
A Review on Autophagy in Orofacial Neuropathic Pain

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Orofacial Neuropathic Pain
2.1. Characteristics and Types of Orofacial Neuropathic Pain
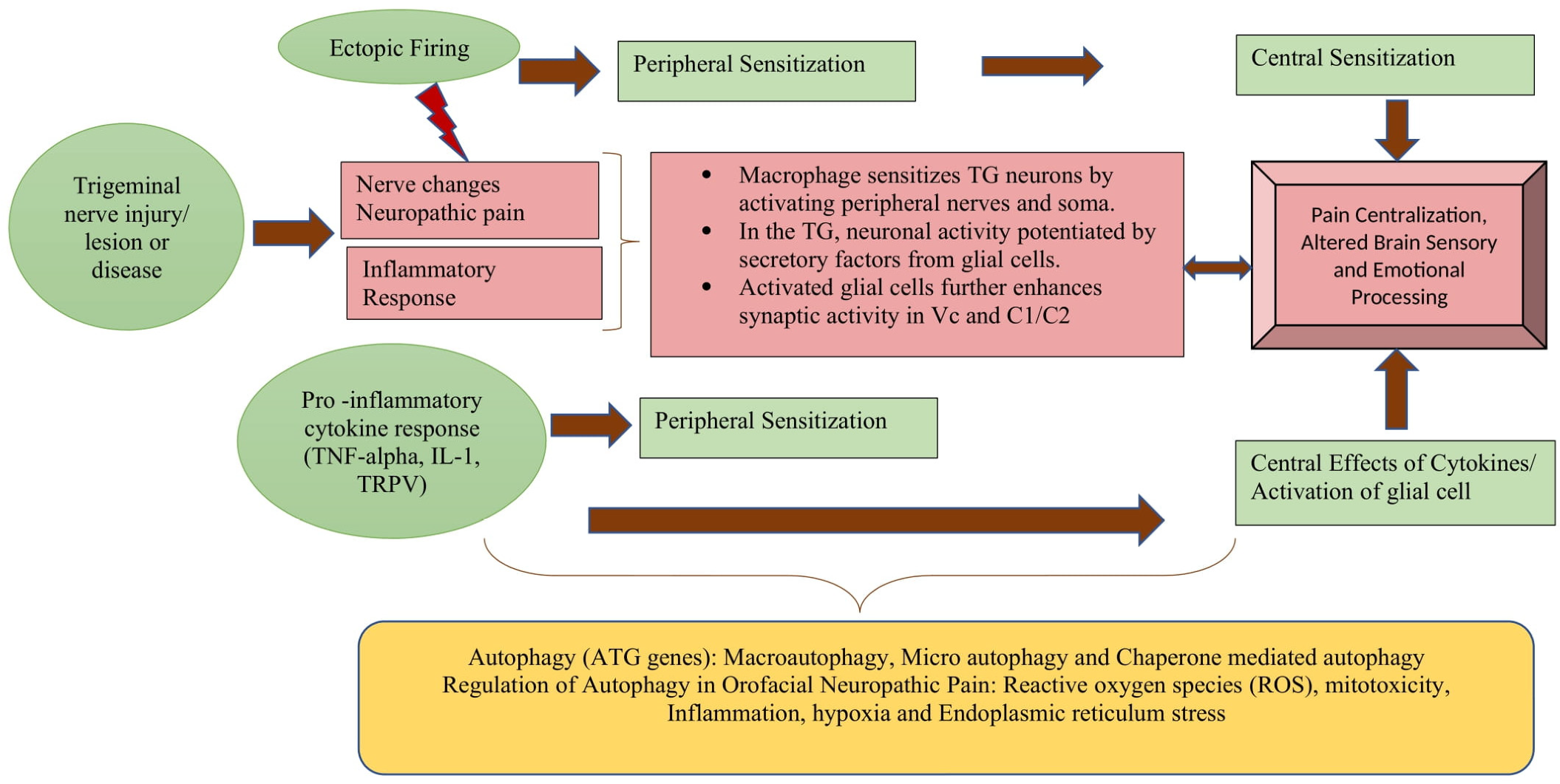
2.2. Pathophysiology of Orofacial Neuropathic Pain
Neurobiology of Orofacial Neuropathic Pain
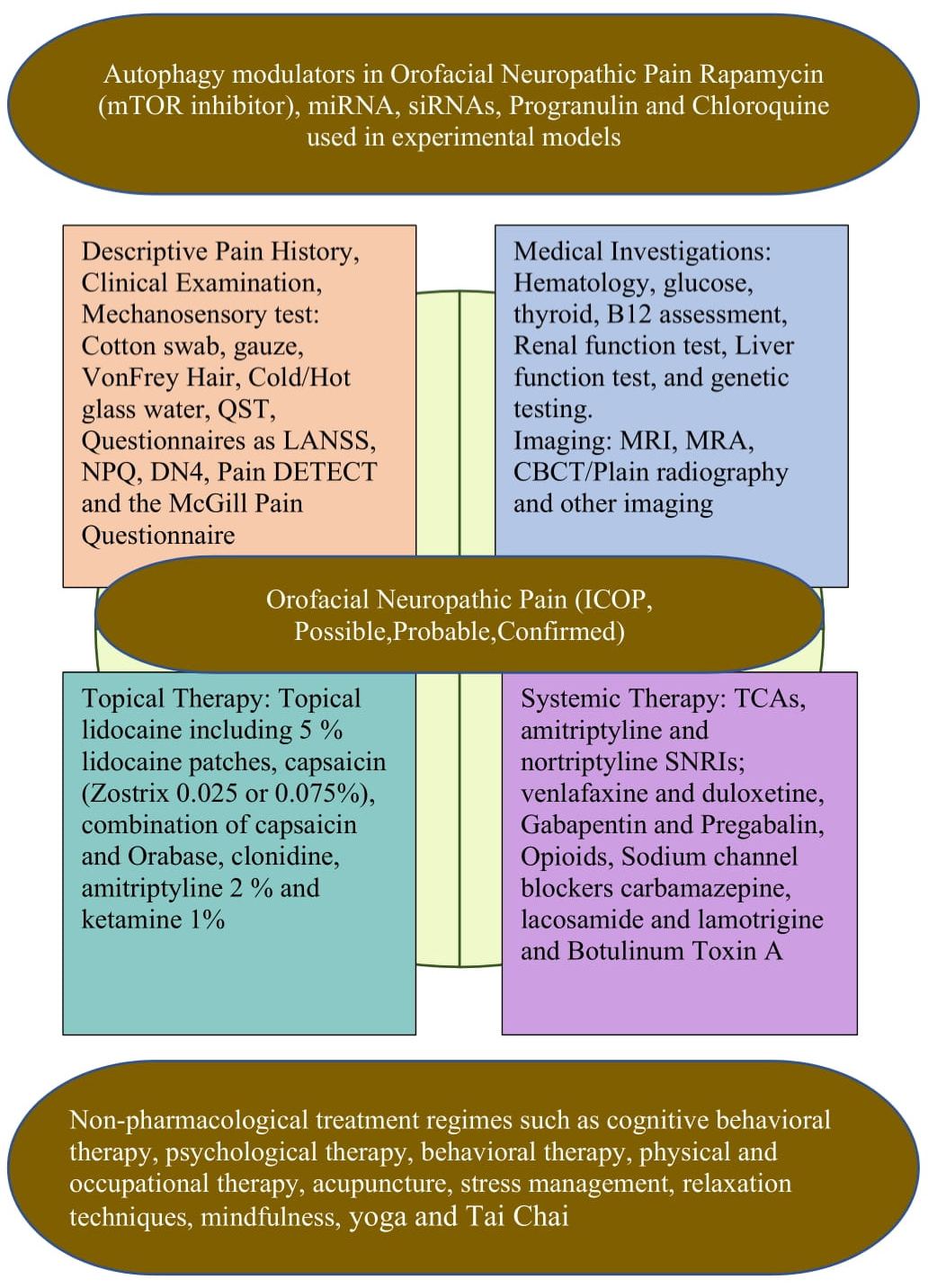
2.3. Assessment of Orofacial Neuropathic Pain
3. Autophagy
3.1. Autophagy in Orofacial Neuropathic Pain Formation
3.2. Therapeutic Potential of Autophagy in the Management of Orofacial Neuropathic Pain
3.2.1. Rapamycin
3.2.2. RNA Agents
3.3. Overview on the Management of Orofacial Neuropathic Pain
4. Conclusions
Funding
Conflicts of Interest
References
- Treede, R.-D.; Rief, W.; Barke, A.; Aziz, Q.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Evers, S.; Finnerup, N.B.; First, M.B.; et al. Chronic pain as a symptom or a disease: The IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain 2019, 160, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrivastava, M.; Battaglino, R.; Ye, L. A comprehensive review on biomarkers associated with painful temporomandibular disorders. Int. J. Oral Sci. 2021, 13, 23. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.P.; Vase, L.; Hooten, W.M. Chronic pain: An update on burden, best practices, and new advances. Lancet 2021, 397, 2082–2097. [Google Scholar] [CrossRef] [PubMed]
- Dahlhamer, J.; Lucas, J.; Zelaya, C.; Nahin, R.; Mackey, S.; DeBar, L.; Kerns, R.; Von Korff, M.; Porter, L.; Helmick, C. Prevalence of Chronic Pain and High-Impact Chronic Pain Among Adults—United States, 2016. MMWR Morb. Mortal. Wkly. Rep. 2018, 67, 1001–1006. [Google Scholar] [CrossRef]
- Haggman-Henrikson, B.; Liv, P.; Ilgunas, A.; Visscher, C.M.; Lobbezoo, F.; Durham, J.; Lövgren, A. Increasing gender differences in the prevalence and chronification of orofacial pain in the population. Pain 2020, 161, 1768–1775. [Google Scholar] [CrossRef]
- Pogatzki-Zahn, E.M.; Segelcke, D.; Schug, S.A. Postoperative pain—From mechanisms to treatment. Pain Rep. 2017, 2, e588. [Google Scholar] [CrossRef]
- Cohen, S.P.; Mao, J. Neuropathic pain: Mechanisms and their clinical implications. BMJ 2014, 348, f7656. [Google Scholar] [CrossRef] [Green Version]
- Meints, S.; Edwards, R. Evaluating psychosocial contributions to chronic pain outcomes. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 87, 168–182. [Google Scholar] [CrossRef]
- Edwards, R.R.; Dworkin, R.H.; Sullivan, M.D.; Turk, D.C.; Wasan, A.D. The Role of Psychosocial Processes in the Development and Maintenance of Chronic Pain. J. Pain 2016, 17, T70–T92. [Google Scholar] [CrossRef] [Green Version]
- Samoborec, S.; Ruseckaite, R.; Ayton, D.; Evans, S. Biopsychosocial factors associated with non-recovery after a minor transport-related injury: A systematic review. PLoS ONE 2018, 13, e0198352. [Google Scholar] [CrossRef]
