
Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology


Abstract
:1. Introduction
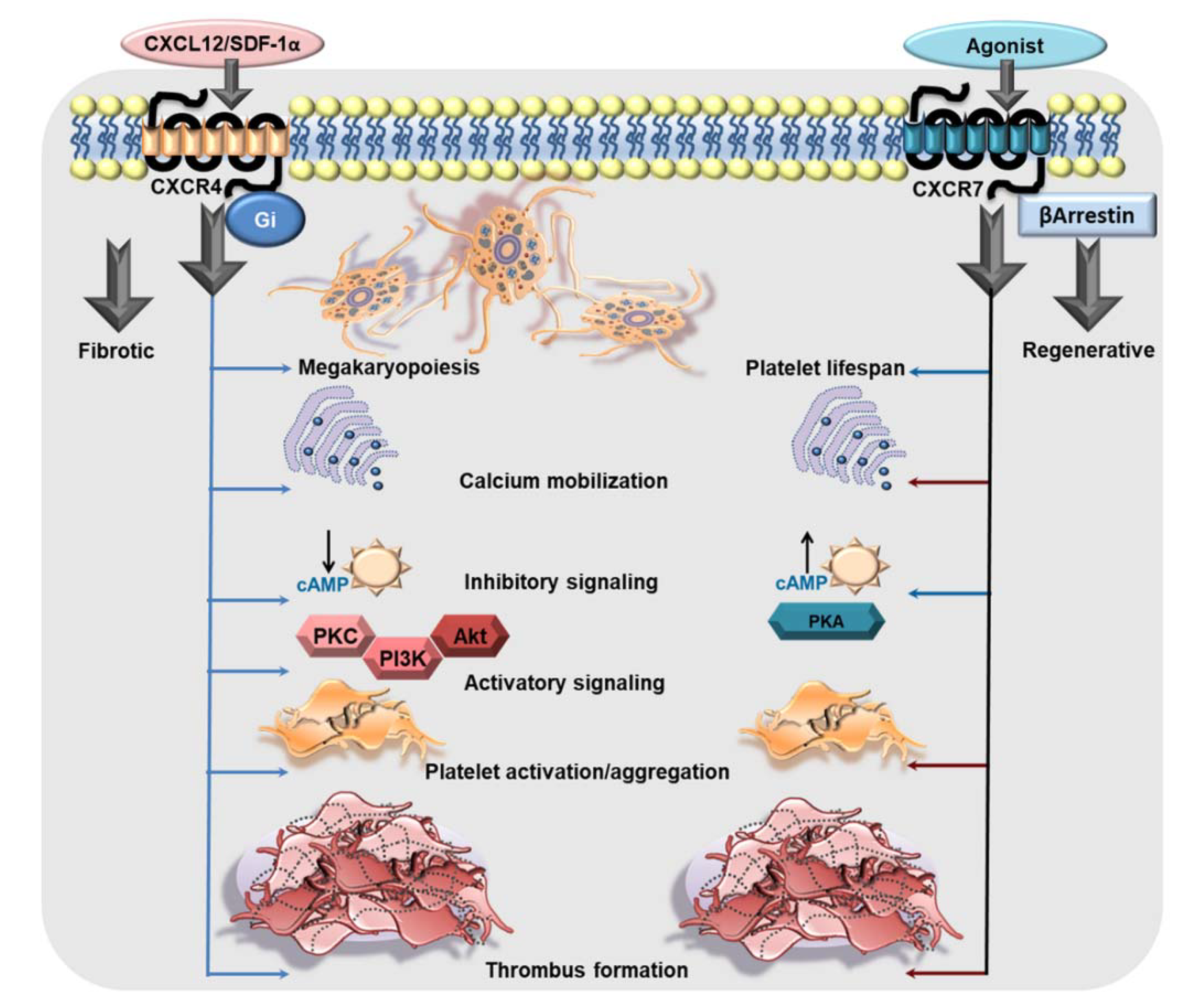
2. The Dichotomy of CXCR4 and ACKR3/CXCR7 in Platelets
2.1. The Typical and the Atypical: Differential Trafficking of CXCR4-CXCR7 in Platelets
2.2. The Pro-Thrombotic Attributes of Canonical CXCR4
2.3. ACKR3/CXCR7 Boosts Platelet Survival
2.4. Atypical Influence of ACKR3/CXCR7 on Thrombotic and Thrombo-Inflammatory Platelet Response
2.4.1. Physiological CXCR7-Agonist MIF
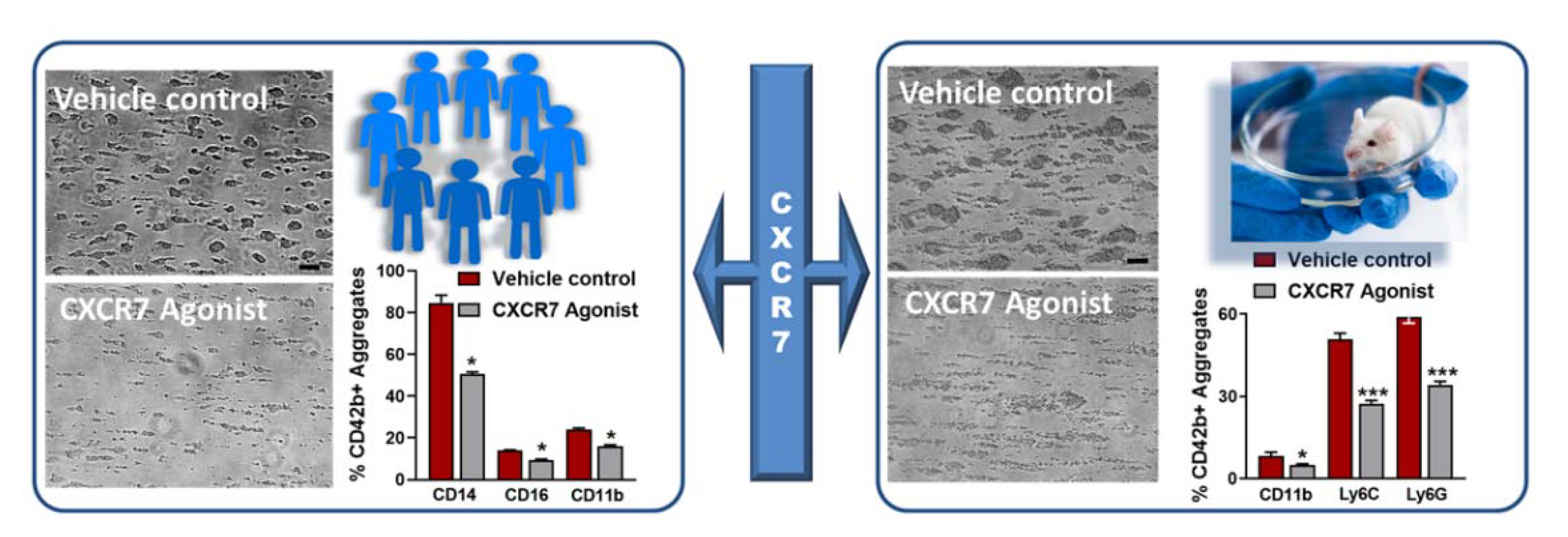
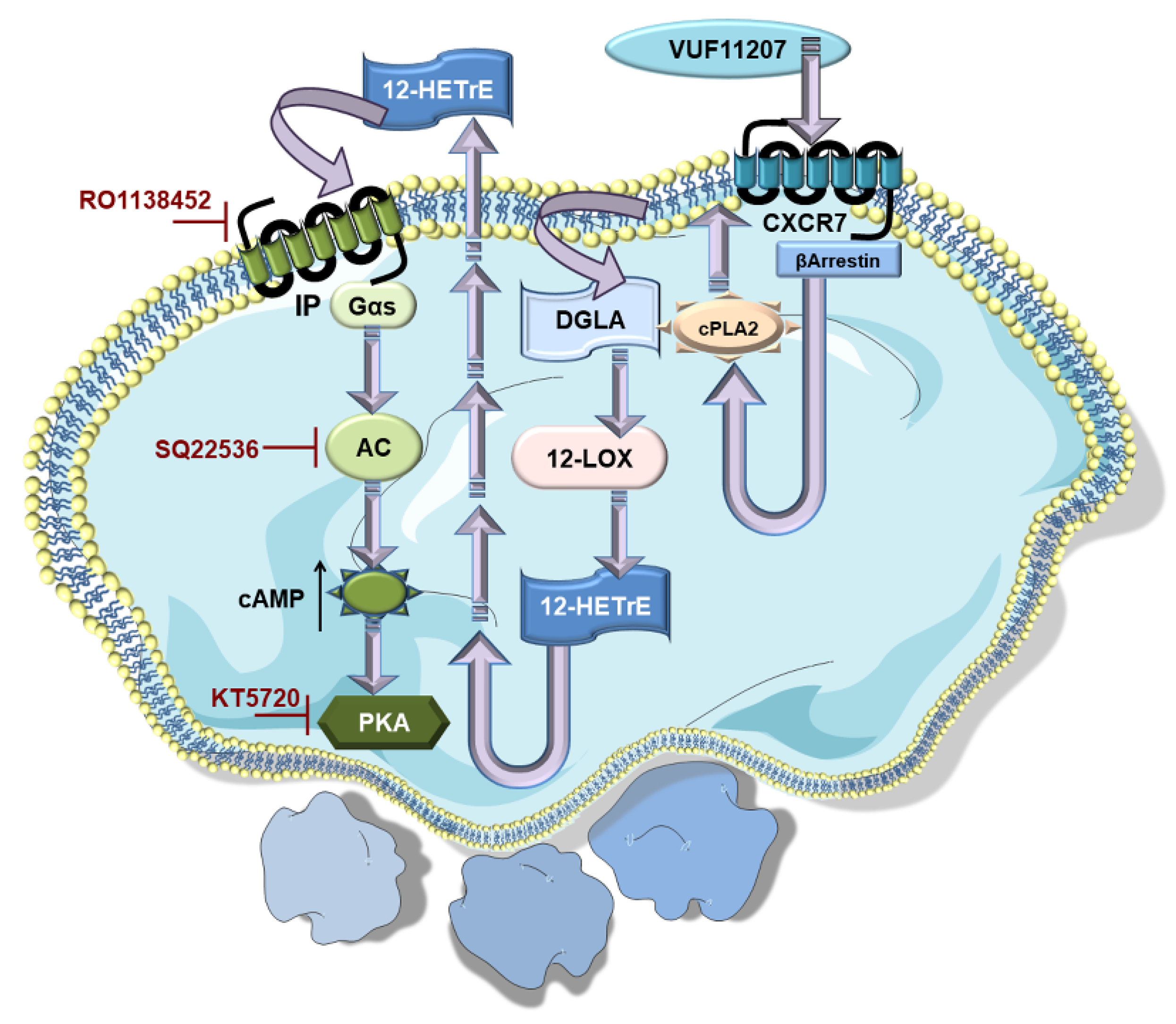
2.4.2. Anti-Thrombotic Effects of Pharmacological Agonist VUF11207
2.4.3. VUF11207 Counteracts Thrombo-Inflammatory Platelet Functions
2.4.4. Unexpected Influence of ACKR3/CXCR7 on the Platelet Lipidome
3. Therapeutic Implication of ACKR3/CXCR7 in Platelet-Associated Cardiovascular Disease
3.1. Therapeutic Potential of ACKR3/CXCR7 against Atheroprogression
3.2. Therapeutic Efficacy of ACKR3/CXCR7 in Limiting Fibrosis and Promoting Tissue Regeneration
3.2.1. Myocardial Regeneration and Functional Recovery
3.2.2. Pulmonary Fibrosis
3.2.3. Hepatic Regeneration
4. Discussion
ACKR3/CXCR7 as an Anti-Thrombotic Drug Target: Considering the Pros and Cons
- ACKR3/CXCR7 is ubiquitously expressed to various extent by circulating immune (monocytes, macrophages, [28] T-lymphocytes [190], B-lymphocytes [191], neutrophils [90,91,92]) and vascular cells (endothelium [27]) and also cells constituting the organs that these blood vessels perfuse (e.g., myocardium [26], brain [30,32,192,193]). Therefore, ACKR3/CXCR7 may elicit a cell- and organ-specific functional response [46], which cannot be restricted specifically to platelets. This necessitates a thorough functional characterization of anticipated cellular targets under the influence of a CXCR7-agonist, to prevent undesirable off-target deleterious effects other than the one intended as an anti-platelet agent.
