
The Role of the miR-17-92 Cluster in Autophagy and Atherosclerosis Supports Its Link to Lysosomal Storage Diseases

 ,
, 
 ,
,  and
and 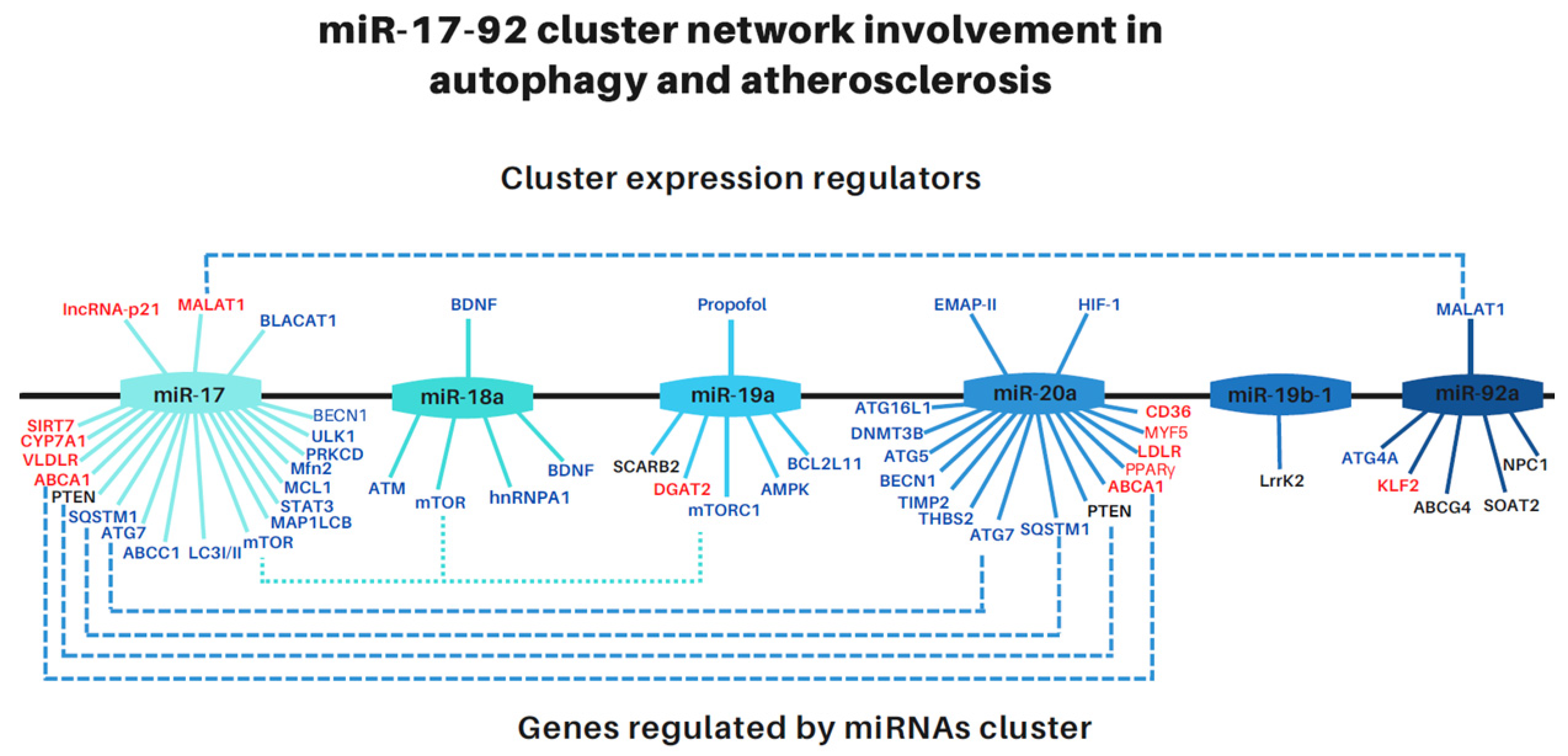{kind=link}
Abstract
:1. Introduction
2. Role of miR-17-92 Cluster Members in Regulating Cellular Energetic Metabolism
2.1. MiR-17-92 Family Members Have Been Identified as Key Regulators in Atherosclerosis
2.2. MiR-17-92 Family Members Participate in the Regulation of Autophagy
2.3. MALAT1, a Long Non-Coding RNA That Binds miRNAs of the miR-17-92 Cluster, Is Also Involved in the Regulation of Atherosclerosis and Autophagy
3. Lysosomes Are Fundamental Organelles for Cellular Metabolism and Autophagy
3.1. Lysosomal Storage Diseases Affect Cellular Metabolism
3.2. MiRNAs of miR-17-92 Cluster Involved in LSD and NPC
3.3. Niemann–Picks Disease Type C and the Possible Involvement of miR-17-92 Cluster in It
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Saliminejad, K.; Khorram Khorshid, H.R.; Soleymani Fard, S.; Ghaffari, S.H. An overview of microRNAs: Biology, functions, therapeutics, and analysis methods. J. Cell. Physiol. 2019, 234, 5451–5465. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Lai, X.; Vera, J. MicroRNA Clusters. In Encyclopedia of Systems Biology; Dubitzky, W., Wolkenhauer, O., Cho, K.-H., Yokota, H., Eds.; Springer: New York, NY, USA, 2013; pp. 1310–1314. [Google Scholar]
- Mendell, J.T. miRiad Roles for the miR-17-92 Cluster in Development and Disease. Cell 2008, 133, 217–222. [Google Scholar] [CrossRef]
- Tan, W.; Li, Y.; Lim, S.G.; Tan, T.M. miR-106b-25/miR-17-92 clusters: Polycistrons with oncogenic roles in hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 5962–5972. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the miR-17-92 Cluster of miRNAs in Cancer. Front. Med. 2015, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Bonauer, A.; Dimmeler, S. The microRNA-17~92 cluster: Still a miRacle? Cell Cycle 2009, 8, 3866–3873. [Google Scholar] [CrossRef]
- Yang, P.; Cai, L.; Zhang, G.; Bian, Z.; Han, G. The role of the miR-17-92 cluster in neurogenesis and angiogenesis in the central nervous system of adults. J. Neurosci. Res. 2017, 95, 1574–1581. [Google Scholar] [CrossRef] [PubMed]
- Tréguer, K.; Heinrich, E.-M.; Ohtani, K.; Bonauer, A.; Dimmeler, S. Role of the MicroRNA-17–92 Cluster in the Endothelial Differentiation of Stem Cells. J. Vasc. Res. 2012, 49, 447–460. [Google Scholar] [CrossRef]
- Mogilyansky, E.; Rigoutsos, I. The miR-17/92 cluster: A comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Izreig, S.; Samborska, B.; Johnson, R.M.; Sergushichev, A.; Ma, E.H.; Lussier, C.; Loginicheva, E.; Donayo, A.O.; Poffenberger, M.C.; Sagan, S.M.; et al. The miR-17∼92 microRNA Cluster Is a Global Regulator of Tumor Metabolism. Cell Rep. 2016, 16, 1915–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rencelj, A.; Gvozdenovic, N.; Cemazar, M. MitomiRs: Their roles in mitochondria and importance in cancer cell metabolism. Radiol. Oncol. 2021, 55, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-L.; Jia, Y.-J.; Xing, B.-H.; Shi, D.-D.; Dong, X.-J. Plasma microRNA-16-5p, -17-5p and -20a-5p: Novel diagnostic biomarkers for gestational diabetes mellitus. J. Obstet. Gynaecol. Res. 2017, 43, 974–981. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhang, X.; Zhou, H. Role of cell free microRNA-19a and microRNA-19b in gestational diabetes mellitus patients. 3 Biotech 2019, 9, 406. [Google Scholar] [CrossRef]
