modulates NAD(+)-Dependent Enzymes and Reduces DNA Damage in Hippocampus. Front. Cell Neurosci. 2018, 12, 263. [Google Scholar] [CrossRef]
- Pinto, A.; Bonucci, A.; Maggi, E.; Corsi, M.; Businaro, R. Anti-Oxidant and Anti-Inflammatory Activity of Ketogenic Diet: New Perspectives for Neuroprotection in Alzheimer’s Disease. Antioxidants 2018, 7, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosinski, C.; Jornayvaz, F.R. Effects of Ketogenic Diets on Cardiovascular Risk Factors: Evidence from Animal and Human Studies. Nutrients 2017, 9, 517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhuang, X.; Lin, X.; Zhong, X.; Zhou, H.; Sun, X.; Xiong, Z.; Huang, Y.; Fan, Y.; Guo, Y.; et al. Low-Carbohydrate Diets and Risk of Incident Atrial Fibrillation: A Prospective Cohort Study. J. Am. Heart Assoc. 2019, 8, e011955. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, S.; Zhou, H.; Du, Z.; Liao, X. U-Shaped Relationship between Carbohydrate Intake Proportion and Incident Atrial Fibrillation. J. Am. Coll. Cardiol. 2019, 73, 4. [Google Scholar] [CrossRef]
- Lagiou, P.; Sandin, S.; Lof, M.; Trichopoulos, D.; Adami, H.O.; Weiderpass, E. Low carbohydrate-high protein diet and incidence of cardiovascular diseases in Swedish women: Prospective cohort study. BMJ 2012, 344, e4026. [Google Scholar] [CrossRef]
- Wang, P.; Tate, J.M.; Lloyd, S.G. Low carbohydrate diet decreases myocardial insulin signaling and increases susceptibility to myocardial ischemia. Life Sci. 2008, 83, 836–844. [Google Scholar] [CrossRef]
- Arsyad, A.; Idris, I.; Rasyid, A.A.; Usman, R.A.; Faradillah, K.R.; Latif, W.O.U.; Lubis, Z.I.; Aminuddin, A.; Yustisia, I.; Djabir, Y.Y. Long-Term Ketogenic Diet Induces Metabolic Acidosis, Anemia, and Oxidative Stress in Healthy Wistar Rats. J. Nutr. Metab. 2020, 2020, 3642035. [Google Scholar] [CrossRef]
- Basnet, S.; Tachamo, N.; Nazir, S.; Dhital, R.; Jehangir, A.; Donato, A. Severe anion gap metabolic acidosis associated with initiation of a very low-carbohydrate diet. J. Community Hosp. Intern. Med. Perspect. 2019, 9, 165–167. [Google Scholar] [CrossRef]
- Slade, S.; Ashurst, J. Diet-induced Ketoacidosis in a Non-diabetic: A Case Report. Clin. Pract. Cases Emerg. Med. 2020, 4, 259–262. [Google Scholar] [CrossRef]
- Sikder, K.; Shukla, S.K.; Patel, N.; Singh, H.; Rafiq, K. High Fat Diet Upregulates Fatty Acid Oxidation and Ketogenesis via Intervention of PPAR-gamma. Cell Physiol. Biochem. 2018, 48, 1317–1331. [Google Scholar] [CrossRef]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Joseph, L.C.; Barca, E.; Subramanyam, P.; Komrowski, M.; Pajvani, U.; Colecraft, H.M.; Hirano, M.; Morrow, J.P. Inhibition of NAPDH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS ONE 2016, 11, e0145750. [Google Scholar]