- It is absolutely essential to consider that ACKR3/CXCR7 exhibits a ligand-specific functional response [194] even in platelets. Although CXCL11, CXCL12/SDF1-α and MIF execute an anti-apoptotic effect through ACKR3/CXCR7, only MIF demonstrates an anti-thrombotic influence [37,40], although this is without affecting other platelet functions like spreading, degranulation and aggregation [74,75]. CXCL12/SDF1-α evidently promotes a pro-thrombotic response but through CXCR4 [21,59,64]. Such discrepancies may arise due to ligand-specific conformational changes upon ACKR3/CXCR7 engagement [194]. A pharmacological CXCR7-agonist intended as an anti-platelet agent will have to be validated for several platelet functions, their interaction with circulatory and vascular cells, and those in the organs (e.g., myocardium) that are infiltrated by activated platelets [5,7,34], to fully apprehend its scope for therapeutic application in cardiovascular pathologies.
- Needless to say, that a detailed mechanistic insight into the anti-platelet mode of action is mandatory to convincingly attribute the observed anti-thrombotic benefits of existing CXCR7-agonists (Table 2) [106,107,109,110,189] or those in development, to be specifically mediated through ACKR3/CXCR7 and not as a pleiotropic effect.
- Over 90% of publications concerning ACKR3/CXCR7 enlisted in PubMed stem from research in the context of various cancers, where ACKR3/CXCR7 has been shown to play a decisive role in cancer progression, angiogenesis and even prognostic outcome [46,195,196,197]. Given the level of our current scientific understanding on ACKR3/CXCR7, it is perhaps neither advisable nor reasonable to continuously trigger this receptor by the prolonged use of an agonist. Therefore, it is essential to ascertain the therapeutic ‘time-window’ during which availability of ACKR3/CXCR7 is observed as a potential anti-platelet drug target in cardiovascular diseases, and to categorically define a dosage and treatment regimen. Our aim should be to utilize the therapeutic benefits of the ACKR3/CXCR7 agonist as an anti-thrombotic agent without inflicting neoplastic changes, and while doing so, to substantiate a functional recovery of the afflicted organs (e.g., myocardium) following thrombo-inflammatory and thrombo-ischemic damage, as effectively demonstrated for myocardial infarction [27,28,76]. The prospect of a ligand and cell-specific response, also biased signaling through ACKR3/CXCR7, can be exploited to fulfill this objective. However, fundamental research on the molecular mechanisms driving signaling events and functional response downstream of ACKR3/CXCR7 needs to be attended to in parallel with translational research initiatives to make productive progress in this direction.
5. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McFadyen, J.D.; Schaff, M.; Peter, K. Current and future antiplatelet therapies: Emphasis on preserving haemostasis. Nat. Rev. Cardiol. 2018, 15, 181–191. [Google Scholar] [CrossRef]
- Rayes, J.; Bourne, J.H.; Brill, A.; Watson, S.P. The dual role of platelet-innate immune cell interactions in thrombo-inflammation. Res. Pract. Thromb. Haemost. 2020, 4, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coenen, D.M.; Heinzmann, A.C.A.; Karel, M.F.A.; Cosemans, J.; Koenen, R.R. The multifaceted contribution of platelets in the emergence and aftermath of acute cardiovascular events. Atherosclerosis 2021, 319, 132–141. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Wang, X.; Peter, K. Platelets in cardiac ischaemia/reperfusion injury: A promising therapeutic target. Cardiovasc. Res. 2019, 115, 1178–1188. [Google Scholar] [CrossRef]
- McFadyen, J.D.; Peter, K. Novel Antithrombotic Drugs on the Horizon: The Ultimate Promise to Prevent Clotting While Avoiding Bleeding. Circ. Res. 2017, 121, 1133–1135. [Google Scholar] [CrossRef]
- Walsh, T.G.; Poole, A.W. Do platelets promote cardiac recovery after myocardial infarction: Roles beyond occlusive ischemic damage. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H1043–H1048. [Google Scholar] [CrossRef]
- Chatterjee, M.; Geisler, T. Inflammatory Contribution of Platelets Revisited: New Players in the Arena of Inflammation. Semin. Thromb. Hemost 2016, 42, 205–214. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair After Myocardial Infarction: From Inflammation to Fibrosis. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Walsh, T.G.; Poole, A.W. Platelets Protect Cardiomyocytes from Ischaemic Damage. TH Open 2017, 1, e24–e32. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Wang, W.; Qu, J.; Croft, L.; Degen, J.L.; Coller, B.S.; Ahamed, J. Platelet TGF-beta1 contributions to plasma TGF-beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 2012, 119, 1064–1074. [Google Scholar] [CrossRef] [Green Version]
- Geisler, T.; Fekecs, L.; Wurster, T.; Chiribiri, A.; Schuster, A.; Nagel, E.; Miller, S.; Gawaz, M.; Stellos, K.; Bigalke, B. Association of platelet-SDF-1 with hemodynamic function and infarct size using cardiac MR in patients with AMI. Eur. J. Radiol. 2012, 81, e486–e490. [Google Scholar] [CrossRef]
- Stellos, K.; Langer, H.; Daub, K.; Schoenberger, T.; Gauss, A.; Geisler, T.; Bigalke, B.; Mueller, I.; Schumm, M.; Schaefer, I.; et al. Platelet-derived stromal cell-derived factor-1 regulates adhesion and promotes differentiation of human CD34+ cells to endothelial progenitor cells. Circulation 2008, 117, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Stellos, K.; Seizer, P.; Bigalke, B.; Daub, K.; Geisler, T.; Gawaz, M. Platelet aggregates-induced human CD34+ progenitor cell proliferation and differentiation to macrophages and foam cells is mediated by stromal cell derived factor 1 in vitro. Semin. Thromb. Hemost. 2010, 36, 139–145. [Google Scholar] [CrossRef]
- Chatterjee, M.; Gawaz, M. Platelet-derived CXCL12 (SDF-1alpha): Basic mechanisms and clinical implications. J. Thromb. Haemost. 2013, 11, 1954–1967. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Buttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Bigalke, B.; Langer, H.; Geisler, T.; Schad, A.; Kogel, A.; Pfaff, F.; Stakos, D.; Seizer, P.; Muller, I.; et al. Expression of stromal-cell-derived factor-1 on circulating platelets is increased in patients with acute coronary syndrome and correlates with the number of CD34+ progenitor cells. Eur. Heart J. 2009, 30, 584–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Bigalke, B.; Borst, O.; Pfaff, F.; Elskamp, A.; Sachsenmaier, S.; Zachmann, R.; Stamatelopoulos, K.; Schonberger, T.; Geisler, T.; et al. Circulating platelet-progenitor cell coaggregate formation is increased in patients with acute coronary syndromes and augments recruitment of CD34+ cells in the ischaemic microcirculation. Eur. Heart J. 2013, 34, 2548–2556. [Google Scholar] [CrossRef]
- Stellos, K.; Gawaz, M. Platelet interaction with progenitor cells: Potential implications for regenerative medicine. Thromb. Haemost. 2007, 98, 922–929. [Google Scholar] [CrossRef]
- Stellos, K.; Gawaz, M. Platelets and stromal cell-derived factor-1 in progenitor cell recruitment. Semin. Thromb. Hemost. 2007, 33, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Rath, D.; Gawaz, M. Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem. Soc. Trans. 2015, 43, 720–726. [Google Scholar] [CrossRef]
- Wurster, T.; Stellos, K.; Haap, M.; Seizer, P.; Geisler, T.; Otton, J.; Indermuehle, A.; Ishida, M.; Schuster, A.; Nagel, E.; et al. Platelet expression of stromal-cell-derived factor-1 (SDF-1): An indicator for ACS? Int. J. Cardiol. 2013, 164, 111–115. [Google Scholar] [CrossRef] [PubMed]
- Rath, D.; Chatterjee, M.; Borst, O.; Muller, K.; Stellos, K.; Mack, A.F.; Bongartz, A.; Bigalke, B.; Langer, H.; Schwab, M.; et al. Expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. Eur. Heart J. 2014, 35, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Rath, D.; Chatterjee, M.; Meyer, L.; Tekath, N.; Olma, C.; Krumm, P.; Adams, C.; Borst, O.; Muller, K.; Droppa, M.; et al. Relative survival potential of platelets is associated with platelet CXCR4/CXCR7 surface exposure and functional recovery following STEMI. Atherosclerosis 2018, 278, 269–277. [Google Scholar] [CrossRef]