- Zhu, Y.; Tian, F.; Li, H.; Zhou, Y.; Lu, J.; Ge, Q. Profiling maternal plasma microRNA expression in early pregnancy to predict gestational diabetes mellitus. Int. J. Gynecol. Obstet. 2015, 130, 49–53. [Google Scholar] [CrossRef]
- Liu, F.; Li, R.; Zhang, Y.; Qiu, J.; Ling, W. Association of Plasma MiR-17-92 With Dyslipidemia in Patients With Coronary Artery Disease. Medicine 2014, 93, e98. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Lv, X.; Liu, F. MiRNA-17 encoded by the miR-17-92 cluster increases the potential for steatosis in hepatoma cells by targeting CYP7A1. Cell. Mol. Biol. Lett. 2018, 23, 16. [Google Scholar] [CrossRef]
- Bobryshev, Y.V.; Shchelkunova, T.A.; Morozov, I.A.; Rubtsov, P.M.; Sobenin, I.A.; Orekhov, A.N.; Smirnov, A.N. Changes of lysosomes in the earliest stages of the development of atherosclerosis. J. Cell. Mol. Med. 2013, 17, 626–635. [Google Scholar] [CrossRef]
- Torzewski, M. The Initial Human Atherosclerotic Lesion and Lipoprotein Modification—A Deep Connection. Int. J. Mol. Sci. 2021, 22, 11488. [Google Scholar] [CrossRef]
- Chen, J.; Xu, L.; Hu, Q.; Yang, S.; Zhang, B.; Jiang, H. MiR-17-5p as circulating biomarkers for the severity of coronary atherosclerosis in coronary artery disease. Int. J. Cardiol. 2015, 197, 123–124. [Google Scholar] [CrossRef]
- Tan, L.; Liu, L.; Jiang, Z.; Hao, X. Inhibition of microRNA-17-5p reduces the inflammation and lipid accumulation, and up-regulates ATP-binding cassette transporterA1 in atherosclerosis. J. Pharmacol. Sci. 2019, 139, 280–288. [Google Scholar] [CrossRef]
- Westerterp, M.; Bochem, A.E.; Yvan-Charvet, L.; Murphy, A.J.; Wang, N.; Tall, A.R. ATP-Binding Cassette Transporters, Atherosclerosis, and Inflammation. Circ. Res. 2014, 114, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Fisher, E.A.; Feig, J.E.; Hewing, B.; Hazen, S.L.; Smith, J.D. High-Density Lipoprotein Function, Dysfunction, and Reverse Cholesterol Transport. Arter. Thromb. Vasc. Biol. 2012, 32, 2813–2820. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tan, L.; Yao, J.; Yang, L. Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Mol. Med. Rep. 2020, 21, 1761–1770. [Google Scholar] [CrossRef]
- Tan, L.; Meng, L.; Shi, X.; Yu, B. Knockdown of microRNA-17-5p ameliorates atherosclerotic lesions in ApoE−/− mice and restores the expression of very low density lipoprotein receptor. Biotechnol. Lett. 2017, 39, 967–976. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; He, F.; Liang, B.; Jing, Y.; Zhang, P.; Liu, W.; Zhao, H. p53-Dependent LincRNA-p21 Protects Against Proliferation and Anti-apoptosis of Vascular Smooth Muscle Cells in Atherosclerosis by Upregulating SIRT7 via MicroRNA-17-5p. J. Cardiovasc. Transl. Res. 2021, 14, 426–440. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Wang, X.; Song, X.; Bai, R.; Yang, H.; Yang, Z.; Xiao, C.; Bian, Y. MicroRNA-20a/b regulates cholesterol efflux through post-transcriptional repression of ATP-binding cassette transporter A1. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, Y.; Ma, J.; Wang, W.; Sun, B.; Zheng, T.; Wei, M.; Sun, Y. MicroRNA-20a participates in the aerobic exercise-based prevention of coronary artery disease by targeting PTEN. Biomed. Pharmacother. 2017, 95, 756–763. [Google Scholar] [CrossRef]
- Güller, I.; McNaughton, S.; Crowley, T.; Gilsanz, V.; Kajimura, S.; Watt, M.; Russell, A.P. Comparative analysis of microRNA expression in mouse and human brown adipose tissue. BMC Genom. 2015, 16, 820. [Google Scholar] [CrossRef]