- Finnerup, N.B.; Haroutounian, S.; Kamerman, P.; Baron, R.; Bennett, D.L.; Bouhassira, D.; Cruccu, G.; Freeman, R.; Hansson, P.; Nurmikko, T.; et al. Neuropathic pain: An updated grading system for research and clinical practice. Pain 2016, 157, 1599–1606. [Google Scholar] [CrossRef] [Green Version]
- Saavedra-Hernández, M.; Castro-Sánchez, A.M.; Cuesta-Vargas, A.I.; Cleland, J.A.; Fernández-de-las-Peñas, C.; Arroyo-Morales, M. The contribution of previous episodes of pain, pain intensity, physical impairment, and pain-related fear to disability in patients with chronic mechanical neck pain. Am. J. Phys. Med. Rehabil 2012, 91, 1070–1076. [Google Scholar] [CrossRef]
- Spahr, N.; Hodkinson, D.; Jolly, K.; Williams, S.; Howard, M.; Thacker, M. Distinguishing between nociceptive and neuropathic components in chronic low back pain using behavioural evaluation and sensory examination. Musculoskelet. Sci. Pract. 2016, 27, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Fitzcharles, M.-A.; Cohen, S.P.; Clauw, D.J.; Littlejohn, G.; Usui, C.; Häuser, W. Nociplastic pain: Towards an understanding of prevalent pain conditions. Lancet 2021, 397, 2098–2110. [Google Scholar] [CrossRef]
- DiBonaventura, M.D.; Sadosky, A.; Concialdi, K.; Hopps, M.; Kudel, I.; Parsons, B.; Cappelleri, J.C.; Hlavacek, P.; Alexander, A.H.; Stacey, B.R.; et al. The prevalence of probable neuropathic pain in the US: Results from a multimodal general-population health survey. J. Pain Res. 2017, 10, 2525–2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Hecke, O.; Austin, S.K.; Khan, R.A.; Smith, B.H.; Torrance, N. Neuropathic pain in the general population: A systematic review of epidemiological studies. Pain 2014, 155, 654–662, Erratum in Pain 2014, 155, 1907. [Google Scholar] [CrossRef]
- Renton, T. Chronic orofacial pain. Oral Dis. 2017, 23, 566–571. [Google Scholar] [CrossRef]
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Ma, Z.; Han, Q.; Wang, X.; Ai, Z.-S.; Zheng, Y. Galectin-3 Inhibition Is Associated with Neuropathic Pain Attenuation after Peripheral Nerve Injury. PLoS ONE 2016, 11, e0148792. [Google Scholar] [CrossRef] [Green Version]
- Rangaraju, S.; Verrier, J.D.; Madorsky, I.; Nicks, J.; Dunn Jr, W.A.; Notterpek, L. Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J. Neurosci. 2010, 30, 11388–11397. [Google Scholar] [CrossRef]
- Bar-Yosef, T.; Damri, O.; Agam, G. Dual Role of Autophagy in Diseases of the Central Nervous System. Front. Cell. Neurosci. 2019, 13, 196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagi, H.M.; Ahmadi, S.; Tarighat, F.; Rahbarghazi, R.; Soleimanpour, H. Interplay between exosomes and autophagy machinery in pain management: State of the art. Neurobiol. Pain 2022, 12, 100095. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, V.V.; Maday, S. Compartment-specific dynamics and functions of autophagy in neurons. Dev. Neurobiol. 2018, 78, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Lüningschrör, P.; Sendtner, M. Autophagy in the presynaptic compartment. Curr. Opin. Neurobiol. 2018, 51, 80–85. [Google Scholar] [CrossRef]