- Rath, D.; Chatterjee, M.; Borst, O.; Muller, K.; Langer, H.; Mack, A.F.; Schwab, M.; Winter, S.; Gawaz, M.; Geisler, T. Platelet surface expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 is associated with clinical outcomes in patients with coronary artery disease. J. Thromb. Haemost. 2015, 13, 719–728. [Google Scholar] [CrossRef]
- Ishizuka, M.; Harada, M.; Nomura, S.; Ko, T.; Ikeda, Y.; Guo, J.; Bujo, S.; Yanagisawa-Murakami, H.; Satoh, M.; Yamada, S.; et al. CXCR7 ameliorates myocardial infarction as a beta-arrestin-biased receptor. Sci. Rep. 2021, 11, 3426. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Hu, S.; Chen, H.; Bu, D.; Zhu, L.; Xu, C.; Chu, F.; Huo, X.; Tang, Y.; Sun, X.; et al. Loss of Endothelial CXCR7 Impairs Vascular Homeostasis and Cardiac Remodeling After Myocardial Infarction: Implications for Cardiovascular Drug Discovery. Circulation 2017, 135, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Yue, J.; Ge, Z.; Xie, Y.; Zhang, M.; Jiang, L. Activation of CXCR7 alleviates cardiac insufficiency after myocardial infarction by promoting angiogenesis and reducing apoptosis. Biomed. Pharmacother. 2020, 127, 110168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Lin, S.; Chen, X.; Yao, Y.; Mao, X.; Shao, B.; Zhuge, Q.; Jin, K. SDF-1/CXCR7 Chemokine Signaling is Induced in the Peri-Infarct Regions in Patients with Ischemic Stroke. Aging Dis. 2018, 9, 287–295. [Google Scholar] [CrossRef] [Green Version]
- Dong, B.C.; Li, M.X.; Wang, X.Y.; Cheng, X.; Wang, Y.; Xiao, T.; Jolkkonen, J.; Zhao, C.S.; Zhao, S.S. Effects of CXCR7-neutralizing antibody on neurogenesis in the hippocampal dentate gyrus and cognitive function in the chronic phase of cerebral ischemia. Neural Regen. Res. 2020, 15, 1079–1085. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, Y.; Xin, S.; Wu, M.; Zhang, Y.; Sun, L. CXCR7 suppression modulates macrophage phenotype and function to ameliorate post-myocardial infarction injury. Inflamm. Res. 2020, 69, 523–532. [Google Scholar] [CrossRef]
- Cheng, X.; Wang, H.; Zhang, X.; Zhao, S.; Zhou, Z.; Mu, X.; Zhao, C.; Teng, W. The Role of SDF-1/CXCR4/CXCR7 in Neuronal Regeneration after Cerebral Ischemia. Front. Neurosci. 2017, 11, 590. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Wang, X.; Lim, B.; Leitner, E.; Klingberg, F.; Ching, V.; Yao, Y.; Huang, D.; Gao, X.M.; Kiriazis, H.; et al. Platelet-Targeted Delivery of Peripheral Blood Mononuclear Cells to the Ischemic Heart Restores Cardiac Function after Ischemia-Reperfusion Injury. Theranostics 2017, 7, 3192–3206. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, M.; Hohmann, J.D.; Searle, A.K.; Abraham, M.K.; Nandurkar, H.H.; Wang, X.; Peter, K. A single-chain antibody-CD39 fusion protein targeting activated platelets protects from cardiac ischaemia/reperfusion injury. Eur. Heart J. 2018, 39, 111–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Xia, Y.; Zuo, K.; Wang, Y.; Zhang, S.; Kuang, D.; Duan, Y.; Zhao, X.; Wang, G. Crosstalk between SDF-1/CXCR4 and SDF-1/CXCR7 in cardiac stem cell migration. Sci. Rep. 2015, 5, 16813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Tong, X.; Xia, W.; Chen, L.; Zhang, X.; Yu, B.; Yang, Z.; Tao, J. CXCR7/p-ERK-Signaling Is a Novel Target for Therapeutic Vasculogenesis in Patients with Coronary Artery Disease. PLoS ONE 2016, 11, e0161255. [Google Scholar] [CrossRef]
- Chatterjee, M.; Seizer, P.; Borst, O.; Schonberger, T.; Mack, A.; Geisler, T.; Langer, H.F.; May, A.E.; Vogel, S.; Lang, F.; et al. SDF-1alpha induces differential trafficking of CXCR4-CXCR7 involving cyclophilin A, CXCR7 ubiquitination and promotes platelet survival. FASEB J. 2014, 28, 2864–2878. [Google Scholar] [CrossRef]
- Stellos, K.; Ruf, M.; Sopova, K.; Kilias, A.; Rahmann, A.; Stamatelopoulos, K.; Jorbenadze, R.; Geisler, T.; Gawaz, M.; Bigalke, B. Plasma levels of stromal cell-derived factor-1 in patients with coronary artery disease: Effect of clinical presentation and cardiovascular risk factors. Atherosclerosis 2011, 219, 913–916. [Google Scholar] [CrossRef]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Muller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; Borst, O.; Walker, B.; Fotinos, A.; Vogel, S.; Seizer, P.; Mack, A.; Alampour-Rajabi, S.; Rath, D.; Geisler, T.; et al. Macrophage migration inhibitory factor limits activation-induced apoptosis of platelets via CXCR7-dependent Akt signaling. Circ. Res. 2014, 115, 939–949. [Google Scholar] [CrossRef]
- Kowalska, M.A.; Ratajczak, J.; Hoxie, J.; Brass, L.F.; Gewirtz, A.; Poncz, M.; Ratajczak, M.Z. Megakaryocyte precursors, megakaryocytes and platelets express the HIV co-receptor CXCR4 on their surface: Determination of response to stromal-derived factor-1 by megakaryocytes and platelets. Br. J. Haematol. 1999, 104, 220–229. [Google Scholar] [CrossRef]
- Canals, M.; Scholten, D.J.; de Munnik, S.; Han, M.K.; Smit, M.J.; Leurs, R. Ubiquitination of CXCR7 controls receptor trafficking. PLoS ONE 2012, 7, e34192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, V.; Lue, H.; Kraemer, S.; Korbiel, J.; Krohn, R.; Ohl, K.; Bucala, R.; Weber, C.; Bernhagen, J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009, 583, 2749–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rath, D.; Chatterjee, M.; Bongartz, A.; Muller, K.; Droppa, M.; Stimpfle, F.; Borst, O.; Zuern, C.; Vogel, S.; Gawaz, M.; et al. Platelet surface expression of SDF-1 is associated with clinical outcomes in the patients with cardiovascular disease. Platelets 2017, 28, 34–39. [Google Scholar] [CrossRef]
- Muller, I.I.; Muller, K.A.; Schonleber, H.; Karathanos, A.; Schneider, M.; Jorbenadze, R.; Bigalke, B.; Gawaz, M.; Geisler, T. Macrophage migration inhibitory factor is enhanced in acute coronary syndromes and is associated with the inflammatory response. PLoS ONE 2012, 7, e38376. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.; Engele, J. Functions of the CXCL12 Receptor ACKR3/CXCR7-What Has Been Perceived and What Has Been Overlooked. Mol. Pharmacol. 2020, 98, 577–585. [Google Scholar] [CrossRef]
- Kawaguchi, N.; Zhang, T.T.; Nakanishi, T. Involvement of CXCR4 in Normal and Abnormal Development. Cells 2019, 8, 185. [Google Scholar] [CrossRef] [Green Version]
- Janssens, R.; Struyf, S.; Proost, P. Pathological roles of the homeostatic chemokine CXCL12. Cytokine Growth Factor Rev. 2018, 44, 51–68. [Google Scholar] [CrossRef]
- Teixido, J.; Martinez-Moreno, M.; Diaz-Martinez, M.; Sevilla-Movilla, S. The good and bad faces of the CXCR4 chemokine receptor. Int. J. Biochem. Cell Biol. 2018, 95, 121–131. [Google Scholar] [CrossRef]
- Riviere, C.; Subra, F.; Cohen-Solal, K.; Cordette-Lagarde, V.; Letestu, R.; Auclair, C.; Vainchenker, W.; Louache, F. Phenotypic and functional evidence for the expression of CXCR4 receptor during megakaryocytopoiesis. Blood 1999, 93, 1511–1523. [Google Scholar] [CrossRef]
- Wang, J.F.; Liu, Z.Y.; Groopman, J.E. The alpha-chemokine receptor CXCR4 is expressed on the megakaryocytic lineage from progenitor to platelets and modulates migration and adhesion. Blood 1998, 92, 756–764. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Celeghini, C.; Cutroneo, G.; Di Baldassarre, A.; Rana, R.; Zauli, G. Differential effects of stromal derived factor-1 alpha (SDF-1 alpha) on early and late stages of human megakaryocytic development. Anat. Rec. 2000, 260, 141–147. [Google Scholar] [CrossRef]
- Hamada, T.; Mohle, R.; Hesselgesser, J.; Hoxie, J.; Nachman, R.L.; Moore, M.A.; Rafii, S. Transendothelial migration of megakaryocytes in response to stromal cell-derived factor 1 (SDF-1) enhances platelet formation. J. Exp. Med. 1998, 188, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, N.; Mott, K.; Upcin, B.; Stegner, D.; Schulze, H.; Ergun, S. CXCL12-Abundant Reticular (CAR) Cells Direct Megakaryocyte Protrusions across the Bone Marrow Sinusoid Wall. Cells 2021, 10, 722. [Google Scholar] [CrossRef]