- Ding, Y.; Zhu, S.; Wu, C.; Qian, L.; Li, D.; Wang, L.; Wang, Y.; Zhang, W.; Yang, M.; Ding, J.; et al. Relationship between porcine miR-20a and its putative target low-density lipoprotein receptor based on dual luciferase reporter gene assays. Asian-Australas. J. Anim. Sci. 2019, 32, 922–929. [Google Scholar] [CrossRef]
- Angerstein, C.; Hecker, M.; Paap, B.K.; Koczan, D.; Thamilarasan, M.; Thiesen, H.-J.; Zettl, U.K. Integration of MicroRNA Databases to Study MicroRNAs Associated with Multiple Sclerosis. Mol. Neurobiol. 2012, 45, 520–535. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Behling, C.; Newbury, R.; Deutsch, R.; Nievergelt, C.; Schork, N.J.; Lavine, J.E. Histopathology of pediatric nonalcoholic fatty liver disease. Hepatology 2005, 42, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ma, Y.; Yang, L.-Y.; Zhao, D. MicroRNA-20a-5p Ameliorates Non-alcoholic Fatty Liver Disease via Inhibiting the Expression of CD36. Front. Cell Dev. Biol. 2020, 8, 596329. [Google Scholar] [CrossRef]
- Dijk, W.; Mattijssen, F.; De la Rosa Rodriguez, M.; Loza Valdes, A.; Loft, A.; Mandrup, S.; Kalkhoven, E.; Qi, L.; Borst, J.W.; Kersten, S. Hypoxia-Inducible Lipid Droplet–Associated Is Not a Direct Physiological Regulator of Lipolysis in Adipose Tissue. Endocrinology 2017, 158, 1231–1251. [Google Scholar] [CrossRef]
- Cheng, J.; Song, Q.; Yang, Y.; Sun, Z.; Tian, X.; Tian, X.; Feng, L. Lipolysis by downregulating miR-92a activates the Wnt/β-catenin signaling pathway in hypoxic rats. Biomed. Rep. 2020, 13, 33. [Google Scholar] [CrossRef]
- Zhong, H.; Zhou, Y.; Xu, Q.; Yan, J.; Zhang, X.; Zhang, H.; Tang, Z.; Xiao, J.; Guo, Z.; Luo, Y.; et al. Low expression of miR-19a-5p is associated with high mRNA expression of diacylglycerol O-acyltransferase 2 (DGAT2) in hybrid tilapia. Genomics 2021, 113, 2392–2399. [Google Scholar] [CrossRef]
- Semo, J.; Chernin, G.; Jonas, M.; Shimoni, S.; George, J. Deletion of the Mir-106b~ 25 MicroRNA cluster attenuates atherosclerosis in Apolipoprotein E knockout mice. Lipids Health Dis. 2019, 18, 208. [Google Scholar] [CrossRef]
- He, Y.; Lin, L.; Cao, J.; Mao, X.; Qu, Y.; Xi, B. Up-regulated miR-93 contributes to coronary atherosclerosis pathogenesis through targeting ABCA1. Int. J. Clin. Exp. Med. 2015, 8, 674–681. [Google Scholar]
- Kim, J.; Yoon, H.; Ramírez, C.M.; Lee, S.-M.; Hoe, H.-S.; Fernández-Hernando, C.; Kim, J. miR-106b impairs cholesterol efflux and increases Aβ levels by repressing ABCA1 expression. Exp. Neurol. 2012, 235, 476–483. [Google Scholar] [CrossRef]
- Borzi, C.; Calzolari, L.; Ferretti, A.M.; Caleca, L.; Pastorino, U.; Sozzi, G.; Fortunato, O. c-Myc shuttled by tumour-derived extracellular vesicles promotes lung bronchial cell proliferation through miR-19b and miR-92a. Cell Death Dis. 2019, 10, 759. [Google Scholar] [CrossRef]
- Kaur, S.; Abu-Shahba, A.G.; Paananen, R.O.; Hongisto, H.; Hiidenmaa, H.; Skottman, H.; Seppänen-Kaijansinkko, R.; Mannerström, B. Small non-coding RNA landscape of extracellular vesicles from human stem cells. Sci. Rep. 2018, 8, 15503. [Google Scholar] [CrossRef]
- Ning, J.; Ye, Y.; Bu, D.; Zhao, G.; Song, T.; Liu, P.; Yu, W.; Wang, H.; Li, H.; Ren, X.; et al. Imbalance of TGF-β1/BMP-7 pathways induced by M2-polarized macrophages promotes hepatocellular carcinoma aggressiveness. Mol. Ther. 2021, 29, 2067–2087. [Google Scholar] [CrossRef]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef]
- Dellago, H.; Bobbili, M.R.; Grillari, J. MicroRNA-17-5p: At the Crossroads of Cancer and Aging—A Mini-Review. Gerontology 2017, 63, 20–28. [Google Scholar] [CrossRef]