- Liu, J.; Chang, F.; Li, F.; Fu, H.; Wang, J.; Zhang, S.; Zhao, J.; Yin, D. Palmitate promotes autophagy and apoptosis through ROS-dependent JNK and p38 MAPK. Biochem. Biophys. Res. Commun. 2015, 463, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.K.; Ha, C.M.; Thoudam, T.; Cha, S.K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.K.; Park, K.S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, I.B.; O’Toole, P.W. Diet-microbiota interactions and their implications for healthy living. Nutrients 2013, 5, 234–252. [Google Scholar] [CrossRef]
- Crosby, L.; Davis, B.; Joshi, S.; Jardine, M.; Paul, J.; Neola, M.; Barnard, N.D. Ketogenic Diets and Chronic Disease: Weighing the Benefits Against the Risks. Front. Nutr. 2021, 8, 403. [Google Scholar] [CrossRef]
- Best, T.H.; Franz, D.N.; Gilbert, D.L.; Nelson, D.P.; Epstein, M.R. Cardiac complications in pediatric patients on the ketogenic diet. Neurology 2000, 54, 2328–2330. [Google Scholar] [CrossRef]
- Khandelwal, D.; Kishor, K.; Kalra, S. Cardiac Rhythm Vigilance in Ketogenic Diet. Eur. J. Arrhythmia Electrophysiol. 2018, 4, 51–52. [Google Scholar]
- Nielsen, R.; Moller, N.; Gormsen, L.C.; Tolbod, L.P.; Hansson, N.H.; Sorensen, J.; Harms, H.J.; Frokiaer, J.; Eiskjaer, H.; Jespersen, N.R.; et al. Cardiovascular Effects of Treatment With the Ketone Body 3-Hydroxybutyrate in Chronic Heart Failure Patients. Circulation 2019, 139, 2129–2141. [Google Scholar] [CrossRef]
- Monzo, L.; Sedlacek, K.; Hromanikova, K.; Tomanova, L.; Borlaug, B.A.; Jabor, A.; Kautzner, J.; Melenovsky, V. Myocardial ketone body utilization in patients with heart failure: The impact of oral ketone ester. Metabolism 2021, 115, 154452. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Svart, M.; Thomsen, H.H.; Sondergaard, E.; Vendelbo, M.H.; Christensen, N.; Tolbod, L.P.; Harms, H.J.; Nielsen, R.; Wiggers, H.; et al. Ketone Body Infusion With 3-Hydroxybutyrate Reduces Myocardial Glucose Uptake and Increases Blood Flow in Humans: A Positron Emission Tomography Study. J. Am. Heart Assoc. 2017, 6, e005066. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.; Soto-Mota, A.; Willis, D.R.; Ellis, J.; Procter, N.E.K.; Greenwood, R.; Saunders, N.; Schulte, R.F.; Vassiliou, V.S.; Tyler, D.J.; et al. Evaluation of Acute Supplementation With the Ketone Ester (R)-3-Hydroxybutyl-(R)-3-Hydroxybutyrate (deltaG) in Healthy Volunteers by Cardiac and Skeletal Muscle (31)P Magnetic Resonance Spectroscopy. Front. Physiol. 