- Liu, X.; Zhu, M.; Ju, Y.; Li, A.; Sun, X. Autophagy dysfunction in neuropathic pain. Neuropeptides 2019, 75, 41–48. [Google Scholar] [CrossRef]
- Lüningschrör, P.; Binotti, B.; Dombert, B.; Heimann, P.; Perez-Lara, A.; Slotta, C.; Thau-Habermann, N.; von Collenberg, C.R.; Karl, F.; Damme, M.; et al. Plekhg5-regulated autophagy of synaptic vesicles reveals a pathogenic mechanism in motoneuron disease. Nat. Commun. 2017, 8, 678. [Google Scholar] [CrossRef] [Green Version]
- Marinelli, S.; Nazio, F.; Tinari, A.; Ciarlo, L.; D’Amelio, M.; Pieroni, L.; Vacca, V.; Urbani, A.; Cecconi, F.; Malorni, W.; et al. Schwann cell autophagy counteracts the onset and chronification of neuropathic pain. Pain 2014, 155, 93–107. [Google Scholar] [CrossRef]
- Guo, J.-S.; Jing, P.-B.; Wang, J.-A.; Zhang, R.; Jiang, B.-C.; Gao, Y.-J.; Zhang, Z.-J. Increased autophagic activity in dorsal root ganglion attenuates neuropathic pain following peripheral nerve injury. Neurosci. Lett. 2015, 599, 158–163. [Google Scholar] [CrossRef]
- Jang, S.Y.; Shin, Y.K.; Park, S.Y.; Park, J.Y.; Lee, H.J.; Yoo, Y.H.; Kim, J.K.; Park, H.T. Autophagic myelin destruction by schwann cells during wallerian degeneration and segmental demyelination. Glia 2016, 64, 730–742. [Google Scholar] [CrossRef]
- Liao, M.-F.; Lu, K.-T.; Hsu, J.-L.; Lee, C.-H.; Cheng, M.-Y.; Ro, L.-S. The Role of Autophagy and Apoptosis in Neuropathic Pain Formation. Int. J. Mol. Sci. 2022, 23, 2685. [Google Scholar] [CrossRef]
- Brosius Lutz, A.; Chung, W.S.; Sloan, S.A.; Carson, G.A.; Zhou, L.; Lovelett, E.; Posada, S.; Zuchero, J.B.; Barres, B.A. Schwann cells use TAM receptor-mediated pha-gocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc. Natl. Acad. Sci. USA 2017, 114, E8072–E8080. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Sanchez, J.A.; Carty, L.; Iruarrizaga-Lejarreta, M.; Palomo-Irigoyen, M.; Varela-Rey, M.; Griffith, M.; Hantke, J.; Macias-Camara, N.; Azkargorta, M.; Aurrekoetxea, I.; et al. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J. Cell Biol. 2015, 210, 153–168. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Kuner, R.; Jensen, T.S. Neuropathic Pain: From Mechanisms to Treatment. Physiol. Rev. 2021, 101, 259–301. [Google Scholar] [CrossRef]
- Woolf, C.J.; Bennett, G.J.; Doherty, M.; Dubner, R.; Kidd, B.; Koltzenburg, M.; Lipton, R.; Loeser, J.D.; Payne, R.; Torebjork, E. Towards a mechanism-based classification of pain? Pain 1998, 77, 227–229. [Google Scholar] [CrossRef]
- Scholz, J.; Finnerup, N.B.; Attal, N.; Aziz, Q.; Baron, R.; Bennett, M.I.; Benoliel, R.; Cohen, M.; Cruccu, G.; Davis, K.D.; et al. The IASP classification of chronic pain for ICD-11: Chronic neuropathic pain. Pain 2019, 160, 53–59. [Google Scholar] [CrossRef] [Green Version]
- International Classification of Orofacial Pain, 1st edition (ICOP). Cephalalgia 2020, 40, 129–221. [CrossRef] [Green Version]