- Comincini, S.; Allavena, G.; Palumbo, S.; Morini, M.; Durando, F.; Angeletti, F.; Pirtoli, L.; Miracco, C. microRNA-17 regulates the expression of ATG7 and modulates the autophagy process, improving the sensitivity to temozolomide and low-dose ionizing radiation treatments in human glioblastoma cells. Cancer Biol. Ther. 2013, 14, 574–586. [Google Scholar] [CrossRef]
- Weidberg, H.; Shvets, E.; Elazar, Z. Biogenesis and Cargo Selectivity of Autophagosomes. Annu. Rev. Biochem. 2011, 80, 125–156. [Google Scholar] [CrossRef]
- Huang, F.X.; Chen, H.J.; Zheng, F.X.; Gao, Z.Y.; Sun, P.F.; Peng, Q.; Liu, Y.; Deng, X.; Huang, Y.H.; Zhao, C.; et al. LncRNA BLACAT1 is involved in chemoresistance of non-small cell lung cancer cells by regulating autophagy. Int. J. Oncol. 2019, 54, 339–347. [Google Scholar] [CrossRef]
- Duan, X.; Zhang, T.; Ding, S.; Wei, J.; Su, C.; Liu, H.; Xu, G. microRNA-17-5p Modulates Bacille Calmette-Guerin Growth in RAW264.7 Cells by Targeting ULK1. PLoS ONE 2015, 10, e0138011. [Google Scholar] [CrossRef]
- Kumar, R.; Sahu, S.K.; Kumar, M.; Jana, K.; Gupta, P.; Gupta, U.D.; Kundu, M.; Basu, J. MicroRNA 17-5p regulates autophagy in Mycobacterium tuberculosis-infected macrophages by targeting Mcl-1 and STAT3. Cell. Microbiol. 2016, 18, 679–691. [Google Scholar] [CrossRef]
- Li, S.; Zhang, J.; Wang, Z.; Wang, T.; Yu, Y.; He, J.; Zhang, H.; Yang, T.; Shen, Z. MicroRNA-17 regulates autophagy to promote hepatic ischemia/reperfusion injury via suppression of signal transductions and activation of transcription-3 expression. Liver Transplant. 2016, 22, 1697–1709. [Google Scholar] [CrossRef]
- Hou, W.; Song, L.; Zhao, Y.; Liu, Q.; Zhang, S. Inhibition of Beclin-1-Mediated Autophagy by MicroRNA-17-5p Enhanced the Radiosensitivity of Glioma Cells. Oncol. Res. 2017, 25, 43–53. [Google Scholar] [CrossRef]
- Sun, Y.; Nie, W.; Qiu, B.; Guo, X.; Zhang, J.; Wei, J. Inhibition of microRNA-17 enhances cisplatin-induced apoptosis of human tongue squamous carcinoma cell. J. Bioenerg. Biomembr. 2021, 53, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Miao, D.; Zhu, Q.; Huang, J.; Lu, G.; Xu, W. MicroRNA-17-5p contributes to osteoarthritis progression by binding p62/SQSTM1. Exp. Ther. Med. 2018, 15, 1789–1794. [Google Scholar] [CrossRef] [PubMed]
- He, J.M.; Liu, P.Y.; Wang, J. MicroRNA-17-5p regulates the growth, migration and invasion of the human osteosarcoma cells by modulating the expression of PTEN. J. BUON 2020, 25, 1028–1034. [Google Scholar]
- Xu, X.; Su, Y.-L.; Shi, J.-Y.; Lu, Q.; Chen, C. MicroRNA-17-5p Promotes Cardiac Hypertrophy by Targeting Mfn2 to Inhibit Autophagy. Cardiovasc. Toxicol. 2021, 21, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Yang, Y.; Wu, J.; Song, J.; Lu, J. microRNA-17-5p downregulation inhibits autophagy and myocardial remodelling after myocardial infarction by targeting STAT3. Autoimmunity 2022, 55, 43–51. [Google Scholar] [CrossRef]
- Du, W.W.; Yang, W.; Fang, L.; Xuan, J.; Li, H.; Khorshidi, A.; Gupta, S.; Li, X.; Yang, B.B. miR-17 extends mouse lifespan by inhibiting senescence signaling mediated by MKP7. Cell Death Dis. 2014, 5, e1355. [Google Scholar] [CrossRef]
- Wu, H.; Wang, F.; Hu, S.; Yin, C.; Li, X.; Zhao, S.; Wang, J.; Yan, X. MiR-20a and miR-106b negatively regulate autophagy induced by leucine deprivation via suppression of ULK1 expression in C2C12 myoblasts. Cell. Signal. 2012, 24, 2179–2186. [Google Scholar] [CrossRef]
- Sun, K.-T.; Chen, M.Y.C.; Tu, M.-G.; Wang, I.-K.; Chang, S.-S.; Li, C.-Y. MicroRNA-20a regulates autophagy related protein-ATG16L1 in hypoxia-induced osteoclast differentiation. Bone 2015, 73, 145–153. [Google Scholar] [CrossRef]