2022, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Takahara, S.; Soni, S.; Phaterpekar, K.; Kim, T.T.; Maayah, Z.H.; Levasseur, J.L.; Silver, H.L.; Freed, D.H.; Ferdaoussi, M.; Dyck, J.R.B. Chronic exogenous ketone supplementation blunts the decline of cardiac function in the failing heart. ESC Heart Fail. 2021, 8, 5606–5612. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.; Tang, Y.; Yue, X.; Gao, R.; Yao, W.; Zhou, Y.; Zhang, H. β-Hydroxybutyrate Mitigated Heart Failure with Preserved Ejection Fraction by Increasing Treg Cells via Nox2/GSK-3β. J. Inflamm. Res. 2021, 14, 4697–4706. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by beta-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Kober, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Belohlavek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef]
- Nishinarita, R.; Niwano, S.; Niwano, H.; Nakamura, H.; Saito, D.; Sato, T.; Matsuura, G.; Arakawa, Y.; Kobayashi, S.; Shirakawa, Y.; et al. Canagliflozin Suppresses Atrial Remodeling in a Canine Atrial Fibrillation Model. J. Am. Heart Assoc. 2021, 10, e017483. [Google Scholar] [CrossRef]
- Li, H.L.; Lip, G.Y.H.; Feng, Q.; Fei, Y.; Tse, Y.K.; Wu, M.Z.; Ren, Q.W.; Tse, H.F.; Cheung, B.Y.; Yiu, K.H. Sodium-glucose cotransporter 2 inhibitors (SGLT2i) and cardiac arrhythmias: A systematic review and meta-analysis. Cardiovasc. Diabetol. 2021, 20, 100. [Google Scholar] [CrossRef]
- Okunrintemi, V.; Mishriky, B.M.; Powell, J.R.; Cummings, D.M. Sodium-glucose co-transporter-2 inhibitors and atrial fibrillation in the cardiovascular and renal outcome trials. Diabetes Obes. Metab. 2021, 23, 276–280. [Google Scholar] [CrossRef]
- Zelniker, T.A.; Bonaca, M.P.; Furtado, R.H.M.; Mosenzon, O.; Kuder, J.F.; Murphy, S.A.; Bhatt, D.L.; Leiter, L.A.; McGuire, D.K.; Wilding, J.P.H.; et al. Effect of Dapagliflozin on Atrial Fibrillation in Patients With Type 2 Diabetes Mellitus: Insights From the DECLARE-TIMI 58 Trial. Circulation 2020, 141, 1227–1234. [Google Scholar] [CrossRef]
- Yurista, S.R.; Sillje, H.H.W.; Oberdorf-Maass, S.U.; Schouten, E.M.; Giani, M.G.P.; Hillebrands, J.L.; van Goor, H.; van Veldhuisen, D.J.; de Boer, R.A.; Westenbrink, B.D. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur. J. Heart Fail. 2019, 21, 862–873. [Google Scholar] [CrossRef] [PubMed]
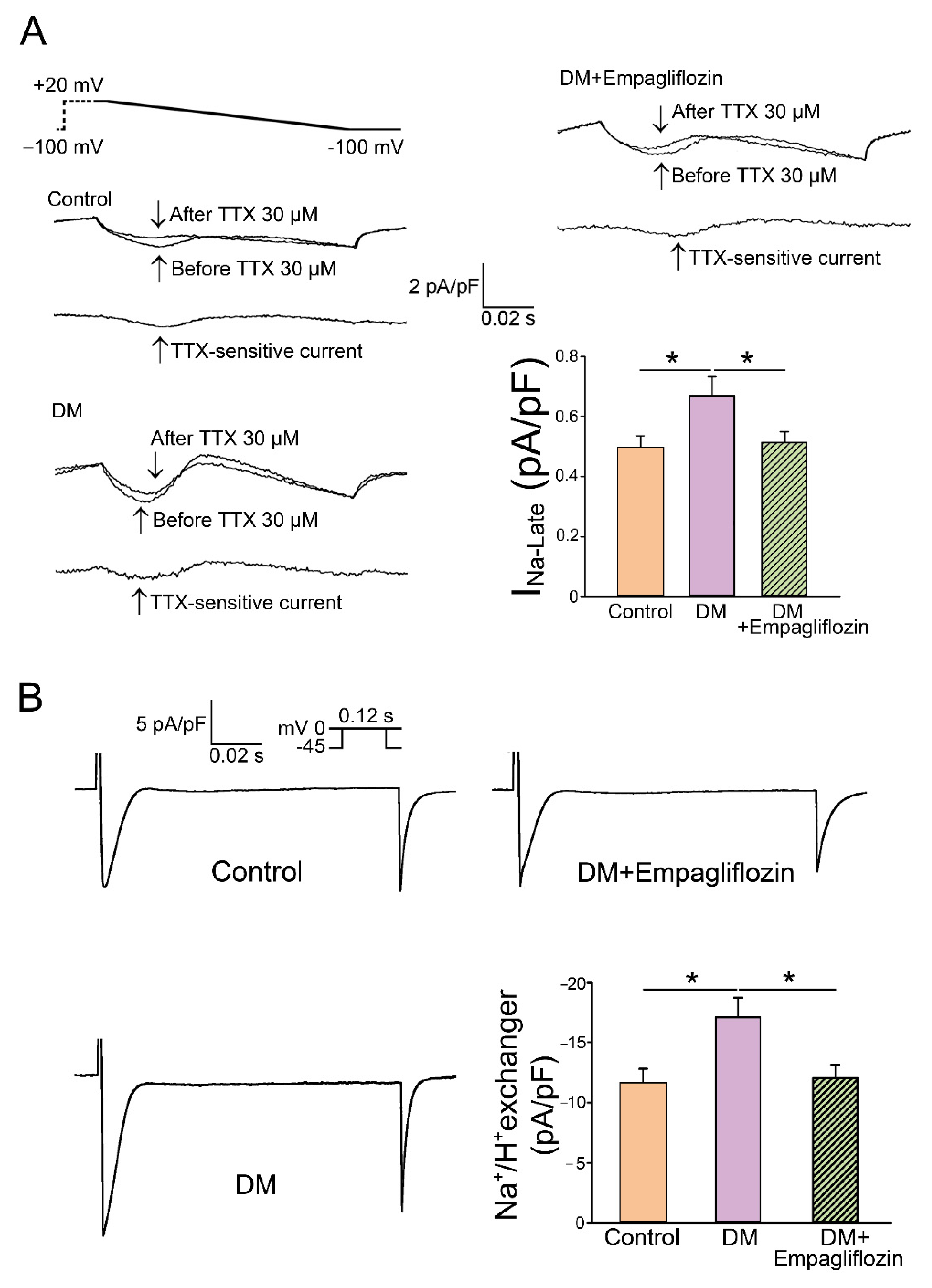