- Pigg, M.; Nixdorf, D.R.; Law, A.S.; Renton, T.; Sharav, Y.; Baad-Hansen, L.; List, T. New International Classification of Orofacial Pain: What Is in It for Endodontists? J. Endod. 2020, 47, 345–357. [Google Scholar] [CrossRef]
- Amir, R.; Kocsis, J.D.; Devor, M. Multiple interacting sites of ectopic spike electrogenesis in primary sensory neurons. J. Neurosci. 2005, 25, 2576–2585. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Ringkamp, M.; Murinson, B.B.; Pogatzki, E.M.; Hartke, T.V.; Weerahandi, H.M.; Campbell, J.N.; Griffin, J.W.; Meyer, R.A. Degeneration of myelinated efferent fibers induces spontaneous activity in uninjured C-fiber afferents. J. Neurosci. 2002, 22, 7746–7753. [Google Scholar] [CrossRef]
- Bostock, H.; Campero, M.; Serra, J.; Ochoa, J.L. Temperature-dependent double spikes in C-nociceptors of neuropathic pain patients. Brain 2005, 128, 2154–2163. [Google Scholar] [CrossRef]
- Matsuda, M.; Huh, Y.; Ji, R.-R. Roles of inflammation, neurogenic inflammation, and neuroinflammation in pain. J. Anesth. 2019, 33, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Caterina, M.J.; Julius, D. The vanilloid receptor: A molecular gateway to the pain pathway. Annu. Rev. Neurosci. 2001, 24, 487–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.; Zhang, Y.; Bantel, C.; Eisenach, J.C. Medium and large injured dorsal root ganglion cells increase TRPV-1, accompanied by increased alpha2C-adrenoceptor co-expression and functional inhibition by clonidine. Pain 2005, 113, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Serra, J.; Solà, R.; Quiles, C.; Casanova-Molla, J.; Pascual, V.; Bostock, H.; Valls-Solé, J. C-nociceptors sensitized to cold in a patient with small-fiber neuropathy and cold allodynia. Pain 2009, 147, 46–53. [Google Scholar] [CrossRef]
- Shinoda, M.; Takeda, M.; Honda, K.; Maruno, M.; Katagiri, A.; Satoh-Kuriwada, S.; Shoji, N.; Tsuchiya, M.; Iwata, K. Involvement of peripheral artemin signaling in tongue pain: Possible mechanism in burning mouth syndrome. Pain 2015, 156, 2528–2537. [Google Scholar] [CrossRef]
- Kanamori, H.; Matsubara, T.; Mima, A.; Sumi, E.; Nagai, K.; Takahashi, T.; Abe, H.; Iehara, N.; Fukatsu, A.; Okamoto, H.; et al. Inhibition of MCP-1/CCR2 pathway ameliorates the development of diabetic nephropathy. Biochem. Biophys. Res. Commun. 2007, 360, 772–777. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; van Rooijen, N.; Tracey, D.J. Depletion of macrophages reduces axonal degeneration and hyperalgesia following nerve injury. Pain 2000, 86, 25–32. [Google Scholar] [CrossRef]
- Chu, L.W.; Cheng, K.I.; Chen, J.Y.; Cheng, Y.C.; Chang, Y.C.; Yeh, J.L.; Hsu, J.H.; Dai, Z.K.; Wu, B.N. Loganin prevents chronic constriction injury-provoked neuropathic pain by reducing TNF-alpha/IL-1beta-mediated NF-kappaB activation and Schwann cell de-myelination. Phytomedicine 2000, 67, 153166. [Google Scholar] [CrossRef]
- Lai, J.; Hunter, J.C.; Porreca, F. The role of voltage-gated sodium channels in neuropathic pain. Curr. Opin. Neurobiol. 2003, 13, 291–297. [Google Scholar] [CrossRef]