- Chen, J.; Liu, L.; Liu, Y.; Liu, X.; Qu, C.; Meng, F.; Ma, J.; Lin, Y.; Xue, Y. Low-Dose Endothelial-Monocyte-Activating Polypeptide-II Induced Autophagy by Down-Regulating miR-20a in U-87 and U-251 Glioma Cells. Front. Cell. Neurosci. 2016, 10, 128. [Google Scholar] [CrossRef]
- Zhao, S.; Yao, D.; Chen, J.; Ding, N.; Ren, F. MiR-20a Promotes Cervical Cancer Proliferation and Metastasis In Vitro and In Vivo. PLoS ONE 2015, 10, e0120905. [Google Scholar] [CrossRef]
- Zhou, Q.; Dong, J.; Luo, R.; Zhou, X.; Wang, J.; Chen, F. MicroRNA-20a regulates cell proliferation, apoptosis and autophagy by targeting thrombospondin 2 in cervical cancer. Eur. J. Pharmacol. 2019, 844, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Mou, Q.; Wang, Y.; Zhu, Z.; Cheng, M. Resveratrol contributes to the inhibition of liver fibrosis by inducing autophagy via the microRNA-20a-mediated activation of the PTEN/PI3K/AKT signaling pathway. Int. J. Mol. Med. 2020, 46, 2035–2046. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhao, J.; Qu, Y.; Yin, R.; Gao, Q.; Ding, S.; Zhang, Y.; Wei, J.; Xu, G. microRNA-20a Inhibits Autophagic Process by Targeting ATG7 and ATG16L1 and Favors Mycobacterial Survival in Macrophage Cells. Front. Cell. Infect. Microbiol. 2016, 6, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; He, J.; Wei, X.; Wan, G.; Lao, Y.; Xu, W.; Li, Z.; Hu, H.; Hu, Z.; Luo, X.; et al. MicroRNA-20a-mediated loss of autophagy contributes to breast tumorigenesis by promoting genomic damage and instability. Oncogene 2017, 36, 5874–5884. [Google Scholar] [CrossRef]
- Li, H.; Lei, Y.; Li, S.; Li, F.; Lei, J. MicroRNA-20a-5p inhibits the autophagy and cisplatin resistance in ovarian cancer via regulating DNMT3B-mediated DNA methylation of RBP1. Reprod. Toxicol. 2022, 109, 93–100. [Google Scholar] [CrossRef]
- Qased, A.B.; Yi, H.; Liang, N.; Ma, S.; Qiao, S.; Liu, X. MicroRNA-18a upregulates autophagy and ataxia telangiectasia mutated gene expression in HCT116 colon cancer cells. Mol. Med. Rep. 2013, 7, 559–564. [Google Scholar] [CrossRef]
- Xu, X.-H.; Ding, D.-F.; Yong, H.-J.; Dong, C.-L.; You, N.; Ye, X.-L.; Pan, M.-L.; Ma, J.-H.; You, Q.; Lu, Y.-B. Resveratrol transcriptionally regulates miRNA-18a-5p expression ameliorating diabetic nephropathy via increasing autophagy. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4952–4965. [Google Scholar]
- Fujiya, M.; Konishi, H.; Mohamed Kamel, M.K.; Ueno, N.; Inaba, Y.; Moriichi, K.; Tanabe, H.; Ikuta, K.; Ohtake, T.; Kohgo, Y. microRNA-18a induces apoptosis in colon cancer cells via the autophagolysosomal degradation of oncogenic heterogeneous nuclear ribonucleoprotein A1. Oncogene 2014, 33, 4847–4856. [Google Scholar] [CrossRef]
- Liang, C.; Zhang, X.; Wang, H.-M.; Liu, X.-M.; Zhang, X.-J.; Zheng, B.; Qian, G.-R.; Ma, Z.-L. MicroRNA-18a-5p functions as an oncogene by directly targeting IRF2 in lung cancer. Cell Death Dis. 2017, 8, e2764. [Google Scholar] [CrossRef]
- Lin, B.; Feng, D.; Xu, J. Cardioprotective effects of microRNA-18a on acute myocardial infarction by promoting cardiomyocyte autophagy and suppressing cellular senescence via brain derived neurotrophic factor. Cell Biosci. 2019, 9, 38. [Google Scholar] [CrossRef]
- Gao, Y.-H.; Qian, J.-Y.; Chen, Z.-W.; Fu, M.-Q.; Xu, J.-F.; Xia, Y.; Ding, X.-F.; Yang, X.-D.; Cao, Y.-Y.; Zou, Y.-Z.; et al. Suppression of Bim by microRNA-19a may protect cardiomyocytes against hypoxia-induced cell death via autophagy activation. Toxicol. Lett. 2016, 257, 72–83. [Google Scholar] [CrossRef]