- Uthman, L.; Baartscheer, A.; Bleijlevens, B.; Schumacher, C.A.; Fiolet, J.W.T.; Koeman, A.; Jancev, M.; Hollmann, M.W.; Weber, N.C.; Coronel, R.; et al. Class effects of SGLT2 inhibitors in mouse cardiomyocytes and hearts: Inhibition of Na(+)/H(+) exchanger, lowering of cytosolic Na(+) and vasodilation. Diabetologia 2018, 61, 722–726. [Google Scholar] [CrossRef] [PubMed]
- Trum, M.; Riechel, J.; Lebek, S.; Pabel, S.; Sossalla, S.T.; Hirt, S.; Arzt, M.; Maier, L.S.; Wagner, S. Empagliflozin inhibits Na(+) /H(+) exchanger activity in human atrial cardiomyocytes. ESC Heart Fail. 2020, 7, 4429–4437. [Google Scholar] [CrossRef] [PubMed]
- Bay, J.; Kohlhaas, M.; Maack, C. Intracellular Na(+) and cardiac metabolism. J. Mol. Cell Cardiol. 2013, 61, 20–27. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Study Subjects | Intervention | Duration of Intervention | Main Outcomes |
|---|---|---|---|---|
| Luca Monzo et al. [160] | 11 HFrEF patients 6 controls | Oral administration of 25 g of KE drink, 38% solution in water | 80-min | 12.9-fold increase of βOHB in peripheral venous Increased utilization of βOHB in HF group Fractional extraction of βOHB directly correlated with HF severity No relationship between enhanced βOHB extraction and FFA or glucose extraction Positive correlation between enhanced βOHB and lactate fractional extraction |
| Roni Nielsen et al. [159] | 24 HFrEF patients 10 controls | 7.5% Na-3-OHB infusion | 3-h | Dose-dependent increase of circulating plasma βOHB levels Improved cardiac output by 40% and LVEF by 8% Improved myocardial oxygen consumption without worsening mechanoenergetic coupling |
| Lars C Gormsen et al. [161] | 8 healthy individuals | 7.5% Na-3-OHB infusion | 390-min | Increased circulating βOHB levels Increased myocardial blood flow by 75% Circulating lactate levels were increased by 35% Halved myocardial glucose uptake No changes in FFA uptake and oxidative capacity |
| Donnie Cameron et al. [162] | 28 healthy individuals | Oral administration of 25 g of KE drink | 30-min | Mild ketosis Decreased blood glucose, lactate, and fatty acid concentrations |
| Study | Study Subjects | Intervention | Duration of Intervention | Main Outcomes |
|---|---|---|---|---|
| Shengen Liao et al. [164] | HFpEF mice induced by HFD + L-NAME fed | Intraperitoneal injections of βOHB at a dose of 10 mmol/kg | Once per week for 15 weeks | Mitigated diastolic dysfunction, reduced interstitial fibrosis, and cardiomyocyte sizes Prevented cardiomyocyte apoptosis, inflammation and oxidative stress |