- Kraemer, B.F.; Borst, O.; Gehring, E.M.; Schoenberger, T.; Urban, B.; Ninci, E.; Seizer, P.; Schmidt, C.; Bigalke, B.; Koch, M.; et al. PI3 kinase-dependent stimulation of platelet migration by stromal cell-derived factor 1 (SDF-1). J. Mol. Med. 2010, 88, 1277–1288. [Google Scholar] [CrossRef]
- Kraemer, B.F.; Schmidt, C.; Urban, B.; Bigalke, B.; Schwanitz, L.; Koch, M.; Seizer, P.; Schaller, M.; Gawaz, M.; Lindemann, S. High shear flow induces migration of adherent human platelets. Platelets 2011, 22, 415–421. [Google Scholar] [CrossRef]
- Abi-Younes, S.; Sauty, A.; Mach, F.; Sukhova, G.K.; Libby, P.; Luster, A.D. The stromal cell-derived factor-1 chemokine is a potent platelet agonist highly expressed in atherosclerotic plaques. Circ. Res. 2000, 86, 131–138. [Google Scholar] [CrossRef]
- Gear, A.R.; Suttitanamongkol, S.; Viisoreanu, D.; Polanowska-Grabowska, R.K.; Raha, S.; Camerini, D. Adenosine diphosphate strongly potentiates the ability of the chemokines MDC, TARC, and SDF-1 to stimulate platelet function. Blood 2001, 97, 937–945. [Google Scholar] [CrossRef] [PubMed]
- Walsh, T.G.; Harper, M.T.; Poole, A.W. SDF-1alpha is a novel autocrine activator of platelets operating through its receptor CXCR4. Cell Signal. 2015, 27, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Gear, A.R.; Camerini, D. Platelet chemokines and chemokine receptors: Linking hemostasis, inflammation, and host defense. Microcirculation 2003, 10, 335–350. [Google Scholar] [CrossRef] [PubMed]
- Kowalska, M.A.; Ratajczak, M.Z.; Majka, M.; Jin, J.; Kunapuli, S.; Brass, L.; Poncz, M. Stromal cell-derived factor-1 and macrophage-derived chemokine: 2 chemokines that activate platelets. Blood 2000, 96, 50–57. [Google Scholar] [CrossRef]
- Salim, J.P.; Goette, N.P.; Lev, P.R.; Chazarreta, C.D.; Heller, P.G.; Alvarez, C.; Molinas, F.C.; Marta, R.F. Dysregulation of stromal derived factor 1/CXCR4 axis in the megakaryocytic lineage in essential thrombocythemia. Br. J. Haematol. 2009, 144, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Muller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, H.; Iguchi, T.; Hayashi, M.; Kaneda, M.; Iida, K.; Shimonaka, M.; Hara, T.; Arai, M.; Koike, Y.; Yamamoto, N.; et al. SDF-1alpha/CXCR4 Signaling in Lipid Rafts Induces Platelet Aggregation via PI3 Kinase-Dependent Akt Phosphorylation. PLoS ONE 2017, 12, e0169609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stellos, K.; Sauter, R.; Fahrleitner, M.; Grimm, J.; Stakos, D.; Emschermann, F.; Panagiota, V.; Gnerlich, S.; Perk, A.; Schonberger, T.; et al. Binding of oxidized low-density lipoprotein on circulating platelets is increased in patients with acute coronary syndromes and induces platelet adhesion to vascular wall in vivo—Brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2017–2020. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Tannous, B.A.; Poznansky, M.C.; Chen, H. CXCR4 antagonist AMD3100 (plerixafor): From an impurity to a therapeutic agent. Pharmacol. Res. 2020, 159, 105010. [Google Scholar] [CrossRef] [PubMed]
- Bobkov, V.; Arimont, M.; Zarca, A.; De Groof, T.W.M.; van der Woning, B.; de Haard, H.; Smit, M.J. Antibodies Targeting Chemokine Receptors CXCR4 and ACKR3. Mol. Pharmacol. 2019, 96, 753–764. [Google Scholar] [CrossRef]
- Mujic-Delic, A.; de Wit, R.H.; Verkaar, F.; Smit, M.J. GPCR-targeting nanobodies: Attractive research tools, diagnostics, and therapeutics. Trends Pharmacol. Sci. 2014, 35, 247–255. [Google Scholar] [CrossRef]
- Kaplan, J.E.; Saba, T.M. Platelet removal from the circulation by the liver and spleen. Am. J. Physiol. 1978, 235, H314–H320. [Google Scholar] [CrossRef]
- Montenont, E.; Rondina, M.T.; Campbell, R.A. Altered functions of platelets during aging. Curr. Opin. Hematol. 2019, 26, 336–342. [Google Scholar] [CrossRef]
- McArthur, K.; Chappaz, S.; Kile, B.T. Apoptosis in megakaryocytes and platelets: The life and death of a lineage. Blood 2018, 131, 605–610. [Google Scholar] [CrossRef]
- Davizon-Castillo, P.; Rowley, J.W.; Rondina, M.T. Megakaryocyte and Platelet Transcriptomics for Discoveries in Human Health and Disease. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1432–1440. [Google Scholar] [CrossRef]
- Handtke, S.; Steil, L.; Greinacher, A.; Thiele, T. Toward the Relevance of Platelet Subpopulations for Transfusion Medicine. Front. Med. 2018, 5, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtz, T.H.; Tillmann, S.; Strussmann, T.; Kraemer, S.; Heemskerk, J.W.; Grottke, O.; Gawaz, M.; von Hundelshausen, P.; Bernhagen, J. Platelet-derived MIF: A novel platelet chemokine with distinct recruitment properties. Atherosclerosis 2015, 239, 1–10. [Google Scholar] [CrossRef]
- Strussmann, T.; Tillmann, S.; Wirtz, T.; Bucala, R.; von Hundelshausen, P.; Bernhagen, J. Platelets are a previously unrecognised source of MIF. Thromb. Haemost. 2013, 110, 1004–1013. [Google Scholar] [CrossRef]
- Cebo, M.; Dittrich, K.; Fu, X.; Manke, M.C.; Emschermann, F.; Rheinlaender, J.; von Eysmondt, H.; Ferreiros, N.; Sudmann-Innerhofer, J.; Witte, A.; et al. Platelet ACKR3/CXCR7 Favors Anti-Platelet Lipids over an Atherothrombotic Lipidome and Regulates Thrombo-inflammation. Blood 2021, in press. [Google Scholar] [CrossRef]
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, E.; Montecucco, F.; Portincasa, P.; Sahebkar, A.; Mollazadeh, H.; Carbone, F. Update on pathological platelet activation in coronary thrombosis. J. Cell Physiol. 2019, 234, 2121–2133. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef]
- Oikonomou, E.; Leopoulou, M.; Theofilis, P.; Antonopoulos, A.S.; Siasos, G.; Latsios, G.; Mystakidi, V.C.; Antoniades, C.; Tousoulis, D. A link between inflammation and thrombosis in atherosclerotic cardiovascular diseases: Clinical and therapeutic implications. Atherosclerosis 2020, 309, 16–26. [Google Scholar] [CrossRef] [PubMed]
- d’Alessandro, E.; Becker, C.; Bergmeier, W.; Bode, C.; Bourne, J.H.; Brown, H.; Buller, H.R.; Ten Cate-Hoek, A.J.; Ten Cate, V.; van Cauteren, Y.J.M.; et al. Thrombo-Inflammation in Cardiovascular Disease: An Expert Consensus Document from the Third Maastricht Consensus Conference on Thrombosis. Thromb. Haemost. 2020, 120, 538–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaid, Y.; Puhm, F.; Allaeys, I.; Naya, A.; Oudghiri, M.; Khalki, L.; Limami, Y.; Zaid, N.; Sadki, K.; Ben El Haj, R.; et al. Platelets Can Associate with SARS-Cov-2 RNA and Are Hyperactivated in COVID-19. Circ. Res. 2020, 127, 1404–1418. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Rondina, M.T. The Era of Thromboinflammation: Platelets Are Dynamic Sensors and Effector Cells During Infectious Diseases. Front. Immunol. 2019, 10, 2204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.; Badimon, L.; Mach, F.; van der Vorst, E.P.C. Therapeutic strategies for atherosclerosis and atherothrombosis: Past, present and future. Thromb. Haemost. 2017, 117, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- McFadyen, J.D.; Stevens, H.; Peter, K. The Emerging Threat of (Micro)Thrombosis in COVID-19 and Its Therapeutic Implications. Circ. Res. 2020, 127, 571–587. [Google Scholar] [CrossRef]
- Althaus, K.; Marini, I.; Zlamal, J.; Pelzl, L.; Singh, A.; Haberle, H.; Mehrlander, M.; Hammer, S.; Schulze, H.; Bitzer, M.; et al. Antibody-induced procoagulant platelets in severe COVID-19 infection. Blood 2021, 137, 1061–1071. [Google Scholar] [CrossRef]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, S.; Jin, Y.; Qin, G.; Yu, L.; Zhang, J. Elevated levels of platelet-monocyte aggregates and related circulating biomarkers in patients with acute coronary syndrome. Int. J. Cardiol. 2007, 115, 361–365. [Google Scholar] [CrossRef]
- Salah, H.M.; Mehta, J.L. Meta-Analysis of the Effect of Aspirin on Mortality in COVID-19. Am. J. Cardiol. 2021, 142, 158–159. [Google Scholar] [CrossRef] [PubMed]