- Yu, S.; Xin, W.; Jiang, Q.; Li, A. Propofol exerts neuroprotective functions by down-regulating microRNA-19a in glutamic acid-induced PC12 cells. BioFactors 2020, 46, 934–942. [Google Scholar] [CrossRef]
- Rogg, E.-M.; Abplanalp, W.T.; Bischof, C.; John, D.; Schulz, M.H.; Krishnan, J.; Fischer, A.; Poluzzi, C.; Schaefer, L.; Bonauer, A.; et al. Analysis of Cell Type-Specific Effects of MicroRNA-92a Provides Novel Insights Into Target Regulation and Mechanism of Action. Circulation 2018, 138, 2545–2558. [Google Scholar] [CrossRef]
- Xia, W.; Chen, H.; Xie, C.; Hou, M. Long-noncoding RNA MALAT1 sponges microRNA-92a-3p to inhibit doxorubicin-induced cardiac senescence by targeting ATG4a. Aging 2020, 12, 8241–8260. [Google Scholar] [CrossRef] [PubMed]
- Josefs, T.; Boon, R.A. The Long Non-coding Road to Atherosclerosis. Curr. Atheroscler. Rep. 2020, 22, 55. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Maleki, S.S.; Nazari-Jahantigh, M. Regulatory Non-coding RNAs in Atherosclerosis. Handb. Exp. Pharmacol. 2022, 270, 463–492. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Song, D.; Song, X.; Song, C. The role of lncRNA MALAT1 in cardiovascular disease. IUBMB Life 2020, 72, 334–342. [Google Scholar] [CrossRef]
- Fu, S.; Wang, Y.; Li, H.; Chen, L.; Liu, Q. Regulatory Networks of LncRNA MALAT-1 in Cancer. Cancer Manag. Res. 2020, 12, 10181–10198. [Google Scholar] [CrossRef]
- Xu, W.; Ding, M.; Wang, B.; Cai, Y.; Guo, C.; Yuan, C. Molecular Mechanism of the Canonical Oncogenic lncRNA MALAT1 in Gastric Cancer. Curr. Med. Chem. 2021, 28, 8800–8809. [Google Scholar] [CrossRef]
- Sun, Q.; Xu, H.; Xue, J.; Yang, Q.; Chen, C.; Yang, P.; Han, A.; Tu, Q.; Lu, J.; Gao, X.; et al. MALAT1 via microRNA-17 regulation of insulin transcription is involved in the dysfunction of pancreatic β-cells induced by cigarette smoke extract. J. Cell. Physiol. 2018, 233, 8862–8873. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, Q.; Lin, F.; Zhu, L.; Huang, F.; Zhao, L.; Ou, R. Casiopeina II-gly acts on lncRNA MALAT1 by miR-17-5p to inhibit FZD2 expression via the Wnt signaling pathway during the treatment of cervical carcinoma. Oncol. Rep. 2019, 42, 1365–1379. [Google Scholar] [CrossRef]
- Shyu, K.-G.; Wang, B.-W.; Pan, C.-M.; Fang, W.-J.; Lin, C.-M. Hyperbaric oxygen boosts long noncoding RNA MALAT1 exosome secretion to suppress microRNA-92a expression in therapeutic angiogenesis. Int. J. Cardiol. 2019, 274, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.-G.; Wang, B.-W.; Fang, W.-J.; Pan, C.-M.; Lin, C.-M. Hyperbaric oxygen-induced long non-coding RNA MALAT1 exosomes suppress MicroRNA-92a expression in a rat model of acute myocardial infarction. J. Cell. Mol. Med. 2020, 24, 12945–12954. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal Storage Diseases: From Pathophysiology to Therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef]
- Platt, F.M.; D’Azzo, A.; Davidson, B.L.; Neufeld, E.F.; Tifft, C.J. Lysosomal storage diseases. Nat. Rev. Dis. Prim. 2018, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Meikle, P.J.; Hopwood, J.J.; Clague, A.E.; Carey, W.F. Prevalence of Lysosomal Storage Disorders. JAMA 1999, 281, 249–254. [Google Scholar] [CrossRef]
- Platt, F.M.; Boland, B.; van der Spoel, A.C. Lysosomal storage disorders: The cellular impact of lysosomal dysfunction. J. Cell Biol. 2012, 199, 723–734. [Google Scholar] [CrossRef]