| Yan Deng et al. [165] | HFpEF mice induced by HFD + DOCP injection | KE gavage of 1 mg/g body weight | Once per day for 30 days | Elevated myocardial βOHB levels Lowered mitochondrial hyperacetylation, suppressed NLRP3 inflammasome formation, elevated citrate synthase activity, inhibited fatty acid uptake |
| Shingo Takahara et al. [163] | HF mice induced by TAC surgery | 20% KE (8.0 g ketones/kg/day) via drinking water or acute infusions of βOHB | 2 weeks supplementation or 1 h infusions | Elevated blood βOHB levels, improved cardiac EF Reduced cardiac fibroblasts and collagen deposition Greater cardiac output No changes in blood glucose, fatty acid, and insulin levels |
| Salva R Yurista et al. [33] | HF mice induced by TAC/MI and in rats by Post-MI | Preventive KE-1 (10% w/w) or treatment KE-2 (3.4 ± 1 g per day) diet | KE-1 for 5 weeks KE-2 for 4 to 6 weeks | Induced sustained ketonemia Improved LVEF, reduced LV-ESV and LV-EDV Induction of genes involved in the myocardial uptake and oxidation of βOHB Restored myocardial ATP production. |
| Julie L Horton et al. [34] | Canine dilated cardiomyopathy induced by cardiac tachypacing | 5 μmol/kg/min infusions of sodium-βOHB, 55 mL/day | 14 days | 2.5-fold higher plasma concentration of βOHB with increasing ketone body uptake Suppressed myocardial glucose uptake and its oxidation rates, reduced net lactate uptake Prevented myocardial oxygen consumption, increased mechanical efficiency by 30% Prevented changes in LVEDP, cardiac output, LV dilatation, |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lkhagva, B.; Lee, T.-W.; Lin, Y.-K.; Chen, Y.-C.; Chung, C.-C.; Higa, S.; Chen, Y.-J. Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention. Cells 2022, 11, 2915. https://doi.org/10.3390/cells11182915
Lkhagva B, Lee T-W, Lin Y-K, Chen Y-C, Chung C-C, Higa S, Chen Y-J. Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention. Cells. 2022; 11(18):2915. https://doi.org/10.3390/cells11182915
Chicago/Turabian StyleLkhagva, Baigalmaa, Ting-Wei Lee, Yung-Kuo Lin, Yao-Chang Chen, Cheng-Chih Chung, Satoshi Higa, and Yi-Jen Chen. 2022. "Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention" Cells 11, no. 18: 2915. https://doi.org/10.3390/cells11182915
APA StyleLkhagva, B., Lee, T. -W., Lin, Y. -K., Chen, Y. -C., Chung, C. -C., Higa, S., & Chen, Y. -J. (2022). Disturbed Cardiac Metabolism Triggers Atrial Arrhythmogenesis in Diabetes Mellitus: Energy Substrate Alternate as a Potential Therapeutic Intervention. Cells, 11(18), 2915. https://doi.org/10.3390/cells11182915