- Black, J.A.; Nikolajsen, L.; Kroner, K.; Jensen, T.S.; Waxman, S.G. Multiple sodium channel isoforms and mitogen-activated protein kinases are present in painful human neuromas. Ann. Neurol. 2008, 64, 644–653. [Google Scholar] [CrossRef]
- Siqueira, S.R.; Alves, B.; Malpartida, H.M.; Teixeira, M.J.; Siqueira, J.T. Abnormal expression of voltage-gated sodium channels Nav1.7, Nav1.3 and Nav1.8 in trigeminal neuralgia. Neuroscience 2009, 164, 573–577. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Waxman, S.G. Sodium channel expression and the molecular pathophysiology of pain after SCI. Prog. Brain Res. 2007, 161, 195–203. [Google Scholar]
- Meacham, K.; Shepherd, A.; Mohapatra, D.P.; Haroutounian, S. Neuropathic Pain: Central vs. Peripheral Mechanisms. Curr. Pain Headache Rep. 2017, 21, 28. [Google Scholar] [CrossRef]
- Ultenius, C.; Linderoth, B.; Meyerson, B.A.; Wallin, J. Spinal NMDA receptor phosphorylation correlates with the presence of neuropathic signs following peripheral nerve injury in the rat. Neurosci. Lett. 2006, 399, 85–90. [Google Scholar] [CrossRef]
- Hains, B.C.; Saab, C.Y.; Klein, J.P.; Craner, M.J.; Waxman, S.G. Altered sodium channel expression in second-order spinal sensory neurons contributes to pain after peripheral nerve injury. J. Neurosci. 2004, 24, 4832–4839. [Google Scholar] [CrossRef] [Green Version]
- Finnerup, N.B.; Jensen, T.S. Spinal cord injury pain—Mechanisms and treatment. Eur. J. Neurol. 2004, 11, 73–82. [Google Scholar] [CrossRef]
- Ducreux, D.; Attal, N.; Parker, F.; Bouhassira, D. Mechanisms of central neuropathic pain: A combined psychophysical and fMRI study in syringomyelia. Brain 2006, 128, 963–976. [Google Scholar] [CrossRef] [Green Version]
- Wasner, G.; Lee, B.B.; Engel, S.; McLachlan, E. Residual spinothalamic tract pathways predict development of central pain after spinal cord injury. Brain 2008, 131, 2387–2400. [Google Scholar] [CrossRef]
- Shinoda, M.; Imamura, Y.; Hayashi, Y.; Noma, N.; Okada-Ogawa, A.; Hitomi, S.; Iwata, K. Orofacial Neuropathic Pain-Basic Research and Their Clinical Relevancies. Front. Mol. Neurosci. 2021, 14, 121. [Google Scholar] [CrossRef]
- Dubner, R.; Ren, K. Brainstem mechanisms of persistent pain following injury. J. Orofac. Pain 2004, 18, 299–305. [Google Scholar]
- Liu, X.-J.; Liu, T.; Chen, G.; Wang, B.; Yu, X.-L.; Yin, C.; Ji, R.-R. TLR signaling adaptor protein MyD88 in primary sensory neurons contributes to persistent inflammatory and neuropathic pain and neuroinflammation. Sci. Rep. 2016, 6, 28188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristoiu, V. Contribution of macrophages to peripheral neuropathic pain pathogenesis. Life Sci. 2013, 93, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Shibuta, K.; Suzuki, I.; Shinoda, M.; Tsuboi, Y.; Honda, K.; Shimizu, N.; Sessle, B.J.; Iwata, K. Organization of hyperactive microglial cells in trigeminal spinal subnucleus caudalis and upper cervical spinal cord associated with orofacial neuropathic pain. Brain Res. 2012, 1451, 74–86. [Google Scholar] [CrossRef]