- Hassan, S.; Sidransky, E.; Tayebi, N. The role of epigenetics in lysosomal storage disorders: Uncharted territory. Mol. Genet. Metab. 2017, 122, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, M.T.; Pereira, V.G.; do Nascimento, C.C.; D’Almeida, V. The Underexploited Role of Non-Coding RNAs in Lysosomal Storage Diseases. Front. Endocrinol. 2016, 7, 133. [Google Scholar] [CrossRef]
- Goedeke, L.; Fernández-Hernando, C. microRNAs: A connection between cholesterol metabolism and neurodegeneration. Neurobiol. Dis. 2014, 72, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Gleason, A.M.; Woo, E.G.; McKinney, C.; Sidransky, E. The Role of Exosomes in Lysosomal Storage Disorders. Biomolecules 2021, 11, 576. [Google Scholar] [CrossRef]
- Ozsait, B.; Komurcu-Bayrak, E.; Levula, M.; Erginel-Unaltuna, N.; Kähönen, M.; Rai, M.; Lehtimäki, T.; Laaksonen, R. Niemann–Pick type C fibroblasts have a distinct microRNA profile related to lipid metabolism and certain cellular components. Biochem. Biophys. Res. Commun. 2010, 403, 316–321. [Google Scholar] [CrossRef]
- Niculescu, L.S.; Simionescu, N.; Fuior, E.V.; Stancu, C.S.; Carnuta, M.G.; Dulceanu, M.D.; Raileanu, M.; Dragan, E.; Sima, A.V. Inhibition of miR-486 and miR-92a decreases liver and plasma cholesterol levels by modulating lipid-related genes in hyperlipidemic hamsters. Mol. Biol. Rep. 2018, 45, 497–509. [Google Scholar] [CrossRef]
- Futerman, A.H.; Zimran, A. (Eds.) Gaucher Disease; CRC Press: Boca Raton, FL, USA, 2006; ISBN 978-0-429-11455-7. [Google Scholar]
- Siebert, M.; Westbroek, W.; Chen, Y.-C.; Moaven, N.; Li, Y.; Velayati, A.; Saraiva-Pereira, M.L.; Martin, S.E.; Sidransky, E. Identification of miRNAs that modulate glucocerebrosidase activity in Gaucher disease cells. RNA Biol. 2014, 11, 1291–1300. [Google Scholar] [CrossRef]
- Dasgupta, N.; Xu, Y.-H.; Li, R.; Peng, Y.; Pandey, M.K.; Tinch, S.L.; Liou, B.; Inskeep, V.; Zhang, W.; Setchell, K.D.; et al. Neuronopathic Gaucher disease: Dysregulated mRNAs and miRNAs in brain pathogenesis and effects of pharmacologic chaperone treatment in a mouse model. Hum. Mol. Genet. 2015, 24, 7031–7048. [Google Scholar] [CrossRef]
- Neufeld, E.F.; Muenzer, J. The Mucopolysaccharidoses. In The Online Metabolic and Molecular Bases of Inherited Disease; Valle, D.L., Antonarakis, S., Ballabio, A., Beaudet, A.L., Mitchell, G.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2019. [Google Scholar]
- Kakkis, E.D.; Muenzer, J.; Tiller, G.E.; Waber, L.; Belmont, J.; Passage, M.; Izykowski, B.; Phillips, J.; Doroshow, R.; Walot, I.; et al. Enzyme-Replacement Therapy in Mucopolysaccharidosis I. N. Engl. J. Med. 2001, 344, 182–188. [Google Scholar] [CrossRef]
- Pereira, V.G.; Queiroz, M.T.; D’Almeida, V. Differential expression of microRNAs from miR-17 family in the cerebellum of mucopolysaccharidosis type I mice. Gene 2016, 595, 207–211. [Google Scholar] [CrossRef]
- Xiao, K.; Lu, D.; Hoepfner, J.; Santer, L.; Gupta, S.; Pfanne, A.; Thum, S.; Lenders, M.; Brand, E.; Nordbeck, P.; et al. Circulating microRNAs in Fabry Disease. Sci. Rep. 2019, 9, 15277. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson–Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef]
- Morena, F.; Oikonomou, V.; Argentati, C.; Bazzucchi, M.; Emiliani, C.; Gritti, A.; Martino, S. Integrated Computational Analysis Highlights unique miRNA Signatures in the Subventricular Zone and Striatum of GM2 Gangliosidosis Animal Models. Int. J. Mol. Sci. 2019, 20, 3179. [Google Scholar] [CrossRef]