- Koenen, J.; Bachelerie, F.; Balabanian, K.; Schlecht-Louf, G.; Gallego, C. Atypical Chemokine Receptor 3 (ACKR3): A Comprehensive Overview of its Expression and Potential Roles in the Immune System. Mol. Pharmacol. 2019, 96, 809–818. [Google Scholar] [CrossRef]
- Ngamsri, K.C.; Muller, A.; Bosmuller, H.; Gamper-Tsigaras, J.; Reutershan, J.; Konrad, F.M. The Pivotal Role of CXCR7 in Stabilization of the Pulmonary Epithelial Barrier in Acute Pulmonary Inflammation. J. Immunol. 2017, 198, 2403–2413. [Google Scholar] [CrossRef] [Green Version]
- Pouzol, L.; Sassi, A.; Baumlin, N.; Tunis, M.; Strasser, D.S.; Lehembre, F.; Martinic, M.M. CXCR7 Antagonism Reduces Acute Lung Injury Pathogenesis. Front. Pharmacol. 2021, 12, 748740. [Google Scholar] [CrossRef]
- Greinacher, A.; Thiele, T.; Warkentin, T.E.; Weisser, K.; Kyrle, P.A.; Eichinger, S. Thrombotic Thrombocytopenia after ChAdOx1 nCov-19 Vaccination. N. Engl. J. Med. 2021, 384, 2092–2101. [Google Scholar] [CrossRef]
- Yeung, J.; Tourdot, B.E.; Adili, R.; Green, A.R.; Freedman, C.J.; Fernandez-Perez, P.; Yu, J.; Holman, T.R.; Holinstat, M. 12(S)-HETrE, a 12-Lipoxygenase Oxylipin of Dihomo-gamma-Linolenic Acid, Inhibits Thrombosis via Galphas Signaling in Platelets. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2068–2077. [Google Scholar] [CrossRef] [Green Version]
- Tourdot, B.E.; Ahmed, I.; Holinstat, M. The emerging role of oxylipins in thrombosis and diabetes. Front. Pharmacol. 2014, 4, 176. [Google Scholar] [CrossRef] [PubMed]
- Harm, T.; Bild, A.; Dittrich, K.; Goldschmied, A.; Nestele, J.; Chatterjee, M.; Fu, X.; Kolb, K.; Castor, T.; Borst, O.; et al. Acute coronary syndrome is associated with a substantial change in the platelet lipidome. Cardiovasc. Res. 2021, cvab238. [Google Scholar] [CrossRef] [PubMed]
- Diehl, P.; Nienaber, F.; Zaldivia, M.T.K.; Stamm, J.; Siegel, P.M.; Mellett, N.A.; Wessinger, M.; Wang, X.; McFadyen, J.D.; Bassler, N.; et al. Lysophosphatidylcholine is a Major Component of Platelet Microvesicles Promoting Platelet Activation and Reporting Atherosclerotic Plaque Instability. Thromb. Haemost. 2019, 119, 1295–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tourdot, B.E.; Adili, R.; Isingizwe, Z.R.; Ebrahem, M.; Freedman, J.C.; Holman, T.R.; Holinstat, M. 12-HETrE inhibits platelet reactivity and thrombosis in part through the prostacyclin receptor. Blood Adv. 2017, 1, 1124–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, J.; Tourdot, B.E.; Fernandez-Perez, P.; Vesci, J.; Ren, J.; Smyrniotis, C.J.; Luci, D.K.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; et al. Platelet 12-LOX is essential for FcgammaRIIa-mediated platelet activation. Blood 2014, 124, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burkard, P.; Vogtle, T.; Nieswandt, B. Platelets in Thrombo-Inflammation: Concepts, Mechanisms, and Therapeutic Strategies for Ischemic Stroke. Hamostaseologie 2020, 40, 153–164. [Google Scholar] [CrossRef]
- Stoll, G.; Nieswandt, B. Thrombo-inflammation in acute ischaemic stroke—Implications for treatment. Nat. Rev. Neurol. 2019, 15, 473–481. [Google Scholar] [CrossRef]
- Chatterjee, M.; Ehrenberg, A.; Toska, L.M.; Metz, L.M.; Klier, M.; Krueger, I.; Reusswig, F.; Elvers, M. Molecular Drivers of Platelet Activation: Unraveling Novel Targets for Anti-Thrombotic and Anti-Thrombo-Inflammatory Therapy. Int. J. Mol. Sci. 2020, 21, 7906. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, M.; Penfold, M.E.; Koenen, R.R.; Thiemann, A.; Heyll, K.; Akhtar, S.; Koyadan, S.; Wu, Z.; Gremse, F.; et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 2014, 129, 1244–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Z.; Lis, R.; Ginsberg, M.; Chavez, D.; Shido, K.; Rabbany, S.Y.; Fong, G.H.; Sakmar, T.P.; Rafii, S.; Ding, B.S. Targeting of the pulmonary capillary vascular niche promotes lung alveolar repair and ameliorates fibrosis. Nat. Med. 2016, 22, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.S.; Cao, Z.; Lis, R.; Nolan, D.J.; Guo, P.; Simons, M.; Penfold, M.E.; Shido, K.; Rabbany, S.Y.; Rafii, S. Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 2014, 505, 97–102. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Li, R.; Wu, J.; Jiang, L.; Zhong, H.A. Drug Design Targeting the CXCR4/CXCR7/CXCL12 Pathway. Curr. Top. Med. Chem. 2016, 16, 1441–1451. [Google Scholar] [CrossRef]
- Wijtmans, M.; Maussang, D.; Sirci, F.; Scholten, D.J.; Canals, M.; Mujic-Delic, A.; Chong, M.; Chatalic, K.L.; Custers, H.; Janssen, E.; et al. Synthesis, modeling and functional activity of substituted styrene-amides as small-molecule CXCR7 agonists. Eur. J. Med. Chem. 2012, 51, 184–192. [Google Scholar] [CrossRef]
- Wang, C.; Chen, W.; Shen, J. CXCR7 Targeting and Its Major Disease Relevance. Front. Pharmacol. 2018, 9, 641. [Google Scholar] [CrossRef] [Green Version]
- Gravel, S.; Malouf, C.; Boulais, P.E.; Berchiche, Y.A.; Oishi, S.; Fujii, N.; Leduc, R.; Sinnett, D.; Heveker, N. The peptidomimetic CXCR4 antagonist TC14012 recruits beta-arrestin to CXCR7: Roles of receptor domains. J. Biol. Chem. 2010, 285, 37939–37943. [Google Scholar] [CrossRef] [Green Version]
- Kalatskaya, I.; Berchiche, Y.A.; Gravel, S.; Limberg, B.J.; Rosenbaum, J.S.; Heveker, N. AMD3100 is a CXCR7 ligand with allosteric agonist properties. Mol. Pharmacol. 2009, 75, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Liu, Y.; Ellison, N.; Shen, J. Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J. Biol. Chem. 2013, 288, 15481–15494. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Liu, Y.; Wang, C.; Zhang, L.; Crocker, L.; Shen, J. Atorvastatin inhibits CXCR7 induction to reduce macrophage migration. Biochem. Pharmacol. 2014, 89, 99–108. [Google Scholar] [CrossRef]
- Jiang, C.; Li, R.; Ma, X.; Hu, H.; Guo, J.; Zhao, J. AMD3100 and SDF1 regulate cellular functions of endothelial progenitor cells and accelerate endothelial regeneration in a rat carotid artery injury model. Mol. Med. Rep. 2020, 22, 3201–3212. [Google Scholar] [CrossRef]
- Saaber, F.; Schutz, D.; Miess, E.; Abe, P.; Desikan, S.; Ashok Kumar, P.; Balk, S.; Huang, K.; Beaulieu, J.M.; Schulz, S.; et al. ACKR3 Regulation of Neuronal Migration Requires ACKR3 Phosphorylation, but Not beta-Arrestin. Cell Rep. 2019, 26, 1473–1488.e1479. [Google Scholar] [CrossRef] [Green Version]
- Walter, H.L.; van der Maten, G.; Antunes, A.R.; Wieloch, T.; Ruscher, K. Treatment with AMD3100 attenuates the microglial response and improves outcome after experimental stroke. J. Neuroinflamm. 2015, 12, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Dai, S.; Wu, W.J.; Tan, W.; Zhu, X.; Mu, J.; Guo, Y.; Bolli, R.; Rokosh, G. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: Role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation 2007, 116, 654–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jujo, K.; Hamada, H.; Iwakura, A.; Thorne, T.; Sekiguchi, H.; Clarke, T.; Ito, A.; Misener, S.; Tanaka, T.; Klyachko, E.; et al. CXCR4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc. Natl. Acad. Sci. USA 2010, 107, 11008–11013. [Google Scholar] [CrossRef] [Green Version]
- Thackeray, J.T.; Derlin, T.; Haghikia, A.; Napp, L.C.; Wang, Y.; Ross, T.L.; Schafer, A.; Tillmanns, J.; Wester, H.J.; Wollert, K.C.; et al. Molecular Imaging of the Chemokine Receptor CXCR4 after Acute Myocardial Infarction. JACC Cardiovasc. Imaging 2015, 8, 1417–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel-Axel, D.; Daub, K.; Seizer, P.; Lindemann, S.; Gawaz, M. Platelet lipoprotein interplay: Trigger of foam cell formation and driver of atherosclerosis. Cardiovasc. Res. 2008, 78, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef]
- Wisler, J.W.; DeWire, S.M.; Whalen, E.J.; Violin, J.D.; Drake, M.T.; Ahn, S.; Shenoy, S.K.; Lefkowitz, R.J. A unique mechanism of beta-blocker action: Carvedilol stimulates beta-arrestin signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 16657–16662. [Google Scholar] [CrossRef] [Green Version]