- Asano, S.; Hayashi, Y.; Iwata, K.; Okada-Ogawa, A.; Hitomi, S.; Shibuta, I.; Imamura, Y.; Shinoda, M. Microglia—Astrocyte Communication via C1q Contributes to Orofacial Neuropathic Pain Associated with Infraorbital Nerve Injury. Int. J. Mol. Sci. 2020, 21, 6834. [Google Scholar] [CrossRef]
- Saito, H.; Katagiri, A.; Okada, S.; Mikuzuki, L.; Kubo, A.; Suzuki, T.; Ohara, K.; Lee, J.; Gionhaku, N.; Iinuma, T.; et al. Ascending projections of nociceptive neurons from trigeminal subnucleus caudalis: A population approach. Exp. Neurol. 2007, 293, 124–136. [Google Scholar] [CrossRef]
- Okada, S.; Katagiri, A.; Saito, H.; Lee, J.; Ohara, K.; Iinuma, T.; Bereiter, D.A.; Iwata, K. Differential activation of ascending noxious pathways associated with trigeminal nerve injury. Pain 2019, 160, 1342–1360. [Google Scholar] [CrossRef]
- Okada-Ogawa, A.; Nakaya, Y.; Imamura, Y.; Kobayashi, M.; Shinoda, M.; Kita, K.; Sessle, B.J.; Iwata, K. Involvement of medullary GABAergic system in extraterritorial neuropathic pain mechanisms associated with inferior alveolar nerve transection. Exp. Neurol. 2015, 267, 42–52. [Google Scholar] [CrossRef]
- Okada-Ogawa, A.; Suzuki, I.; Nakaya, Y.; Kobayashi, M.; Ebihara, K.; Imamura, Y.; Iwata, K. Involvement of GABAergic interneurons in orofacial neuropathic pain following trigeminal nerve transection in rats. PAIN Res. 2013, 28, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Antonaci, F.; Arceri, S.; Rakusa, M.; Mitsikostas, D.D.; Milanov, I.; Todorov, V.; Ramusino, M.C.; Costa, A. Pitfals in recognition and management of trigeminal neuralgia. J. Headache Pain 2020, 21, 82. [Google Scholar] [CrossRef]
- Christoforou, J. Neuropathic Orofacial Pain. Dent. Clin. N. Am. 2018, 62, 565–584. [Google Scholar] [CrossRef]
- Baad-Hansen, L.; Benoliel, R. Neuropathic orofacial pain: Facts and fiction. Cephalalgia 2017, 37, 670–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elias, L.A.; Yilmaz, Z.; Smith, J.G.; Bouchiba, M.; van der Valk, R.A.; Page, L.; Barker, S.; Renton, T. PainDETECT: A suitable screening tool for neuropathic pain in patients with painful post-traumatic trigeminal nerve injuries? Int. J. Oral. Maxillofac. Surg. 2014, 43, 120–126. [Google Scholar] [CrossRef]
- Mathieson, S.; Maher, C.G.; Terwee, C.B.; Folly de Campos, T.; Lin, C.W. Neuropathic pain screening questionnaires have limited measurement properties. A systematic review. J. Clin. Epidemiol. 2015, 68, 957–966. [Google Scholar] [CrossRef]
- Wu, J.; Lipinski, M.M. Autophagy in Neurotrauma: Good, Bad, or Dysregulated. Cells 2019, 8, 693. [Google Scholar] [CrossRef] [Green Version]
- Glick, D.; Barth, S.; MacLeod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Xie, Z.; Klionsky, D.J. Autophagosome formation: Core machinery and adaptations. Nat. Cell Biol. 2007, 9, 1102–1109. [Google Scholar] [CrossRef]