- Watson, L.; Keatinge, M.; Gegg, M.; Bai, Q.; Sandulescu, M.C.; Vardi, A.; Futerman, A.H.; Schapira, A.H.; Burton, E.A.; Bandmann, O. Ablation of the pro-inflammatory master regulator miR-155 does not mitigate neuroinflammation or neurodegeneration in a vertebrate model of Gaucher’s disease. Neurobiol. Dis. 2019, 127, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadhukhan, T.; Bagh, M.B.; Sadhukhan, S.; Appu, A.P.; Mondal, A.; Iben, J.; Li, T.; Coon, S.L.; Mukherjee, A.B. Ablation of microRNA-155 and neuroinflammation in a mouse model of CLN1-disease. Biochem. Biophys. Res. Commun. 2021, 571, 137–144. [Google Scholar] [CrossRef]
- Gentner, B.; Visigalli, I.; Hiramatsu, H.; Lechman, E.; Ungari, S.; Giustacchini, A.; Schira, G.; Amendola, M.; Quattrini, A.; Martino, S.; et al. Identification of Hematopoietic Stem Cell–Specific miRNAs Enables Gene Therapy of Globoid Cell Leukodystrophy. Sci. Transl. Med. 2010, 2, 58ra84. [Google Scholar] [CrossRef]
- Vanier, M.T. Niemann-Pick disease type C. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef]
- Burton, B.K.; Ellis, A.G.; Orr, B.; Chatlani, S.; Yoon, K.; Shoaff, J.R.; Gallo, D. Estimating the prevalence of Niemann-Pick disease type C (NPC) in the United States. Mol. Genet. Metab. 2021, 134, 182–187. [Google Scholar] [CrossRef]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Wheeler, S.; Sillence, D.J. Niemann–Pick type C disease: Cellular pathology and pharmacotherapy. J. Neurochem. 2020, 153, 674–692. [Google Scholar] [CrossRef]
- Guix, F.X.; Capitán, A.M.; Casadomé-Perales, Á.; Palomares-Pérez, I.; del Castillo, I.L.; Miguel, V.; Goedeke, L.; Martín, M.G.; Lamas, S.; Peinado, H.; et al. Increased exosome secretion in neurons aging in vitro by NPC1-mediated endosomal cholesterol buildup. Life Sci. Alliance 2021, 4, e202101055. [Google Scholar] [CrossRef] [PubMed]
- Tharkeshwar, A.K.; Trekker, J.; Vermeire, W.; Pauwels, J.; Sannerud, R.; Priestman, D.A.; Te Vruchte, D.; Vints, K.; Baatsen, P.; Decuypere, J.-P.; et al. A novel approach to analyze lysosomal dysfunctions through subcellular proteomics and lipidomics: The case of NPC1 deficiency. Sci. Rep. 2017, 7, 41408. [Google Scholar] [CrossRef]
- Torres, S.; Balboa, E.; Zanlungo, S.; Enrich, C.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Lysosomal and Mitochondrial Liaisons in Niemann-Pick Disease. Front. Physiol. 2017, 8, 982. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortuño-Sahagún, D.; Enterría-Rosales, J.; Izquierdo, V.; Griñán-Ferré, C.; Pallàs, M.; González-Castillo, C. The Role of the miR-17-92 Cluster in Autophagy and Atherosclerosis Supports Its Link to Lysosomal Storage Diseases. Cells 2022, 11, 2991. https://doi.org/10.3390/cells11192991
Ortuño-Sahagún D, Enterría-Rosales J, Izquierdo V, Griñán-Ferré C, Pallàs M, González-Castillo C. The Role of the miR-17-92 Cluster in Autophagy and Atherosclerosis Supports Its Link to Lysosomal Storage Diseases. Cells. 2022; 11(19):2991. https://doi.org/10.3390/cells11192991
Chicago/Turabian StyleOrtuño-Sahagún, Daniel, Julia Enterría-Rosales, Vanesa Izquierdo, Christian Griñán-Ferré, Mercè Pallàs, and Celia González-Castillo. 2022. "The Role of the miR-17-92 Cluster in Autophagy and Atherosclerosis Supports Its Link to Lysosomal Storage Diseases" Cells 11, no. 19: 2991. https://doi.org/10.3390/cells11192991
APA StyleOrtuño-Sahagún, D., Enterría-Rosales, J., Izquierdo, V., Griñán-Ferré, C., Pallàs, M., & González-Castillo, C. (2022). The Role of the miR-17-92 Cluster in Autophagy and Atherosclerosis Supports Its Link to Lysosomal Storage Diseases. Cells, 11(19), 2991. https://doi.org/10.3390/cells11192991