- Ryba, D.M.; Li, J.; Cowan, C.L.; Russell, B.; Wolska, B.M.; Solaro, R.J. Long-Term Biased beta-Arrestin Signaling Improves Cardiac Structure and Function in Dilated Cardiomyopathy. Circulation 2017, 135, 1056–1070. [Google Scholar] [CrossRef] [Green Version]
- Escot, S.; Blavet, C.; Hartle, S.; Duband, J.L.; Fournier-Thibault, C. Misregulation of SDF1-CXCR4 signaling impairs early cardiac neural crest cell migration leading to conotruncal defects. Circ. Res. 2013, 113, 505–516. [Google Scholar] [CrossRef] [Green Version]
- LaRocca, T.J.; Altman, P.; Jarrah, A.A.; Gordon, R.; Wang, E.; Hadri, L.; Burke, M.W.; Haddad, G.E.; Hajjar, R.J.; Tarzami, S.T. CXCR4 Cardiac Specific Knockout Mice Develop a Progressive Cardiomyopathy. Int. J. Mol. Sci. 2019, 20, 2267. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, U.; Ghalayini, W.; Dong, F.; Weber, K.; Zou, Y.R.; Rabbany, S.Y.; Rafii, S.; Penn, M.S. Role of cardiac myocyte CXCR4 expression in development and left ventricular remodeling after acute myocardial infarction. Circ. Res. 2010, 107, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Sierro, F.; Biben, C.; Martinez-Munoz, L.; Mellado, M.; Ransohoff, R.M.; Li, M.; Woehl, B.; Leung, H.; Groom, J.; Batten, M.; et al. Disrupted cardiac development but normal hematopoiesis in mice deficient in the second CXCL12/SDF-1 receptor, CXCR7. Proc. Natl. Acad. Sci. USA 2007, 104, 14759–14764. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Crawford, D.; Tsuchihashi, T.; Behrens, T.W.; Srivastava, D. The chemokine receptor CXCR7 functions to regulate cardiac valve remodeling. Dev. Dyn. 2011, 240, 384–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betterman, K.L.; Harvey, N.L. Decoys and cardiovascular development: CXCR7 and regulation of adrenomedullin signaling. Dev. Cell 2014, 30, 490–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, K.R.; Karpinich, N.O.; Espenschied, S.T.; Willcockson, H.H.; Dunworth, W.P.; Hoopes, S.L.; Kushner, E.J.; Bautch, V.L.; Caron, K.M. Decoy receptor CXCR7 modulates adrenomedullin-mediated cardiac and lymphatic vascular development. Dev. Cell 2014, 30, 528–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerrits, H.; van Ingen Schenau, D.S.; Bakker, N.E.; van Disseldorp, A.J.; Strik, A.; Hermens, L.S.; Koenen, T.B.; Krajnc-Franken, M.A.; Gossen, J.A. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis 2008, 46, 235–245. [Google Scholar] [CrossRef]
- Ceholski, D.K.; Turnbull, I.C.; Pothula, V.; Lecce, L.; Jarrah, A.A.; Kho, C.; Lee, A.; Hadri, L.; Costa, K.D.; Hajjar, R.J.; et al. CXCR4 and CXCR7 play distinct roles in cardiac lineage specification and pharmacologic beta-adrenergic response. Stem Cell Res. 2017, 23, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Ghadge, S.K.; Messner, M.; Seiringer, H.; Maurer, T.; Staggl, S.; Zeller, T.; Muller, C.; Bornigen, D.; Weninger, W.J.; Geyer, S.H.; et al. Smooth Muscle Specific Ablation of CXCL12 in Mice Downregulates CXCR7 Associated with Defective Coronary Arteries and Cardiac Hypertrophy. Int. J. Mol. Sci. 2021, 22, 5908. [Google Scholar] [CrossRef] [PubMed]
- Hess, A.; Derlin, T.; Koenig, T.; Diekmann, J.; Wittneben, A.; Wang, Y.; Wester, H.J.; Ross, T.L.; Wollert, K.C.; Bauersachs, J.; et al. Molecular imaging-guided repair after acute myocardial infarction by targeting the chemokine receptor CXCR4. Eur. Heart J. 2020, 41, 3564–3575. [Google Scholar] [CrossRef] [PubMed]
- Proulx, C.; El-Helou, V.; Gosselin, H.; Clement, R.; Gillis, M.A.; Villeneuve, L.; Calderone, A. Antagonism of stromal cell-derived factor-1alpha reduces infarct size and improves ventricular function after myocardial infarction. Pflugers Arch. 2007, 455, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Jujo, K.; Ii, M.; Sekiguchi, H.; Klyachko, E.; Misener, S.; Tanaka, T.; Tongers, J.; Roncalli, J.; Renault, M.A.; Thorne, T.; et al. CXC-chemokine receptor 4 antagonist AMD3100 promotes cardiac functional recovery after ischemia/reperfusion injury via endothelial nitric oxide synthase-dependent mechanism. Circulation 2013, 127, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roncalli, J.; Renault, M.A.; Tongers, J.; Misener, S.; Thorne, T.; Kamide, C.; Jujo, K.; Tanaka, T.; Ii, M.; Klyachko, E.; et al. Sonic hedgehog-induced functional recovery after myocardial infarction is enhanced by AMD3100-mediated progenitor-cell mobilization. J. Am. Coll. Cardiol. 2011, 57, 2444–2452. [Google Scholar] [CrossRef] [Green Version]
- Lohse, M.J.; Engelhardt, S.; Eschenhagen, T. What is the role of beta-adrenergic signaling in heart failure? Circ. Res. 2003, 93, 896–906. [Google Scholar] [CrossRef]
- Noor, N.; Patel, C.B.; Rockman, H.A. Beta-arrestin: A signaling molecule and potential therapeutic target for heart failure. J. Mol. Cell Cardiol. 2011, 51, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Davidson, S.M.; Andreadou, I.; Barile, L.; Birnbaum, Y.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Downey, J.M.; Girao, H.; Pagliaro, P.; Penna, C.; et al. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc. Res. 2019, 115, 1156–1166. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Andreadou, I.; Cabrera-Fuentes, H.A.; Devaux, Y.; Frangogiannis, N.G.; Frantz, S.; Guzik, T.; Liehn, E.A.; Gomes, C.P.C.; Schulz, R.; Hausenloy, D.J. Immune cells as targets for cardioprotection: New players and novel therapeutic opportunities. Cardiovasc. Res. 2019, 115, 1117–1130. [Google Scholar] [CrossRef] [Green Version]
- Hohmann, J.D.; Wang, X.; Krajewski, S.; Selan, C.; Haller, C.A.; Straub, A.; Chaikof, E.L.; Nandurkar, H.H.; Hagemeyer, C.E.; Peter, K. Delayed targeting of CD39 to activated platelet GPIIb/IIIa via a single-chain antibody: Breaking the link between antithrombotic potency and bleeding? Blood 2013, 121, 3067–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, M.; Haigh, K.; Nguyen, T.; Wang, X.; Lim, B.; Yap, M.L.; Eddy, E.M.; Haigh, J.J.; Peter, K. The pulmonary microvasculature entraps induced vascular progenitor cells (iVPCs) systemically delivered after cardiac ischemia-reperfusion injury: Indication for preservation of heart function via paracrine effects beyond engraftment. Microcirculation 2019, 26, e12493. [Google Scholar] [CrossRef]
- Ziegler, M.; Alt, K.; Paterson, B.M.; Kanellakis, P.; Bobik, A.; Donnelly, P.S.; Hagemeyer, C.E.; Peter, K. Highly Sensitive Detection of Minimal Cardiac Ischemia using Positron Emission Tomography Imaging of Activated Platelets. Sci. Rep. 2016, 6, 38161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Peter, K. Molecular Imaging of Atherothrombotic Diseases: Seeing Is Believing. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Keane, C.; Tilley, D.; Cunningham, A.; Smolenski, A.; Kadioglu, A.; Cox, D.; Jenkinson, H.F.; Kerrigan, S.W. Invasive Streptococcus pneumoniae trigger platelet activation via Toll-like receptor 2. J. Thromb. Haemost. 2010, 8, 2757–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Stoppelaar, S.F.; Claushuis, T.A.; Schaap, M.C.; Hou, B.; van der Poll, T.; Nieuwland, R.; van ’t Veer, C. Toll-Like Receptor Signalling Is Not Involved in Platelet Response to Streptococcus pneumoniae In Vitro or In Vivo. PLoS ONE 2016, 11, e0156977. [Google Scholar] [CrossRef]
- Amison, R.T.; O’Shaughnessy, B.G.; Arnold, S.; Cleary, S.J.; Nandi, M.; Pitchford, S.C.; Bragonzi, A.; Page, C.P. Platelet Depletion Impairs Host Defense to Pulmonary Infection with Pseudomonas aeruginosa in Mice. Am. J. Respir. Cell Mol. Biol. 2018, 58, 331–340. [Google Scholar] [CrossRef] [Green Version]
- Boilard, E.; Pare, G.; Rousseau, M.; Cloutier, N.; Dubuc, I.; Levesque, T.; Borgeat, P.; Flamand, L. Influenza virus H1N1 activates platelets through FcgammaRIIA signaling and thrombin generation. Blood 2014, 123, 2854–2863. [Google Scholar] [CrossRef]
- Hottz, E.D.; Bozza, F.A.; Bozza, P.T. Platelets in Immune Response to Virus and Immunopathology of Viral Infections. Front. Med. 2018, 5, 121. [Google Scholar] [CrossRef]