- Herpin, A.; Lescat, L.; Bobe, J.; Jenny, A.; Seiliez, I. Lighting chaperone-mediated autophagy (CMA) evolution with an ancient LAMP: The existence of a functional CMA activity in fish. Autophagy 2000, 16, 1918–1920. [Google Scholar] [CrossRef]
- Xilouri, M.; Stefanis, L. Chaperone mediated autophagy to the rescue: A new-fangled target for the treatment of neurodegenerative diseases. Mol. Cell. Neurosci. 2015, 66, 29–36. [Google Scholar] [CrossRef]
- Chu, C.; Levine, E.; Gear, R.W.; Bogen, O.; Levine, J.D. Mitochondrial dependence of nerve growth factor-induced mechanical hyperalgesia. Pain 2011, 152, 1832–1837. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, L.F.; Levine, J.D. Alcohol consumption enhances antiretroviral painful peripheral neuropathy by mitochon-drial mechanisms. Eur. J. Neurosci. 2010, 32, 811–818. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.H.; Piao, S.F.; Dey, S.; McAfee, Q.; Karakousis, G.; Villanueva, J.; Hart, L.S.; Levi, S.; Hu, J.; Zhang, G.; et al. Targeting ER stress-induced autophagy overcomes BRAF inhibitor resistance in melanoma. J. Clin. Investig. 2014, 124, 1406–1417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melendez, A.; Neufeld, T.P. The cell biology of autophagy in metazoans: A developing story. Development 2008, 135, 2347–2360. [Google Scholar] [CrossRef] [PubMed]
- Moloudizargari, M.; Asghari, M.H.; Ghobadi, E.; Fallah, M.; Rasouli, S.; Abdollahi, M. Autophagy, its mechanisms and regulation: Implications in neurodegenerative diseases. Ageing Res. Rev. 2017, 40, 64–74. [Google Scholar] [CrossRef]
- Berliocchi, L.; Russo, R.; Maiaru, M.; Levato, A.; Bagetta, G.; Corasaniti, M.T. Autophagy impairment in a mouse model of neuropathic pain. Mol. Pain 2011, 7, 83. [Google Scholar] [CrossRef] [Green Version]
- Inceoglu, B.; Bettaieb, A.; Trindade da Silva, C.A.; Lee, K.S.; Haj, F.G.; Hammock, B.D. Endoplasmic reticulum stress in the peripheral nervous system is a significant driver of neuropathic pain. Proc. Natl. Acad. Sci. USA 2015, 112, 9082–9087. [Google Scholar] [CrossRef] [Green Version]
- Jadli, A.S.; Ballasy, N.; Edalat, P.; Patel, V.B. Inside (sight) of tiny communicator: Exosome biogenesis, secretion, and uptake. Mol. Cell. Biochem. 2020, 467, 77–94. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Zha, H.; Fan, Y.; Yang, L.; Yin, M.; Miao, W.; He, J.; Wang, Y. Autophagy protects against cerebral ischemic reperfusion injury by inhibiting neuroinflammation. Am. J. Transl. Res. 2021, 13, 4726. [Google Scholar]
- Huang, H.C.; Chen, L.; Zhang, H.X.; Li, S.F.; Liu, P.; Zhao, T.Y.; Li, C.X. Autophagy promotes peripheral nerve re-generation and motor recovery following sciatic nerve crush injury in rats. J. Mol. Neurosci. 2016, 58, 416–423. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Morse, L.R.; Falci, S.P.; Olson, J.K.; Shrivastava, M.; Nguyen, N.; Linnman, C.; Troy, K.L.; Battaglino, R.A. hsa-MiR-19a-3p and hsa-MiR-19b-3p Are Associated with Spinal Cord Injury-Induced Neuropathic Pain: Findings from a Genome-Wide MicroRNA Expression Profiling Screen. Neurotrauma Rep. 2021, 2, 424–439. [Google Scholar] [CrossRef]
- Hausser, J.; Zavolan, M. Identification and consequences of miRNA-target inter- actions–beyond repression of gene expression. Nat. Rev. Genet. 2014, 15, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Parisi, C.; Napoli, G.; Amadio, S.; Spalloni, A.; Apolloni, S.; Longone, P.; Volonte, C. MicroRNA-125b regulates microglia activation and motor neuron death in ALS. Cell Death Differ. 2016, 23, 531–541. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Jiang, K.; Li, Z. MiR-145 ameliorates neuropathic pain via inhibiting inflammatory responses and mTOR signaling pathway by targeting Akt3 in a rat model. Neurosci. Res. 2008, 134, 10–17. [Google Scholar] [CrossRef]
- Xie, X.; Ma, L.; Xi, K.; Zhang, W.; Fan, D. MicroRNA-183 suppresses neuropathic pain and expression of AMPA receptors by targeting mTOR/VEGF Signaling pathway. Cell. Physiol. Biochem. 2017, 41, 181–192. [Google Scholar] [CrossRef]
- Altmann, C.; Hardt, S.; Fischer, C.; Heidler, J.; Lim, H.Y.; Haussler, A.; Albuquerque, B.; Zimmer, B.; Moser, C.; Behrends, C.; et al. Progranulin overexpression in sensory neurons attenuates neuropathic pain in mice: Role of autophagy. Neurobiol. Dis. 2016, 96, 294–311. [Google Scholar] [CrossRef]
- Berliocchi, L.; Maiaru, M.; Varano, G.P.; Russo, R.; Corasaniti, M.T.; Bagetta, G.; Tassorelli, C. Spinal autophagy is differently modulated in distinct mouse models of neuropathic pain. Mol. Pain 2015, 11, 3. [Google Scholar] [CrossRef] [Green Version]
- Zakrzewska, J.M. Medical management of trigeminal neuropathic pains. Expert Opin. Pharmacother. 2010, 11, 1239–1254. [Google Scholar] [CrossRef]
- Padilla, M.; Clark, G.T.; Merrill, R.L. Topical medications for orofacial neuropathic pain: A review. J. Am. Dent. Assoc. 2000, 131, 184–195. [Google Scholar] [CrossRef]
- Epstein, J.B.; Grushka, M.; Le, N. Topical clonidine for orofacial pain: A pilot study. J. Orofac. Pain 1997, 11, 346–352. [Google Scholar]
- Sawynok, J.; Zinger, C. Topical amitriptyline and ketamine for post-herpetic neuralgia and other forms of neuropathic pain. Expert Opin. Pharmacother. 2016, 17, 601–609. [Google Scholar] [CrossRef]
- Lewis, M.A.; Sankar, V.; De Laat, A.; Benoliel, R. Management of neuropathic orofacial pain. Oral Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod. 2007, 103 (Suppl. S32), e1–e24. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shrivastava, M.; Ye, L. A Review on Autophagy in Orofacial Neuropathic Pain. Cells 2022, 11, 3842. https://doi.org/10.3390/cells11233842
Shrivastava M, Ye L. A Review on Autophagy in Orofacial Neuropathic Pain. Cells. 2022; 11(23):3842. https://doi.org/10.3390/cells11233842
Chicago/Turabian StyleShrivastava, Mayank, and Liang Ye. 2022. "A Review on Autophagy in Orofacial Neuropathic Pain" Cells 11, no. 23: 3842. https://doi.org/10.3390/cells11233842
APA StyleShrivastava, M., & Ye, L. (2022). A Review on Autophagy in Orofacial Neuropathic Pain. Cells, 11(23), 3842. https://doi.org/10.3390/cells11233842