- Pitchford, S.; Cleary, S.; Arkless, K.; Amison, R. Pharmacological strategies for targeting platelet activation in asthma. Curr. Opin. Pharmacol. 2019, 46, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Takeda, T.; Morita, H.; Saito, H.; Matsumoto, K.; Matsuda, A. Recent advances in understanding the roles of blood platelets in the pathogenesis of allergic inflammation and bronchial asthma. Allergol. Int. 2018, 67, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Mallah, H.; Ball, S.; Sekhon, J.; Parmar, K.; Nugent, K. Platelets in chronic obstructive pulmonary disease: An update on pathophysiology and implications for antiplatelet therapy. Respir. Med. 2020, 171, 106098. [Google Scholar] [CrossRef] [PubMed]
- Middleton, E.A.; Rondina, M.T.; Schwertz, H.; Zimmerman, G.A. Amicus or Adversary Revisited: Platelets in Acute Lung Injury and Acute Respiratory Distress Syndrome. Am. J. Respir. Cell Mol. Biol. 2018, 59, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Chebbo, M.; Duez, C.; Alessi, M.C.; Chanez, P.; Gras, D. Platelets: A potential role in chronic respiratory diseases? Eur. Respir. Rev. 2021, 30, 210062. [Google Scholar] [CrossRef]
- Kornerup, K.N.; Page, C.P. The role of platelets in the pathophysiology of asthma. Platelets 2007, 18, 319–328. [Google Scholar] [CrossRef]
- Laidlaw, T.M.; Kidder, M.S.; Bhattacharyya, N.; Xing, W.; Shen, S.; Milne, G.L.; Castells, M.C.; Chhay, H.; Boyce, J.A. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood 2012, 119, 3790–3798. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Wei, T.; Dong, L.; Wang, Q.; Wu, Z.; Yan, Q.; Zhang, W.; Lu, Y.; Wu, M. Cangrelor alleviates bleomycin-induced pulmonary fibrosis by inhibiting platelet activation in mice. Mol. Immunol. 2020, 120, 83–92. [Google Scholar] [CrossRef]
- Diehl, A.M. Neighborhood watch orchestrates liver regeneration. Nat. Med. 2012, 18, 497–499. [Google Scholar] [CrossRef]
- Caligiuri, A.; Gentilini, A.; Pastore, M.; Gitto, S.; Marra, F. Cellular and Molecular Mechanisms Underlying Liver Fibrosis Regression. Cells 2021, 10, 2759. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Espinoza, L.; Huch, M. The balancing act of the liver: Tissue regeneration versus fibrosis. J. Clin. Investig. 2018, 128, 85–96. [Google Scholar] [CrossRef] [Green Version]
- Khurana, A.; Sayed, N.; Allawadhi, P.; Weiskirchen, R. It’s all about the spaces between cells: Role of extracellular matrix in liver fibrosis. Ann. Transl. Med. 2021, 9, 728. [Google Scholar] [CrossRef] [PubMed]
- DeLeve, L.D.; Wang, X.; Wang, L. VEGF-sdf1 recruitment of CXCR7+ bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G739–G746. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Brunnthaler, L.; Panhuber, A.; Starlinger, P.; Assinger, A. Till Death Do Us Part—The Multifaceted Role of Platelets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 3113. [Google Scholar] [CrossRef]
- Chauhan, A.; Adams, D.H.; Watson, S.P.; Lalor, P.F. Platelets: No longer bystanders in liver disease. Hepatology 2016, 64, 1774–1784. [Google Scholar] [CrossRef]
- Grozovsky, R.; Giannini, S.; Falet, H.; Hoffmeister, K.M. Novel mechanisms of platelet clearance and thrombopoietin regulation. Curr. Opin. Hematol. 2015, 22, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Roberts, L.N.; Bernal, W. Incidence of Bleeding and Thrombosis in Patients with Liver Disease. Semin. Thromb. Hemost. 2020, 46, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbalpha is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Lisman, T.; Porte, R.J. The role of platelets in liver inflammation and regeneration. Semin. Thromb. Hemost. 2010, 36, 170–174. [Google Scholar] [CrossRef]
- Shido, K.; Chavez, D.; Cao, Z.; Ko, J.; Rafii, S.; Ding, B.S. Platelets prime hematopoietic and vascular niche to drive angiocrine-mediated liver regeneration. Signal Transduct. Target. Ther. 2017, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Offermanns, S. Activation of platelet function through G protein-coupled receptors. Circ. Res. 2006, 99, 1293–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thibeault, P.E.; Ramachandran, R. Biased signaling in platelet G-protein coupled receptors. Can. J. Physiol. Pharmacol. 2021, 99, 255–269. [Google Scholar] [CrossRef]
- Meyer zu Heringdorf, D.; Jakobs, K.H. Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim. Biophys. Acta 2007, 1768, 923–940. [Google Scholar] [CrossRef] [Green Version]
- Dowal, L.; Flaumenhaft, R. Targeting platelet G-protein coupled receptors (GPCRs): Looking beyond conventional GPCR antagonism. Curr. Vasc. Pharmacol. 2010, 8, 140–154. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; Insel, P.A. Inflammation and thrombosis in COVID-19 pathophysiology: Proteinase-activated and purinergic receptors as drivers and candidate therapeutic targets. Physiol. Rev. 2021, 101, 545–567. [Google Scholar] [CrossRef]
- Alarabi, A.B.; Karim, Z.A.; Hinojos, V.; Lozano, P.A.; Hernandez, K.R.; Montes Ramirez, J.E.; Ali, H.E.A.; Khasawneh, F.T.; Alshbool, F.Z. The G-protein betagamma subunits regulate platelet function. Life Sci. 2020, 262, 118481. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.A.; Chatterjee, M.; Rath, D.; Geisler, T. Platelets, inflammation and anti-inflammatory effects of antiplatelet drugs in ACS and CAD. Thromb. Haemost. 2015, 114, 498–518. [Google Scholar] [CrossRef]
- Capodanno, D.; Bhatt, D.L.; Eikelboom, J.W.; Fox, K.A.A.; Geisler, T.; Michael Gibson, C.; Gonzalez-Juanatey, J.R.; James, S.; Lopes, R.D.; Mehran, R.; et al. Dual-pathway inhibition for secondary and tertiary antithrombotic prevention in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 242–257. [Google Scholar] [CrossRef] [PubMed]
- Sibbing, D.; Aradi, D.; Jacobshagen, C.; Gross, L.; Trenk, D.; Geisler, T.; Orban, M.; Gori, T.; Hadamitzky, M.; Merkely, B.; et al. A randomised trial on platelet function-guided de-escalation of antiplatelet treatment in ACS patients undergoing PCI. Rationale and design of the Testing Responsiveness to Platelet Inhibition on Chronic Antiplatelet Treatment for Acute Coronary Syndromes (TROPICAL-ACS) Trial. Thromb. Haemost. 2017, 117, 188–195. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-arrestin—But not G protein-mediated signaling by the "decoy" receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naumann, U.; Cameroni, E.; Pruenster, M.; Mahabaleshwar, H.; Raz, E.; Zerwes, H.G.; Rot, A.; Thelen, M. CXCR7 functions as a scavenger for CXCL12 and CXCL11. PLoS ONE 2010, 5, e9175. [Google Scholar] [CrossRef] [Green Version]
- Mahabaleshwar, H.; Tarbashevich, K.; Nowak, M.; Brand, M.; Raz, E. beta-arrestin control of late endosomal sorting facilitates decoy receptor function and chemokine gradient formation. Development 2012, 139, 2897–2902. [Google Scholar] [CrossRef] [Green Version]
- Tobia, C.; Chiodelli, P.; Barbieri, A.; Buraschi, S.; Ferrari, E.; Mitola, S.; Borsani, G.; Guerra, J.; Presta, M. Atypical Chemokine Receptor 3 Generates Guidance Cues for CXCL12-Mediated Endothelial Cell Migration. Front. Immunol. 2019, 10, 1092. [Google Scholar] [CrossRef] [PubMed]
- Mahabaleshwar, H.; Boldajipour, B.; Raz, E. Killing the messenger: The role of CXCR7 in regulating primordial germ cell migration. Cell Adh. Migr. 2008, 2, 69–70. [Google Scholar] [CrossRef] [Green Version]
- Uto-Konomi, A.; McKibben, B.; Wirtz, J.; Sato, Y.; Takano, A.; Nanki, T.; Suzuki, S. CXCR7 agonists inhibit the function of CXCL12 by down-regulation of CXCR4. Biochem. Biophys. Res. Commun. 2013, 431, 772–776. [Google Scholar] [CrossRef]
- Boehm, M.; Beaumont, K.; Jones, R.; Kalgutkar, A.S.; Zhang, L.; Atkinson, K.; Bai, G.; Brown, J.A.; Eng, H.; Goetz, G.H.; et al. Discovery of Potent and Orally Bioavailable Macrocyclic Peptide-Peptoid Hybrid CXCR7 Modulators. J. Med. Chem. 2017, 60, 9653–9663. [Google Scholar] [CrossRef] [PubMed]
- Adlere, I.; Caspar, B.; Arimont, M.; Dekkers, S.; Visser, K.; Stuijt, J.; de Graaf, C.; Stocks, M.; Kellam, B.; Briddon, S.; et al. Modulators of CXCR4 and CXCR7/ACKR3 Function. Mol. Pharmacol. 2019, 96, 737–752. [Google Scholar] [CrossRef] [Green Version]
- Lounsbury, N. Advances in CXCR7 Modulators. Pharmaceuticals 2020, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Tripathi, V.; Ahmad, M.; Nath, N.; Mir, R.A.; Chauhan, S.S.; Luthra, K. CXCR7 mediated Gialpha independent activation of ERK and Akt promotes cell survival and chemotaxis in T cells. Cell Immunol. 2012, 272, 230–241. [Google Scholar] [CrossRef]
- Humpert, M.L.; Pinto, D.; Jarrossay, D.; Thelen, M. CXCR7 influences the migration of B cells during maturation. Eur. J. Immunol. 2014, 44, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Banisadr, G.; Podojil, J.R.; Miller, S.D.; Miller, R.J. Pattern of CXCR7 Gene Expression in Mouse Brain Under Normal and Inflammatory Conditions. J. Neuroimmune Pharmacol. 2016, 11, 26–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schonemeier, B.; Kolodziej, A.; Schulz, S.; Jacobs, S.; Hoellt, V.; Stumm, R. Regional and cellular localization of the CXCl12/SDF-1 chemokine receptor CXCR7 in the developing and adult rat brain. J. Comp. Neurol. 2008, 510, 207–220. [Google Scholar] [CrossRef]
- Montpas, N.; St-Onge, G.; Nama, N.; Rhainds, D.; Benredjem, B.; Girard, M.; Hickson, G.; Pons, V.; Heveker, N. Ligand-specific conformational transitions and intracellular transport are required for atypical chemokine receptor 3-mediated chemokine scavenging. J. Biol. Chem. 2018, 293, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Freitas, C.; Desnoyer, A.; Meuris, F.; Bachelerie, F.; Balabanian, K.; Machelon, V. The relevance of the chemokine receptor ACKR3/CXCR7 on CXCL12-mediated effects in cancers with a focus on virus-related cancers. Cytokine Growth Factor Rev. 2014, 25, 307–316. [Google Scholar] [CrossRef]
- Santagata, S.; Ierano, C.; Trotta, A.M.; Capiluongo, A.; Auletta, F.; Guardascione, G.; Scala, S. CXCR4 and CXCR7 Signaling Pathways: A Focus on the Cross-Talk Between Cancer Cells and Tumor Microenvironment. Front. Oncol. 2021, 11, 591386. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Riese, D.J., 2nd; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef] [PubMed]
- Puchert, M.; Engele, J. The peculiarities of the SDF-1/CXCL12 system: In some cells, CXCR4 and CXCR7 sing solos, in others, they sing duets. Cell Tissue Res. 2014, 355, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Berahovich, R.D.; Zabel, B.A.; Penfold, M.E.; Lewen, S.; Wang, Y.; Miao, Z.; Gan, L.; Pereda, J.; Dias, J.; Slukvin, II; et al. CXCR7 protein is not expressed on human or mouse leukocytes. J. Immunol. 2010, 185, 5130–5139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Martin, L.; Sanchez-Mateos, P.; Cabanas, C. CXCR7 impact on CXCL12 biology and disease. Trends Mol. Med. 2013, 19, 12–22. [Google Scholar] [CrossRef]
- Odemis, V.; Lipfert, J.; Kraft, R.; Hajek, P.; Abraham, G.; Hattermann, K.; Mentlein, R.; Engele, J. The presumed atypical chemokine receptor CXCR7 signals through G(i/o) proteins in primary rodent astrocytes and human glioma cells. Glia 2012, 60, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Reyes-Alcaraz, A.; Yong, H.J.; Nguyen, L.P.; Park, H.K.; Inoue, A.; Lee, C.S.; Seong, J.Y.; Hwang, J.I. CXCR7: A beta-arrestin-biased receptor that potentiates cell migration and recruits beta-arrestin2 exclusively through Gbetagamma subunits and GRK2. Cell Biosci. 2020, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The chemokine SDF-1/CXCL12 binds to and signals through the orphan receptor RDC1 in T lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (Cardio) Vascular Pathophysiology | Animal Models Used | Mouse Line | Functional Evaluation | Ref. |
|---|---|---|---|---|
| Atherosclerosis | Wire injury of carotid arteries | CAG-CreERTMCxcr7flox/flox, CAG-CreERTMCxcr7flox/floxApoe−/− (ubiquitous cxcr7 deletion) | Increased neointima formation, lesional macrophage accumulation after vascular injury. | [103] |
| Increased serum cholesterol levels and hyperlipidemia-induced monocytosis. | ||||
| Neointimal hyperplasia | Endothelial denudation of the femoral artery by angioplasty wire inflicted injury | CXCR7f/fCdh5-CreERT2+ conditional endothelial cxcr7 deficient mice | Increased neointimal hyperplasia and neointima/media thickness ratio | [27] |
| Angiogenesis | Hind-limb ischemia (femoral artery ligation, laser Doppler imaging) | CXCR7f/fCdh5-CreERT2+ | Reduced blood flow recovery, reduced vascular density in the ischemic gastrocnemius | [27] |
| Pulmonary fibrosis | Acute and chronic Intratracheal administration of bleomycin or hydrochloric acid | VE-cadherin–CreERT2Cxcr7loxP/loxP, Cxcr7iΔEC/iΔEC | Intra-tracheal instillation of TC14012 reduces pulmonary fibrosis (SMA, collagen I expression) and notch signaling in control mice | [104] |
| Pulmonary inflammation | Mice exposed to nebulized LPS; CXCR7 antagonist CCX771 (10 mg/kg body weight, s.c.) | C57BL/6 mice | Reduced transepithelial migration, release of neutrophil chemoattractant, neutrophil MPO activity and oxidative burst; decreased microvascular permeability in treated mice | [91] |
| Hepatic fibrosis | Single and repeated injections of CCl4 for acute and chronic injury; bile duct ligation induced cholestasis model | VE-cad-CreERT2Cxcr7loxP/loxP Endothelial cxcr7 knock out | Impaired hepatic regeneration due to diminished Id1-mediated generation of angiocrine factors; increased fibrotic response | [105] |
| Myocardial infarction | Permanent ligation of the left anterior descending (LAD) coronary artery | CXCR7f/fCdh5-CreERT2+ inducible endothelial cxcr7 knockout mice | Increased infarct size, reduced vascular density at the infarcted region, impaired cardiac function and remodeling post-MI, increased mortality | [27] |
| Adenoviral delivery of cxcr7 via left ventricle | C57BL/6 mice | Decreased infarct size, improved cardiac function | [27] | |
| LAD ligation | αMHC-Cre+/− CXCR7flox/flox; cardiomyocyte-specific Cxcr7 knockout mice | Normal heart phenotype but prominent left ventricular enlargement and systolic dysfunction post MI | [26] | |
| LAD ligation | Col1a2-CreERT2+/− CXCR7flox/flox; fibroblast-specific knockout mice | No significant reduction in heart Cxcr7 expression, weight, left ventricular volume, and systolic function under basal condition or after MI | [26] | |
| LAD ligation for 30 min, MI/IR; VUF11207 (i.v. pre-MI) | C57BL/6 mice | Reduced infarct size, less deteriorated LVEF 24 h post MI/IR | [76] |
| CXCR7 Agonist | Type | Tested Therapeutic Potential in | Ref. |
|---|---|---|---|
| VUF11207 | Small molecule agonist | Platelet-inhibition; reduced arterial thrombosis | [76] |
| Reduced thrombo-inflammatory response post MI/IR, arterial injury, that induced by HIT-IgG ex vivo | |||
| Reduced infarct size, less deteriorated LVEF post-MI | |||
| ChemoCentryx CCX771 | Small molecule agonist | Reduced atheroprogression following vascular injury in hyperlipidemic Apoe−/− mice | [103] |
| Reduced serum cholesterol and triglyceride levels in hyperlipidemic Ldlr−/− mice | |||
| TC14012 | Cyclic peptide agonist | Myocardial regeneration, functional recovery post-MI | [27] |
| Neovascularization and myocardial regeneration post-MI | [28] | ||
| Reduced pulmonary fibrosis | [104] | ||
| Improved hepatic regeneration, reduced fibrosis | [105] | ||
| AMD3100 | Small molecule allosteric agonist | Myocardial recovery post-MI | [116,117,118] |
| Reduced microglial activation, improved outcome following experimental ischemic stroke | [115] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, M. Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology. Cells 2022, 11, 213. https://doi.org/10.3390/cells11020213
Chatterjee M. Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology. Cells. 2022; 11(2):213. https://doi.org/10.3390/cells11020213
Chicago/Turabian StyleChatterjee, Madhumita. 2022. "Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology" Cells 11, no. 2: 213. https://doi.org/10.3390/cells11020213
APA StyleChatterjee, M. (2022). Atypical Roles of the Chemokine Receptor ACKR3/CXCR7 in Platelet Pathophysiology. Cells, 11(2), 213. https://doi.org/10.3390/cells11020213